

Abstract
Glioblastoma stem-like cells (GSC) promote tumor initiation, progression and therapeutic resistance. Here we show how GSC can be targeted by the FDA approved drug mibefradil which inhibits the T-type calcium channel Cav3.2. This calcium channel was highly expressed in human GBM specimens and enriched in GSC. Analyses of the TCGA and REMBRANDT databases confirmed upregulation of Cav3.2 in a subset of tumors and showed that overexpression associated with worse prognosis. Mibefradil treatment or RNAi-mediated attenuation of Cav3.2 was sufficient to inhibit the growth, survival and stemness of GSC, and also sensitized them to temozolomide (TMZ) chemotherapy. Proteomic and transcriptomic analyses revealed that Cav3.2 inhibition altered cancer signaling pathways and gene transcription. Cav3.2 inhibition suppressed GSC growth in part by inhibiting pro-survival AKT/mTOR pathways and stimulating pro-apoptotic survivin and BAX pathways. Further, Cav3.2 inhibition decreased expression of oncogenes (PDGFA, PDGFB, and TGFB1) and increased expression of tumor suppressor genes (TNFRSF14 and HSD17B14). Oral administration of mibefradil inhibited growth of GSC-derived GBM murine xenografts, prolonged host survival and sensitized tumors to TMZ treatment. Our results offer a comprehensive characterization of Cav3.2 in GBM tumors and GSC, and provide a preclinical proof of concept for repurposing mibefradil as a mechanism-based treatment strategy for GBM.
Keywords: Calcium channels, glioblastoma, cancer stem cells, mibefradil, Cav3.2
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor. GBM is associated with a dismal prognosis and an average life expectancy of ∼15 months despite optimal therapy consisting of surgery, chemotherapy and radiation (1). Lack of therapeutic success is attributed to various factors including rapid tumor cell infiltration of the brain, inter- and intra-tumoral heterogeneity, limited diffusion of therapeutic drugs across the blood brain barrier and brain/tumor parenchyma as well as to the presence within the tumor of GBM stem cells (GSCs) that are resistant to radio- and chemotherapy and that are capable of tumor generation and unlimited self-renewal (2-5). Overcoming GSC resistance to existing therapies or induction of GSC differentiation could greatly enhance therapeutic outcome.
Calcium signaling plays a ubiquitous role in many cellular regulatory processes (6-9) including proliferation, apoptosis and gene transcription (10) (11). Additionally, alteration of free cytosolic calcium results in the transcription of immediate early genes (12) (13) such as c-fos, c-jun, the cyclic AMP response element and the serum response element. Expression of these genes triggers entry into the cell cycle through expression of cyclins and cyclin-dependent kinases (14-17). Calcium influx is mediated by voltage-gated Ca++ (Cav) channels (18) of which there are five types: L-type, P-type, N-type, R-type and T-type. Aberrant expression and activity of T-type calcium channels (Cav3.2) has been implicated in cancer (19) through their role in the regulation of cell cycle progression. In line with this, an increase in intracellular calcium, regulated by Cav3.2 expression, has been shown to regulate GBM cell proliferation (20, 21). Furthermore, calcium entry participates in the complex network responsible for mouse embryonic stem cell self-renewal (22). A very recent study, published while the present manuscript was in revision, showed that membrane-depolarizing channel blockers induce selective glioma cell death by impairing nutrient transport and unfolded protein/amino acid responses (23).
The present work aimed at studying the expression, functions, mechanisms of action, and therapeutic targeting of calcium channels with mibefradil and shRNA in combination with cytotoxic therapies. Mibefradil (C29H38FN3O3) (Supplementary Figure 1) is a T-type FDA-approved calcium channel blocker previously marketed by Roche as Posicor for the treatment of hypertension. Our data demonstrate that Cav3.2 is highly expressed in human GBM and GSCs and that expression correlates with patient survival. Inhibition of Cav3.2 suppressed both GSC growth and stemness and in vivo xenograft growth and sensitized GSCs to chemotherapy. Mechanistically, mibefradil altered multiple cancer regulatory pathways as well as the expression of several oncogenes and tumor suppressors in GSCs. Mibefradil inhibited HIF1α/HIF2 expression under hypoxic conditions.
Materials and Methods
Cells and tumor specimens
Human GBM cell lines U87, A172, U373, T98G and SNB19, were obtained from American Type Culture Collection (Manassas, VA) within the last four years and used at less than 20 passages. U1242, U251 and SF767 were kind gifts from Dr. Isa Hussaini and Dr. Benjamin Purow (University of Virginia), and Dr. Russel Pieper (UCSF), respectively. They were obtained within the last 10 years and used at less than 35 passages. Primary glioblastoma cells (GBM-6 and GMB-10), a gift from Dr. Jann Sarkaria (Mayo Clinic), were isolated from patients who underwent surgery at the Mayo clinic and were used at less than 10 passages (24). GSCs XO-1, XO-2, XO-3, XO-4, XO-8 and XO-9, kindly offered by Dr. Deric Park, were isolated from GBM specimens obtained from patients undergoing surgery at the University of Pittsburgh and were used at less than 8 passages. GSCs 206, 827 and 578 (used at less than 12 passages) were isolated from patient surgical specimens. GSCs were characterized for in vivo tumorigenesis, pluripotency, self-renewal, stem cell markers, and neurosphere formation (25). Matched CD133-positive and CD133- cells were isolated from GBM surgical specimens (used at one passage) were kindly provided by Dr. Rainer Glass (University Clinics Munich, Germany). All cell lines underwent testing for species and mycoplasma infection using Mycoplasma Detection Kit-QuickTest (Biotool, Houston, TX). GBM surgical specimens were obtained from the University of Virginia Brain Tumor Bank according to procedures that were approved by the Review Board of the University of Virginia. All tumors were characterized by an experienced neuropathologist.
Immunoblotting
Immunoblotting was performed as previously described (26). Antibodies used were Cav3.2, Nestin, Tuj-1 and HIF1α (Santa Cruz Biotechnologies, Santa Cruz, CA), p27, GFAP, BMI1, MAP2, Bax, Sox2, mTOR, HIF2 and PARP (Cell Signaling, Beverly, MA), β-actin and GAPDH (Santa Cruz Biotechnologies, Santa Cruz, CA).
Quantitative RT-PCR
Total RNA was extracted from GSCs using RNeasy extraction kit (Qiagen, Germantown, MD). cDNA was synthesized using the iScript cDNA synthesis kit (BioRad, Hercules, CA) and quantitative PCR analysis was performed using the CFX Connect Real-time System (BioRad Hercules, CA). GAPDH was used as an endogenous control. Primer sequences are listed in the Supplemental Methods.
Cell transfections
Lentiviral control vectors (sh-control) or vectors encoding Cav3.2 shRNA (sh-Cav3.2; pooled) (Santa Cruz Biotechnologies, Santa Cruz, CA) or two additional sh-Cav3.2 (Applied Biological Materials, Richmond, BC) were generated. GSCs were seeded in poly-L-ornithine pre-coated plates and infected with the lentivuruses.
Cell death and cell proliferation assays
Cell death was assessed by trypan blue assay. Cell proliferation was assessed by cell counting for five days as previously described (27). The Alamar Blue assay was utilized according to the manufacturer's instruction. All experiments were performed three times.
TCGA and REMBRANDT data analyses
Differential expression of Cav3.2 was analyzed in GBM (n=607) and normal unmatched brain samples (n=11) from TCGA data. The effect of Cav3.2 expression on patient survival wasassessed using the cBioportal website (www.cBioportal.org). Differential expression of Cav3.2 and its correlation with survival was also assessed in the REMBRANDT database, which used an Affymetrix HG U133 v2.0 Plus platform to analyze 178 GBM samples.
Hypoxia experiments
GSCs were incubated in a hypoxic incubator (Thermo Fisher, Waltham, MA) with a 94:5:1 mixture of N2/CO2/O2. The cells were treated with mibefradil (5 μM) or vehicle (water) in normoxia (5% O2) or hypoxia (1% O2) for 24-48 h. The cells were subsequently subjected to immunoblotting for HIF proteins.
Reverse Phase Protein Arrays
Proteomic screening was performed by reverse phase protein array (RPPA) as previously described (28-30). Protein analytes were chosen for analysis based on their previously described involvement in key aspects of tumor biology. All antibodies were validated for single band specificity and for ligand-induction (phospho-specific antibodies) by immunoblotting prior to use on the arrays as previously described (28-30). Additional details are in the Supplementary Methods.
RNA sequencing
GSCs were treated with mibefradil (5 μM) or vehicle control for 24 h or transfected with sh-Cav3.2 or scrambled shRNA (Santa Cruz, CA) at 37 °C prior to RNA extraction. RNA sequencing was performed by Hudson Alpha (Huntsville, AL) as described in the Supplementary Methods.
Rescue Experiments
Functional rescue experiments were performed to determine if molecules regulated by mibefradil from proteomic and transcriptomic screenings mediate the effects of mibefradil on GSCs. The experimental details are described in the Supplementary Methods.
In vivo experiments
The therapeutic effects of inhibiting Cav3.2 with mibefradil were assessed using an orthotopic GSC-based xenograft mouse model. GSC 827 cells (3 × 105) were stereotactically implanted into the corpus striatum of immunodeficient mice (n = 10 per treatment group). Six days after implantation, the animals were treated with control (H2O) or mibefradil (24 mg/kg) by oral gavage every 6 h and/or TMZ (100 mg/kg body weight) IP once a day for 4 days. Treatment stopped for 7 days. Mice were treated for two cycles. Tumor volumes were visualized and quantified by MRI and animal survival was determined.
Immunohistochemistry
Immunohistochemistry (IHC) was used to assess the expressions of Ki67, cleaved Caspase 3, SOX2 and GFAP in brain tumor xenograft sections. IHC was performed as previously described (31) using antibodies against Ki67, cleaved Caspase 3, SOX2 or GFAP (Cell signaling, Beverly, MA) and secondary antibodies conjugated with the fluorescent dyes Alexa 488 or Alexa 555 (Fisher, Waltham, MA).
Statistical analyses
To evaluate the statistical significance of in vivo animal experiments, we used both two-sample t-test and non-parametric Wilcoxen rank-sum test. The continuous variable reverse phase protein array data generated were subjected to both unsupervised and supervised statistical analyses. Statistical analyses were performed on final microarray intensity values obtained using R version 2.9.2 software (The R Foundation for Statistical Computing). If the distribution of variables for the analyzed groups were normal, a two-sample t-test was performed. If the variances of two groups were equal, two-sample t-test with a pooled variance procedure was used to compare the means of intensity between two groups. Otherwise, two-sample t-test without a pooled variance procedure was adopted. For non-normally distributed variables, the Wilcoxon rank sum test was used. Significance levels were set at p < 0.05.
For RNA-seq, Raw FASTQ sequencing reads were chastity filtered to remove clusters having outlying intensity corresponding to bases other than the called base. Filtered reads were assessed for quality using FastQC. Reads were splice-aware aligned to the Ensembl GRCh38 genome using STAR (32), and reads overlapping GRCh38.82 gene regions were counted using featureCounts (33). The DESeq2 Bioconductor package (34) in the R statistical computing environment was used for normalizing count data, performing exploratory data analysis, estimating dispersion, and fitting a negative binomial model for each gene comparing the expression for each cell line comparison. After obtaining a list of differentially expressed genes, log fold changes, and p-values, Benjamini-Hochberg False Discovery Rate procedure was used to correct p-values for multiple testing.
Results
Cav3.2 is highly expressed in GBM tumor specimens and GSCs, and expression correlates with patient survival
To determine if Cav3.2 is deregulated in GSCs and human tumor specimens, we measured Cav3.2 protein expression by immunoblotting and compared it to expression in commonly used established cell lines and normal brain. The data show that Cav 3.2 is highly expressed in all GSCs and in some GBM cell lines (Fig. 1A). To determine if the expression of Cav3.2 is enriched in the stem cell fraction, we assessed the level of Cav3.2 in CD133 positive and CD133 negative cells derived from the same tumors (elevated SOX2 expression and reduced GFAP expression in CD133 positive cells compared to CD133 negative cells confirmed the stem cell identity of these cells). Cav3.2 expression was several fold higher expressed in the CD133 positive fraction than in the corresponding CD133 negative fraction. This suggests a link between elevated Cav3.2 expression and the stem cell state (Fig. 1B). The majority of GBM tumor samples expressed high levels of Cav3.2 while expression in normal brain was uniformly low (Fig.1C).
Figure 1. Cav3.2 expression in Glioblastoma and correlation with stemness and patient survival.
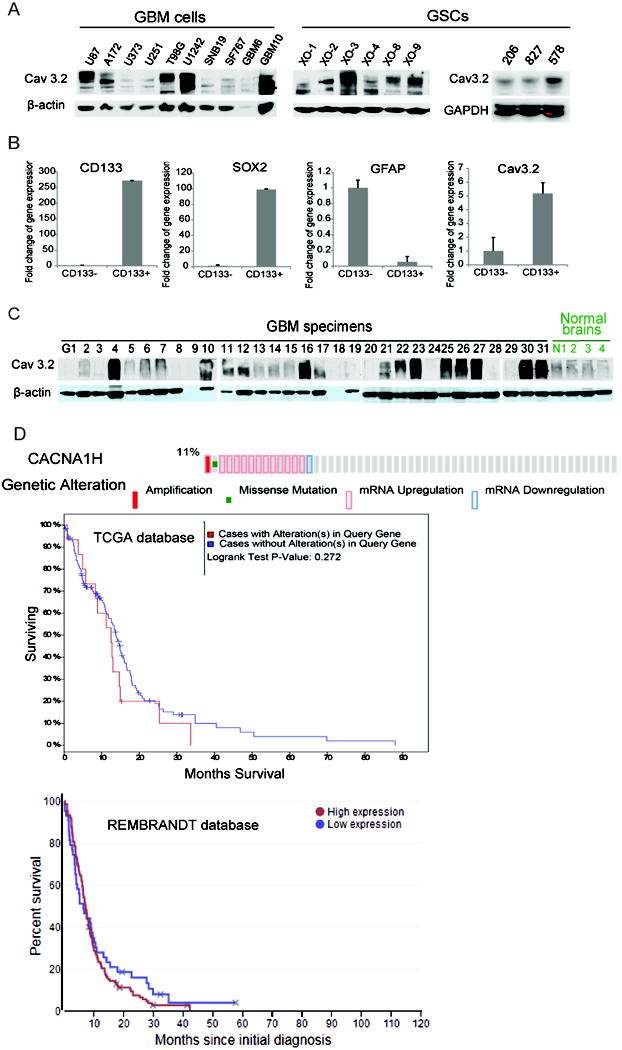
A) The glioblastoma cell lines U87, A172, U373, U251, T98G, U1242, SNB-19, SF-767, primary GBM cells GBM-6 and GBM-10, GBM stem cells XO-1, 2, 3, 4, 8, 9 and 206, 827 and 578 were lysed and immunoblotted for Cav3.2 and/or β-actin/GAPDH loading controls. B) Primary GBM cells (other than the ones of Fig. 1A) were sorted for CD133 expression by FACS and subjected to quantitative RT-PCR to determine the expression of CD133, SOX2, GFAP and Cav3.2. C) GBM human specimens G1-G31 and normal brains N1-4 were subjected to immunoblotting for Cav3.2 and β-Actin. D) TCGA (upper panel) and REMBRANDT (lower panel) data analyses of Cav3.2 (CACNA1H) mRNA expression and correlation with patient survival, Provisional, mRNA Expression z-Scores (RNA Seq V2 RSEM, with a z0score threshold 1.0) in upper panel. The analyses showed worse survival with high expression of Cav3.2. The above data show high expression of Cav3.2 in a subset of GBM and GSC and a trend towards inverse correlation with patient survival.
We also analyzed Cav3.2 expression in the TCGA and REMBRANDT databases. The TCGA analysis showed that 11% of GBM patients display either amplification, mutations or mRNA upregulation of Cav3.2. Patients with an alteration of Cav3.2 demonstrated a trend towards worse survival than patients with normal Cav3.2 (Cav3.2 mRNA Expression z-Scores (RNA-Seq V2 RSEM), with a z-score threshold 1.0) (Fig. 1D upper panel). The REMBRANDT analysis also showed worse survival with high expression of Cav3.2 at thresholds of 25 percentile (Fig. 1D bottom panel).
The above data demonstrate that Cav3.2 is highly expressed in a subset of GBM tumors and GSCs and that high expression may correlate with poor prognosis.
Cav3.2 blockade inhibits cell growth, induces cell death and enhances the effect of temozolomide (TMZ) in GSCs
To assess the effects of Cav3.2 inhibition on GSC function, we first tested the effects of mibefradil on cell growth, proliferation and death, also in combination with TMZ. GSCs (827, 206, 578) were treated with mibefradil (2.5-5 μM) and / or with TMZ (400 nM) and analyzed for growth by Alamar blue assay. The results showed that mibefradil significantly inhibited cell growth and enhanced the inhibition of GSC growth by TMZ (Fig. 2A). To determine the effects of mibefradil on GSC proliferation, cells were treated with mibefradil (5 μM) and/or TMZ (400 nM) and counted for 5 days. Mibefradil significantly inhibited cell proliferation and enhanced the inhibition of GSC proliferation by TMZ (Fig. 2B). To determine whether mibefradil affects cell death, we treated cells as above and performed a trypan blue assay. The results showed that mibefradil significantly enhances cell death also in combination with TMZ (Fig. 2C). The above data show that mibefradil exhibits anti-cancer stem cell effects also in combination with TMZ.
Figure 2. Cav3.2 blocker mibefradil inhibits GSC growth and enhances the effects of temozolomide in GSCs.
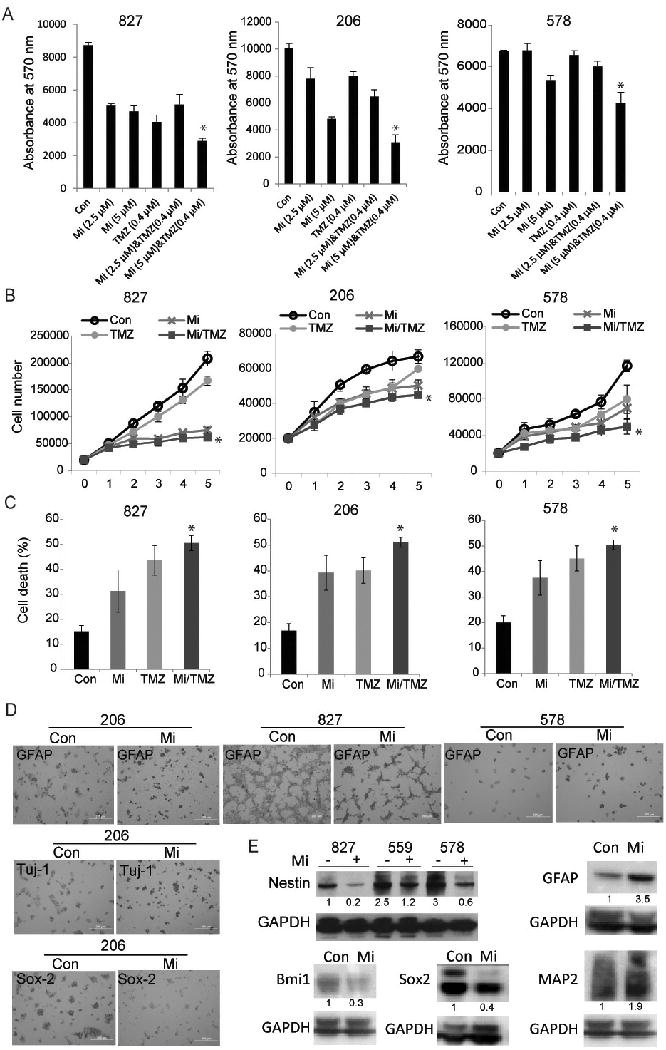
A) GSCs 827, 206, 578 were treated with mibefradil (Mi) and/or temozolomide (TMZ) for 48 h. The cells were subsequently assessed for cell growth by Alamar blue assay. B) GSCs were treated with mibefradil and TMZ or control. The cells were subsequently assessed for proliferation by cell counting over a period of 5 days and growth curves were established. C) GSCs were treated with mibefradil and TMZ or control for 48 h and cell death was assessed by trypan blue assay. These data show that mibefradil induces GSC cell death, which is further enhanced by combinational treatment with TMZ (p<0.05). D) GSCs were seeded in pre-coated dishes with poly-L-ornithine. The cells were treated with mibefradil for 48 h, fixed and immunostained with differentiation markers, GFAP and Tuj-1 and stem cell marker Sox2. E) The GSCs were treated with mibefradil for 48 h then subjected to immunoblotting (quantified: numbers under blots) for the stem cell markers Nestin, Bmi1, Sox2 and astrocyte and neuronal markers GFAP and MAP2 and GAPDH control. These data show that mibefradil induces stem cell differentiation evidenced by the downregulation of Nestin Bmi1 and Sox2 and upregulation of GFAP, Tuj1 or MAP2. *, p<0.05 (Mi vs. combination).
Cav3.2 blockade induces glioblastoma stem cell differentiation
Cav3.2 has been associated with differentiation of mouse embryonic stem cells (35). Based on that, we hypothesized that Cav3.2 might also regulate GSCs. To test this hypothesis, we determined the effects of mibefradil on the in situ expression of stemness and differentiation markers in GSCs using immunocytochemistry for the stemness and differentiation markers Sox2, GFAP and Tuj1. Mibefradil treatment increased the differentiation markers, GFAP (astrocytes) and Tuj1 (neurons) (Fig. 2D) and inhibited expression of stem cell markers Sox2 (Fig. 2D). We confirmed the downregulation of stemness markers Nestin, Bmi1 and Sox2 as well as the upregulation of differentiation markers GFAP and MAP2 through immunoblotting (Fig. 2E). We also found, through RPPA, that mibefradil treatment decreased the protein level of CD133 whilst simultaneously increasing the level of GFAP (Fig. 4D). The above data indicate that mibefradil induces GSC differentiation.
Figure 4. Mibefradil inhibits several oncogenic pathways in GSC.
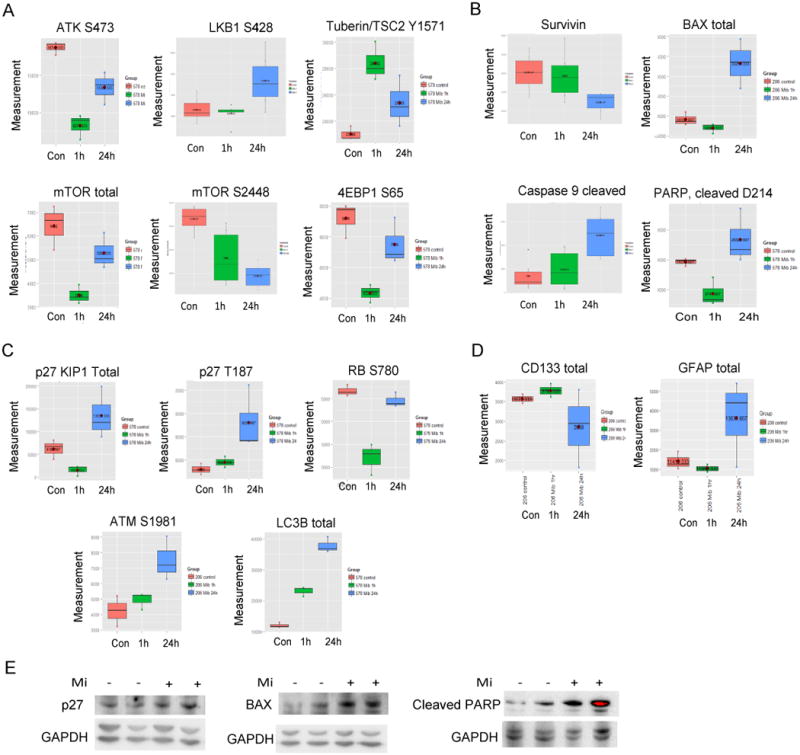
GSCs were treated with vehicle control (in red) or mibefradil for 1 h (in green) or 24 h (in blue) and the cell lysate was subjected to RPPA. Mibefradil downregulated A) the AKT/mTOR pathway whilst simultaneously upregulating LKB1 and TSC2, B) Mibefradil downregulated survivin and upregulated BAX, cleaved caspase 9 and cleaved PARP, C) Mibefradil upregulated p27, ATM and LC3B, D) Mibefradil downregulated CD133 and upregulated GFAP. E) RPPA verification by immunoblotting. These data show that mibefradil inhibits pro-survival pathways whilst inducing cell cycle arrest, apoptosis and DNA damage (p<0.05).
Cav3.2 knock-down induces cell death and inhibits proliferation of GSCs
To confirm that the cell death and growth suppression induced by mibefradil was attributed to inhibition of Cav3.2, we silenced Cav3.2 with shRNA and analyzed the effect on cell proliferation and death. Cells were infected with lentivirus encoding either sh-control or sh-Cav3.2 for 48 h. Cav3.2 knockdown by sh-Cav3.2 was verified by immunoblotiing (Fig. 3A). The cells were counted at 3 and 5 days after lentivirus infection. Cav3.2 silencing significantly inhibited cell proliferation of GSCs (Fig. 3B). To assess whether Cav3.2 silencing affected cell death, cells were infected as above and a trypan blue assay was performed. The data showed that silencing of Cav3.2 significantly enhanced cell death (Fig. 3C). To exclude off target effects of sh-Cav3.2, we used two additional sh-Cav3.2 and found similar effects on cell proliferation and death (Supplementary Fig. 3). The above data show that silencing of Cav3.2 expression leads to comparable anti-cancer effects on GSCs as mibefradil treatment.
Figure 3. Silencing of Cav3.2 induces GSC death and inhibits GSC growth.

A) GSCs 827 and 206 were transfected with sh-Cav3.2 or sh-control for 48 h and subjected to immunoblotting for Cav3.2 and β-Actin. B) GSCs were either transfected with sh-Cav3.2 or sh-control for 48 h or treated with mibefradil for 48 h. The cells were subsequently assessed for proliferation by cell counting over a period of 5 days and growth curves were established. C) Cell death was assessed by trypan blue assay. These data show that silencing Cav3.2 inhibits GSC proliferation and induces cell death in a similar manner to mibefradil. *, p<0.05.
Mibefradil inhibits hypoxia inducible factors HIF1α and HIF2 in GSCs
Hypoxia and HIF levels have been associated with GSC maintenance and resistance to therapy (36) (37). We hypothesized that Cav3.2 inhibition might affect hypoxia-induced HIF1 and HIF2 in GSCs. Thus we determined Cav3.2 expression in a hypoxic environment and the effect of mibefradil on HIF1α and HIF2 expression. GSCs were grown in 1% oxygen for 24 h and the level of Cav3.2 was determined by immunoblotting. Hypoxia induced Cav3.2 expression (Supplementary Fig. 2A). HIF1α and HIF2 were increased in GSCs under hypoxic conditions compared to normoxic conditions (Supplementary Fig. 2B, 2C). HIF1α and HIF2 expressions were significantly suppressed by mibefradil treatment (Supplementary Fig. 2B, 2C), suggesting that mibefradil might overcome GBM resistance to chemotherapy partially through inhibition of HIF1α and HIF2 in GSCs.
Cav3.2 blockade inhibits several major oncogenic pathways in GSC
To investigate the mechanism of action of Cav3.2 blockade with mibefradil in GSCs, we performed proteomic screenings using Reverse Phase Protein Arrays (RPPA) in GSCs treated with or without mibefradil for 1 or 24 h. The arrays measured the expressions and activation of > 300 proteins associated with cancer. mibefradil induced dramatic and significant changes in GSC signaling including inhibition of the AKT/mTOR pro-survival pathway as evidenced by decreased activation of AKT, mTOR and 4EBP1 and upregulation of LKB1 and Tuberin/TSC2 phosphorylation (Fig. 4A). Mibefradil also induced signaling changes associated with the induction of cell cycle arrest and apoptosis including inhibition of survivin and activation of BAX, Caspase 9, PARP, p27 and Rb (Fig. 4B, 4C). Mibefradil also affected molecules associated with DNA damage repair and autophagy (activation of ATM and LC3B) (Fig. 4C). Additionally, mibefradil inhibited the expression of the stemness marker CD133 and increased the expression of the astrocytic marker GFAP (Fig. 4D). Select random RPPA results (changes in p27, BAX, and cleaved PARP) were verified by immunoblotting (Fig. 4E). The above data show that mibefradil-mediated Cav3.2 inhibition inhibits GSC malignancy by acting on multiple key cancer regulatory pathways.
Cav3.2 blockade induces apoptosis and inhibits cell proliferation via Bax, p27 and mTOR
To determine if BAX and p27 mediate the pro-apoptotic effect of mibefradil, we assessed the effects of silencing p27 and BAX on mibefradil-induced cell death. We transfected GSC cells with either si-control, si-BAX or si-p27 before treating them with mibefradil for 48 h. Inhibition of either BAX or p27 abrogated mibefradil-induced cell death (Fig. 5A). Silencing of BAX and p27 was confirmed by immunoblotting (Fig. 5B). In order to deduce the role of mTOR in mibefradil-mediated suppression of proliferation and cell death, we overexpressed mTOR prior to treating GSCs with mibefradil for 48 h. Overexpression of mTOR partially mitigated mibefradil-induced cell death and growth suppression (Fig. 5C). Overexpression of mTOR was confirmed by immunoblotting (Fig. 5D). The above data show that the anti-cancer effects of Cav3.2 inhibition are partly mediated by the induction of apoptosis and inhibition of cell cycle progression via regulation of mTOR, BAX and p27.
Figure 5. Mibefradil induces apoptosis via Bax, p27 and mTOR.
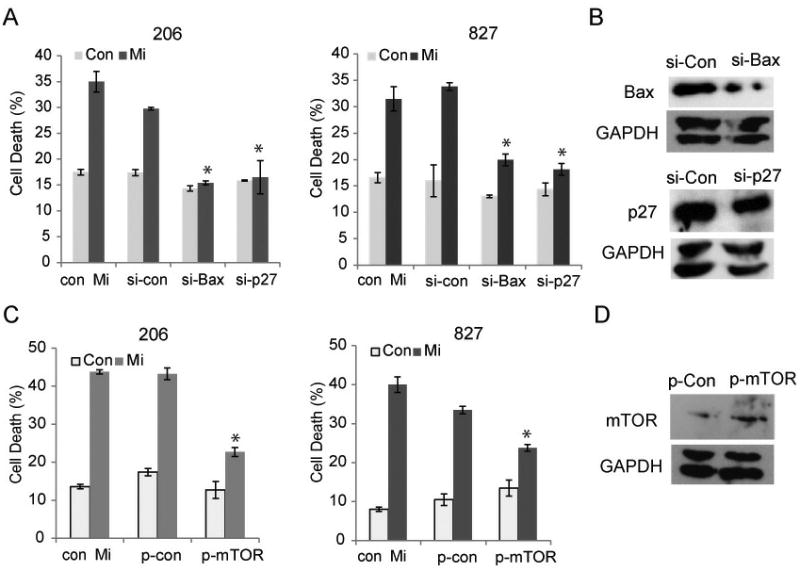
A) GSCs 206 and 827 cells were transiently transfected with either si-control, si-p27 or si-Bax 48 h then treated with mibefradil or control for 48 h. Cell death was then assessed by trypan blue as previously described. Inhibition of either BAX or p27 abrogated mibefradil-induced cell death. These data show mibefradil acts to induce GSC cell death through the induction of apoptosis. B) Immunoblotting was undertaken to verify silencing of BAX and p27. C) GSCs were transfected with either control plasmid (p-con) or plasmid encoding mTOR (p-mTOR) for 48 h then treated with mibefradil or control for 48 h. Cell death was assessed by trypan blue assay. The data show mibefradil induces GSC cell death partly by inhibiting mTOR. D) Immunoblotting was undertaken to verify overexpression of mTOR. *, p<0.05.
Cav3.2 blockade alters gene expression in GSCs
Since calcium is required for gene transcription (38) (39), we hypothesized that mibefradil might alter gene expression in GSCs. To test this hypothesis, we performed RNA deep sequencing (RNA-seq) on GSCs treated with vehicle control or mibefradil for 24 h. Mibefradil significantly altered the expression of many genes (Fig. 6A, 6B). The RNA-seq data were partially verified by qRT-PCR (Fig. 6C). Notably, mibefradil induced the expression of several tumors suppressors such as TNFRSF14 and HSD17B14 whilst simultaneously downregulating the expression of several oncogenes such as PDGFA, PDGFB, TGFB1, METTL7B, EGR3 and TNFRSF12A in GSCs. Interestingly, mibefradil-induced inhibition of METTL7B, a novel oncogene found overexpressed in primary stem-like GBM (40), and EGR3 (41) both of which may partially account for mibefradil-induced differentiation as well as decreased GSC proliferation and survival. RNA-seq data were deposited at the GEO Database (Accession # GSE95106).
Figure 6. Mibefradil alters gene expression in GSCs.
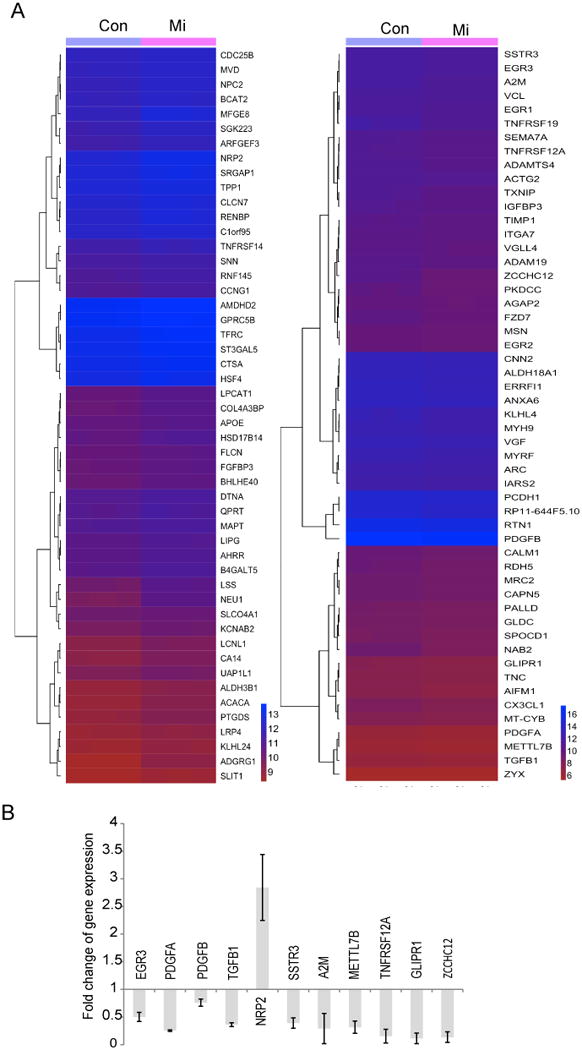
GSCs (827) were treated with mibefradil or control for 24 h prior to total RNA extraction. RNA deep sequencing (RNA-seq) was performed. The data analysis (A) shows blockage of Cav3.2 by mibefradil alters gene expression, including downregulation of several oncogenes such as PDGFA, PDGFB, TGFB1, METTL7B, EGR3 and TNFRSF12A in GSC 827 and upregulation of tumor suppressive NRP2. B) Confirmation of RNA-seq data by quantitative PCR (p < 0.05 for all shown genes).
Cav3.2 blockcade inhibits in vivo tumor growth and enhances the effects of temozolomide
We tested the effects of mibefradil alone and in combination with TMZ on the growth of established GSC xenografts and animal survival. GSC 827 cells (3 × 105) were stereotactically implanted in the striata of immunodeficient mice (n = 10). Six days after implantation, mibefradil (24 mg/kg body weight) was administered per oral gavage every 6 h for 4 days. TMZ (100 mg/kg body weight) was concurrently administered IP once a day for 4 days. The above treatment plan was repeated 7 days later. MRI scans were performed 21 days after surgery and tumor sizes were measured. Animal survival was also assessed. The data show that mibefradil alone significantly inhibited tumor growth. This inhibition was of similar magnitude as TMZ alone. Combined treatment with mibefradil and TMZ inhibited tumor growth in an additive fashion (Fig. 7A, 7B). Similarly, both mibefradil and TMZ alone significantly prolonged animal survival with the combination of both drugs further prolonging animal survival (Fig. 7C). Immunohistochemical analysis of the xenografts revealed downregulation of the proliferation marker Ki67 and upregulation of the apoptosis regulator cleaved caspase 3 in mibefradil-treated xenografts. The expression of stem cell marker SOX2 was decreased, while the astrocyte marker GFAP was elevated in mibefradil-treated tumors (Fig. 7D). These data suggest that mibefradil is a promising drug for GBM therapy.
Figure 7. Mibefradil inhibits GSC xenograft growth and prolongs animal survival, also in combination with TMZ.
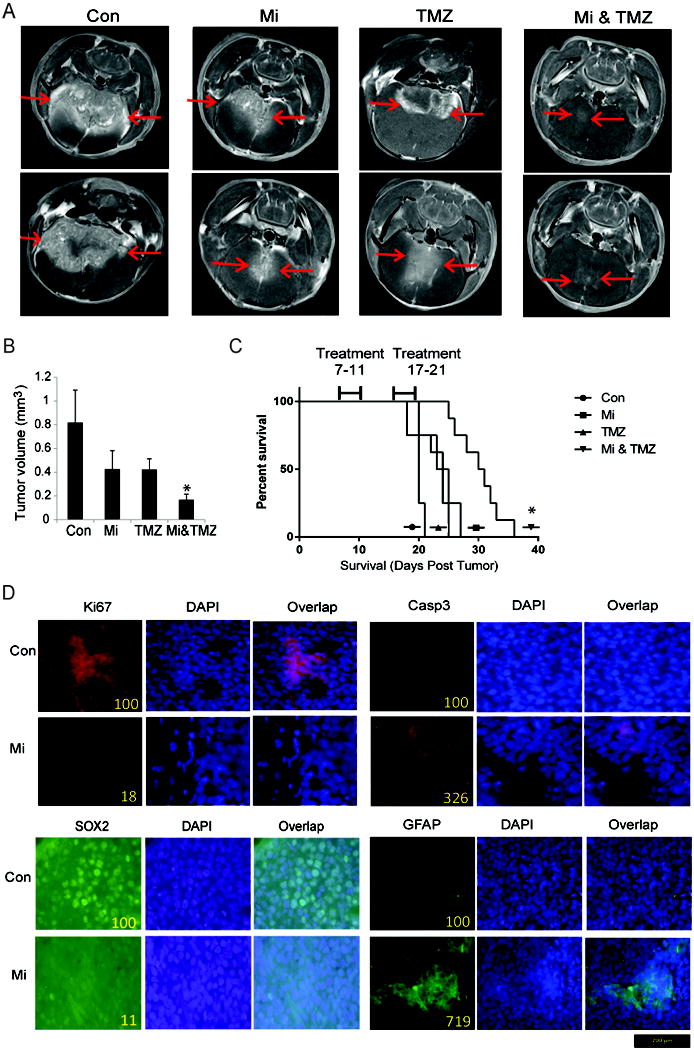
A) GSC 827 cells were stereotactically implanted in the striatum of immunodeficient mice (n=10). Mibefradil or vehicle control were administered by daily oral gavage starting six days post-tumor implantation. The animals were subjected to MRI scan at 3 weeks after tumor implantation and B) tumor volumes were quantified. C) Animals were treated as in (A) and survival was analyzed. The data show that mibefradil significantly inhibits tumor growth and sensitizes tumors to TMZ treatment. D) Immunohistochemical staining of xenograft sections from (A) for the proliferation marker Ki67, the apoptotic marker cleaved-Caspase 3, stem cell marker Sox2 and astrocyte marker GFAP showing significantly reduced Ki67 and increased cleaved-caspase 3, as well as reduced Sox2 and elevated GFAP level in Mibefradil-treated xenografts (Sections at 40× magnification; Staining was quantified on sections with control set at 100). *, p<0.05.
Discussion
Changes in intracellular Ca2+ levels can modulate signaling pathways and gene transcription that control a broad range of cellular events, including those important to tumorigenesis and cancer progression (42). Calcium channels are differentially expressed in malignant cells and alterations in their activity highlight the possible use of calcium channels as targets for new therapies (43) (44). Mibefradil is an orally bioavailable blocker of T and L type calcium channels, marketed by Roche as Posicor, for the treatment of hypertension. Although blood pressure lowering effects are mediated by L channel blockade, mibefradil has end organ protective effects that are mediated by T-type calcium channel inhibition resulting in reduced proliferation (45). Mibefradil was previously tested for effects upon GBM cell growth (46), but its mechanism of action in GBM and GSC is not well understood. In this study, we investigated the expression, function, mechanisms of action, and therapeutic value of targeting of T-type calcium channels in GBM and GSCs.
We showed that expression of Cav3.2 is increased in GBM tumors and GSCs. Increased expression of Cav3.2 in GBM correlated with poor prognosis suggesting the possibility of targeting Cav3.2 for GBM therapy and improved patient survival. We established that mibefradil sensitized GSCs to TMZ treatment, a key chemotherapeutic agent used in the treatment of GBM. GSCs partly mediate resistance to chemo and radiotherapy and have become vital targets for reversal of chemo-resistance. Notably, after surgical intervention and chemotherapy, resistant GSCs survive and are able to initiate and maintain malignant growth of GBM (4). We showed that mibefradil induced the differentiation of GSCs, as evidenced by the downregulation of GSC stemness markers, CD133, Nestin, Bmi1 and Sox2, and upregulation of differentiation markers GFAP, Tuj1 and MAP2. While Cav3.2 inhibition strongly impairs GSC malignant parameters, it likely also affects differentiated bulk GBM cells as has been shown for the U87 cell line (21).
To establish whether the anticancer properties of mibefradil are attributed to inhibition of Cav3.2, we used shRNA to specifically silence Cav3.2. Silencing of Cav3.2 displayed similar anticancer effects as mibefradil treatment. This indicates that inhibition of Cav3.2 is the primary mechanism through which mibefradil exerts its known anticancer effects. We also discovered that Cav3.2 is upregulated in GSCs under hypoxic conditions and, importantly, mibefradil downregulated HIF1α and HIF2. Hypoxia strongly correlates with poor patient survival, therapeutic resistance and an aggressive tumor phenotype. It was also previously demonstrated that GSCs are critically dependent on the HIFs for survival, self-renewal, and growth (47, 48). This suggests that mibefradil can inhibit GSC malignant parameters by reducing hypoxic pressure and inhibiting HIFs.
We investigated the mechanism of action of mibefradil on GSC. Using proteomic screening, immunoblot validation and functional rescue experiments, we found that cell cycle and apoptosis signaling pathways are significantly activated by mibefradil in GSCs, including decreased phosphorylation of AKT, mTOR and 4EBP1 and upregulation of p27 KIP1, BAX, FOXO1 and cleaved PARP leading to decreased tumor cell proliferation through downregulation of key survival pathways and cell cycle arrest, respectively. Furthermore, silencing of p27 or Bax, both of which are upregulated in response to mibefradil treatment, abrogated drug-induced cell death, suggesting a role for p27 and BAX in mediating mibefradil toxicity. Also, overexpression of mTOR, which is downregulated in response to mibefradil treatment, inhibited drug-induced cell death.
To understand the effect of mibefradil on the transcriptome, we performed RNA-seq. We found that mibefradil treatment resulted in a decrease in expression of several oncogenes such as PDGFA, PDGFB, TGFB1, METTL7B, EGR3 and TNFRSF12A, and an increase in the expression of numerous tumor suppressors including TNFRSF14 and HSD17B14, confirmed by qRT-PCR. These data provide insights into the molecular basis of responsiveness to Cav3.2 inhibition in cancer cells suggesting an important role for mibefradil in the regulation of genes implicated in cancer. In addition to the changes in protein function and gene transcription described above, blocking Cav3.2 might also influence GSC function by affecting resting membrane potential. In fact, recently published research has shown that brain tumor stem cells have elevated resting membrane potential that is regulated by connexins and EAG2 potassium channels to influence these cells' malignant functions (49, 50).
Mibefradil significantly reduced in vivo tumor growth and prolonged animal survival. Additionally, combination of mibefradil with TMZ further enhanced therapeutic effect and survival. These data have promising translational potential. Mibefradil, a FDA approved low toxicity therapeutic, could easily translate into clinical trials. Cavion LLC undertook a trial to assess the safety of mibefradil in 30 healthy patients, followed by a second trial (NCT01480050), in conjunction with NCI, to assess the efficiency and optimal dosage of mibefradil sequentially administered in combination with TMZ in patients with recurrent GBM. A third trial (NCT02202993) is currently enrolling recurrent GBM patients to study the effect and safety of mibefradil combined with hypofractionated radiation therapy. This emphasizes the relevance of our study in establishing the molecular and functional basis of Cav3.2 targeting for GBM therapy. Our data also suggest that mibefradil is likely to achieve greater clinical benefits in patients in combination with TMZ.
Altogether, our data show novel roles and mechanisms of action of T type calcium channel Cav3.2 in GSCs and GBM (Supplementary Fig. 4) and support the use of mibefradil in combination with TMZ for GBM therapy.
Supplementary Material
Acknowledgments
Grant support: This work was supported by grants from the Commonwealth Research Commercialization Fund of Virginia (CRCF), the Virginia Biosciences Health Research Corporation (VBHRC), and NIH R01 grants NS045209 (R.A.) and NIH R01 CA134843 (R.A.).
Footnotes
Conflict of interest: Lloyd Gray is a paid consultant of Cavion LLC, who is commercializing the drug mibefradil.
References
- 1.Wen PY, Reardon DA. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nature reviews Neurology. 2016;12:69–70. doi: 10.1038/nrneurol.2015.242. [DOI] [PubMed] [Google Scholar]
- 2.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer research. 2004;64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 4.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–5. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 7.Brink F. The role of calcium ions in neural processes. Pharmacological reviews. 1954;6:243–98. [PubMed] [Google Scholar]
- 8.Brini M, Carafoli E. Calcium signalling: a historical account, recent developments and future perspectives. Cellular and molecular life sciences : CMLS. 2000;57:354–70. doi: 10.1007/PL00000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen H, Rasmussen JE. Calcium as intracellular messenger: from simplicity to complexity. Current topics in cellular regulation. 1990;31:1–109. doi: 10.1016/b978-0-12-152831-7.50003-2. [DOI] [PubMed] [Google Scholar]
- 10.Silver RB. Imaging structured space-time patterns of Ca2+ signals: essential information for decisions in cell division. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1999;13:S209–15. doi: 10.1096/fasebj.13.9002.s209. [DOI] [PubMed] [Google Scholar]
- 11.Deliot N, Constantin B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim Biophys Acta. 2015;1848:2512–22. doi: 10.1016/j.bbamem.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Roche E, Prentki M. Calcium regulation of immediate-early response genes. Cell calcium. 1994;16:331–8. doi: 10.1016/0143-4160(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 13.Rozengurt E. Early signals in the mitogenic response. Science. 1986;234:161–6. doi: 10.1126/science.3018928. [DOI] [PubMed] [Google Scholar]
- 14.Werlen G, Belin D, Conne B, Roche E, Lew DP, Prentki M. Intracellular Ca2+ and the regulation of early response gene expression in HL-60 myeloid leukemia cells. J Biol Chem. 1993;268:16596–601. [PubMed] [Google Scholar]
- 15.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–30. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 16.Lu KP, Means AR. Regulation of the cell cycle by calcium and calmodulin. Endocrine reviews. 1993;14:40–58. doi: 10.1210/edrv-14-1-40. [DOI] [PubMed] [Google Scholar]
- 17.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–4. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 18.Rao VR, Perez-Neut M, Kaja S, Gentile S. Voltage-gated ion channels in cancer cell proliferation. Cancers. 2015;7:849–75. doi: 10.3390/cancers7020813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santoni G, Santoni M, Nabissi M. Functional role of T-type calcium channels in tumour growth and progression: prospective in cancer therapy. British journal of pharmacology. 2012;166:1244–6. doi: 10.1111/j.1476-5381.2012.01908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valerie NC, Dziegielewska B, Hosing AS, Augustin E, Gray LS, Brautigan DL, Larner JM, Dziegielewski J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem Pharmacol. 2013;85:888–97. doi: 10.1016/j.bcp.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Zhang J, Jiang D, Zhang D, Qian Z, Liu C, Tao J. Inhibition of T-type Ca(2)(+) channels by endostatin attenuates human glioblastoma cell proliferation and migration. British journal of pharmacology. 2012;166:1247–60. doi: 10.1111/j.1476-5381.2012.01852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Gomez JA, Levitsky KL, Lopez-Barneo J. T-type Ca2+ channels in mouse embryonic stem cells: modulation during cell cycle and contribution to self-renewal. American journal of physiology Cell physiology. 2012;302:C494–504. doi: 10.1152/ajpcell.00267.2011. [DOI] [PubMed] [Google Scholar]
- 23.Niklasson M, Maddalo G, Sramkova Z, Mutlu E, Wee S, Sekyrova P, Schmidt L, Fritz N, Dehnisch I, Kyriatzis G, et al. Membrane-depolarizing channel blockers induce selective glioma cell death by impairing nutrient transport and unfolded protein/amino acid responses. Cancer research. 2017 doi: 10.1158/0008-5472.CAN-16-2274. [DOI] [PubMed] [Google Scholar]
- 24.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 25.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–52. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Guessous F, DiPierro C, Zhang Y, Mudrick T, Fuller L, Johnson E, Marcinkiewicz L, Engelhardt M, Kefas B, et al. Interactions between PTEN and the c-Met pathway in glioblastoma and implications for therapy. Molecular cancer therapeutics. 2009;8:376–85. doi: 10.1158/1535-7163.MCT-08-0627. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Kim J, Mueller AC, Dey B, Yang Y, Lee DH, Hachmann J, Finderle S, Park DM, Christensen J, et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell death and differentiation. 2014;21:720–34. doi: 10.1038/cdd.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, Warneke J, Krouse R, Stratton SP, Liotta L, Longo C, Pellicani G, et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res (Phila) 5:403–13. doi: 10.1158/1940-6207.CAPR-11-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierobon M, Vanmeter AJ, Moroni N, Galdi F, Petricoin EF., 3rd Reverse-phase protein microarrays. Methods Mol Biol. 823:215–35. doi: 10.1007/978-1-60327-216-2_14. [DOI] [PubMed] [Google Scholar]
- 30.Paweletz CP, Charboneau L, Bichsel VE, Simone NL, Chen T, Gillespie JW, Emmert-Buck MR, Roth MJ, Petricoin IE, Liotta LA. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20:1981–9. doi: 10.1038/sj.onc.1204265. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Schiff D, Park D, Abounader R. MicroRNA-608 and microRNA-34a regulate chordoma malignancy by targeting EGFR, Bcl-xL and MET. PloS one. 2014;9:e91546. doi: 10.1371/journal.pone.0091546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yanagi K, Takano M, Narazaki G, Uosaki H, Hoshino T, Ishii T, Misaki T, Yamashita JK. Hyperpolarization-activated cyclic nucleotide-gated channels and T-type calcium channels confer automaticity of embryonic stem cell-derived cardiomyocytes. Stem Cells. 2007;25:2712–9. doi: 10.1634/stemcells.2006-0388. [DOI] [PubMed] [Google Scholar]
- 36.Pajonk F, Vlashi E, McBride WH. Radiation resistance of cancer stem cells: the 4 R's of radiobiology revisited. Stem Cells. 2010;28:639–48. doi: 10.1002/stem.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crowder SW, Balikov DA, Hwang YS, Sung HJ. Cancer Stem Cells under Hypoxia as a Chemoresistance Factor in Breast and Brain. Current pathobiology reports. 2014;2:33–40. doi: 10.1007/s40139-013-0035-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyons MR, West AE. Mechanisms of specificity in neuronal activity-regulated gene transcription. Progress in neurobiology. 2011;94:259–95. doi: 10.1016/j.pneurobio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. British journal of pharmacology. 2014;171:945–60. doi: 10.1111/bph.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ernst A, Hofmann S, Ahmadi R, Becker N, Korshunov A, Engel F, Hartmann C, Felsberg J, Sabel M, Peterziel H, et al. Genomic and expression profiling of glioblastoma stem cell-like spheroid cultures identifies novel tumor-relevant genes associated with survival. Clin Cancer Res. 2009;15:6541–50. doi: 10.1158/1078-0432.CCR-09-0695. [DOI] [PubMed] [Google Scholar]
- 41.Liu D, Evans I, Britton G, Zachary I. The zinc-finger transcription factor, early growth response 3, mediates VEGF-induced angiogenesis. Oncogene. 2008;27:2989–98. doi: 10.1038/sj.onc.1210959. [DOI] [PubMed] [Google Scholar]
- 42.Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Current opinion in pharmacology. 2016;29:1–6. doi: 10.1016/j.coph.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287:31666–73. doi: 10.1074/jbc.R112.343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta. 2015;1848:2502–11. doi: 10.1016/j.bbamem.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 45.Tzivoni D. End organ protection by calcium-channel blockers. Clinical cardiology. 2001;24:102–6. doi: 10.1002/clc.4960240202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keir ST, Friedman HS, Reardon DA, Bigner DD, Gray LA. Mibefradil, a novel therapy for glioblastoma multiforme: cell cycle synchronization and interlaced therapy in a murine model. Journal of neuro-oncology. 2013;111:97–102. doi: 10.1007/s11060-012-0995-0. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–56. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, Hubaud A, Stadler B, Choi M, Bar M, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–52. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hitomi M, Deleyrolle LP, Mulkearns-Hubert EE, Jarrar A, Li M, Sinyuk M, Otvos B, Brunet S, Flavahan WA, Hubert CG, et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015;11:1031–42. doi: 10.1016/j.celrep.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, Yang H, Wang TA, Stehbens SJ, Younger S, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci. 2015;18:1236–46. doi: 10.1038/nn.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.