

INTRODUCTION
Every year, more than 8000 new cases of chronic myeloid leukemia (CML) are diagnosed in the United States1. BCR-ABL1, a fusion protein kinase derived from a reciprocal translocation between chromosomes 9 and 22, is necessary and sufficient for CML pathogenesis2. Tyrosine kinase inhibitors (TKIs) of BCR-ABL1 have revolutionized CML therapy, with life expectancy now close to that of the general population3. As a result, the prevalence of CML is growing, as patients on TKIs live with what is more and more viewed as a chronic ailment rather than a potentially lethal disease. It is estimated that over 25% of CML patients will switch TKIs at least once during their lifetime due to TKI intolerance or resistance4. Mutations in the kinase domain (KD) of BCR-ABL1 are the most extensively studied mechanism of TKI resistance in CML, but fail to explain anywhere from 20–40% of resistant cases. Activation of alternative, BCR-ABL1-independent survival pathways has been mechanistically implicated in these cases, and may also explain the phenomenon of persistence in responding patients who fail to clear minimal residual disease (MRD) or experience recurrence upon discontinuation of therapy despite achieving deep molecular response (DMR, BCR-ABL1 ≤ 0.01% on the international scale, IS).
DEFINITIONS
The National Comprehensive Cancer Network and the 2013 European LeukemiaNet (ELN) guidelines recommend cytogenetic and/or molecular monitoring at 3, 6 and 12 months into frontline TKI therapy5,6. ELN recommendations categorize the molecular and cytogenetic responses at each time interval as “optimal”, “warning” or “failure”. Optimal responses are associated with a life expectancy similar to that of the general population, whereas failure is associated with TKI resistance and increased risk of disease progression/death, necessitating a change in TKI therapy. Failure to achieve complete hematologic response (CHR; normalization of peripheral blood counts; resolution of splenomegaly and CML-related symptoms) or complete cytogenetic response (CCyR; 0% Ph+ metaphases based on analysis of 20 bone marrow cells) within an allocated duration of time constitutes TKI failure, as does loss of these milestones or progression to accelerated phase (AP-CML) or blastic phase (BP-CML) at any time point. Whether failure to achieve major molecular response (MMR; BCR-ABL1 ≤ 0.1% on the IS) in patients with CCyR defines failure is subject to ongoing debate. Similarly, confirmed loss of MMR while CCyR is maintained does not technically constitute failure, although most of these patients will go on to lose CCyR7.
Overt resistance such as loss of CHR or even progression to AP/BC-CML is associated with unfavorable clinical outcomes and represents a situation very different from persistent low-level disease associated with MRD which is clinically relevant only in the context of TKI discontinuation. Primary resistance implies failure to achieve time-dependent endpoints of CHR, CCyR and MMR upon initiation of TKI therapy, while secondary (acquired) resistance is defined as the loss of response8. At the mechanistic level, we classify TKI resistance as either BCR-ABL1-dependent or BCR-ABL1 independent (Figure 1). Although this distinction seems formalistic, it does have a great degree of clinical relevance, as it informs the strategy required to combat resistance: BCR-ABL1-dependent resistance is reliant upon mechanisms that subvert effective BCR-ABL1 kinase inhibition, such as point mutations in the kinase domain that impair drug binding or cellular/biological processes that interfere with TKI availability and result in suboptimal drug concentrations at the target. In contrast, BCR-ABL1-independent resistance is mediated through alternative survival pathways operating in the context of effective TKI inhibition of BCR-ABL1. Overt clinical resistance is observed via both mechanisms, although acquired resistance is more likely to be BCR-ABL1 dependent, while primary resistance tends to be BCR-ABL1-independent. In BCR-ABL1-dependent resistance, achieving or restoring BCR-ABL1 inhibition is expected to induce or recapture responses, and the most effective approach is the use of alternate TKIs. For obvious reasons this strategy in isolation will not be effective in BCR-ABL1-independent resistance. In this review, we will discuss the mechanisms underlying BCR-ABL1-dependent and independent resistance and therapeutic strategies designed to circumvent them.
Figure 1.
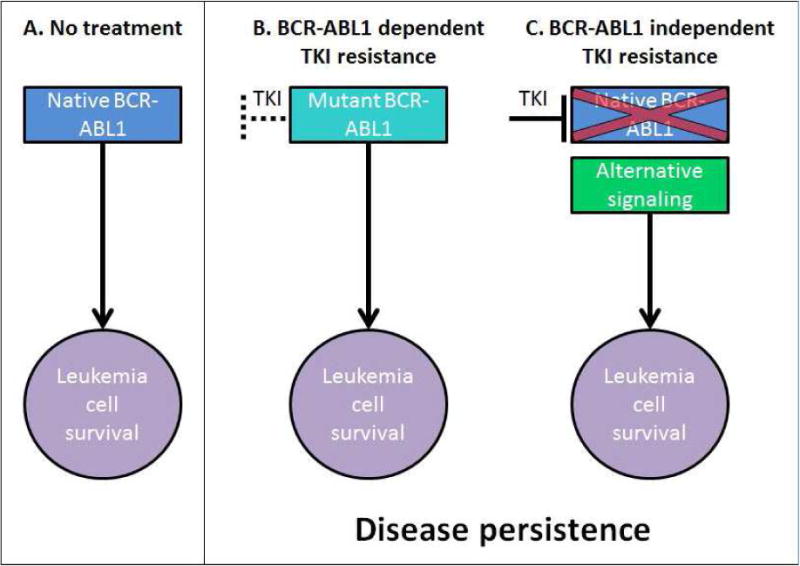
BCR-ABL1-dependent vs. independent resistance. (A) Native BCR-ABL1 signaling in the absence of TKI inhibition is necessary and sufficient for leukemogenesis in CML. (B) Kinase domain mutations in BCR-ABL1 can alter the binding of TKIs and lead to reconstitution of BCR-ABL1 signaling. (C) In the setting of effective BCR-ABL1 inhibition with TKIs, leukemia cells persist due to activation of alternative survival pathways.
BCR-ABL1 DEPENDENT RESISTANCE
BCR-ABL1 KD Mutations
General Considerations
The active sites of tyrosine kinases exist in two principal conformations that are distinct by the position of key structural motifs, including the activation loop (A-loop) that controls access of substrate to the catalytic site, the ATP-binding loop (P-loop) and the highly conserved aspartate-phenylalanine-glycine (DFG) motif that coordinates an adenosine triphosphate (ATP)-bound magnesium ion. In the inactive conformation, the activation loop is in a closed position, and the DFG in an outward (“DFG out”) orientation. In contrast, in active kinases the A-loop is in an open conformation, and the DFG motif is oriented toward the catalytic site (“DFG-in”) (Figure 2)9. Depending on whether they recognize an active or inactive kinase conformation, TKIs are referred to as type I or type II inhibitors, respectively10. Although all active site inhibitors are essentially ATP-competitive, type II inhibitors could be considered as stabilizers of an inactive enzyme conformation, while type I inhibitors compete more directly with ATP for binding. Of the approved BCR-ABL1 TKIs, imatinib, nilotinib and ponatinib are type II inhibitors, dasatinib is a type I inhibitor, and bosutinib exhibits features of both11–15. These general structural distinctions have practical consequences as they inform the number and types of mutations that confer resistance to a given TKI. Generally, type II inhibitors exhibit more stringent binding requirements, exposing more mutational vulnerabilities, but have the advantage of increased selectivity16. Type I inhibitors tend to be more promiscuous, but less prone to mutational escape.
Figure 2.
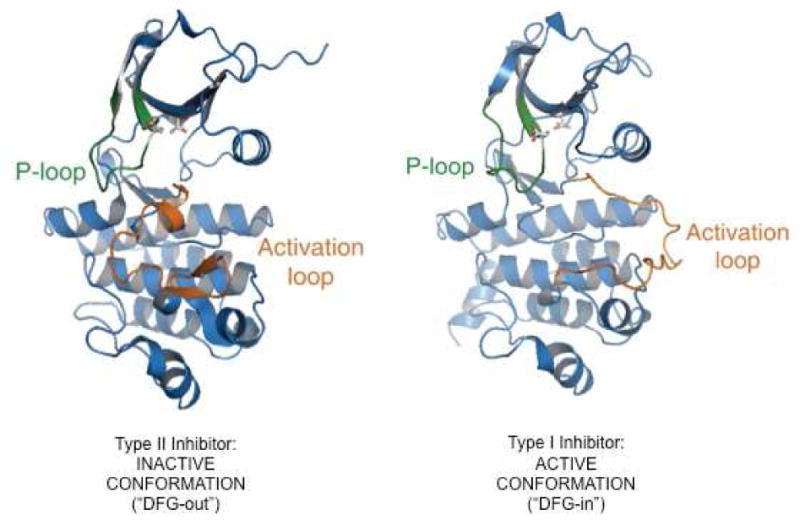
Type I and Type II inhibitors. (A) Type II inhibitors stabilize the inactive conformation of BCR-ABL1 in which the activation loop is closed and the DFG is in an outward (“DFG out”) orientation. (B) Type I inhibitors are ATP-competitive, binding to BCR-ABL1 when the activation loop is in an open position conformation and the DFG motif is oriented toward the catalytic site (“DFG-in”).
Courtesy of T. Clackson, PhD, Cambridge, MA.
Clinically observed BCR-ABL1 KD mutations and structure-function relationships
Anywhere from 50–90% of CML patients who experience hematologic relapse on imatinib have been reported to harbor KD mutations17–20. Point substitutions at just twelve residues (M244, G250, Q252, Y253, E255, V299, F311, T315, F317, M351, F359 and H396) account for most resistance-associated KD mutations (Figure 3A)21. KD mutations develop with greater frequency in AP/BP-CML than in CP-CML18. For instance a study of 297 patients with primary or acquired resistance to imatinib reported KD mutations in 27% of CP patients, 52% AP patients, 75% myeloid BC patients and 83% lymphoid BC patients22. This suggests that reactivation of BCR-ABL1 signaling is critical to conferring an aggressive clinical phenotype. KD mutations can also be detected at low levels in patients at diagnosis, and may in some cases become clinically relevant upon selection of clones by TKI therapy23,24. However, as this is not a predictable development, testing for KD mutations at diagnosis is not generally recommended5,24. Interestingly, the duration of disease prior to initiation of TKI therapy correlates with the frequency of KD mutations, which supports a role for BCR-ABL1 induced self-mutagenesis18. Moreover, advanced phase CML, clonal cytogenetic evolution and KD mutation rate are correlated, suggesting a temporal relationship between uninhibited exposure to BCR-ABL1 kinase activity and degree of genomic instability25.
Figure 3.
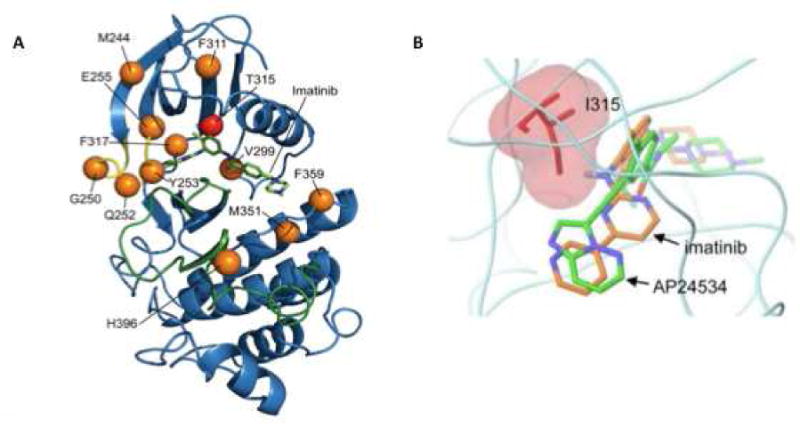
Key residues influence BCR-ABL1-dependent resistance to TKIs. (A) Crystal structure of the ABL1 kinase domain in complex with imatinib. Twelve positions (in orange, T315 in red) account for most clinical BCR-ABL1 TKI resistance. The phosphate-binding (yellow) and activation loops (green) are indicated. (B) Superposition of imatinib and AP24534 (ponatinib) highlighting the effect of the Thr to Ile mutation. High-affinity binding of imatinib and other 2G TKIs to BCR-ABL1 requires a critical hydrogen bond with residue T315, which is eliminated upon the conversion of threonine to isoleucine. Unlike other clinically available TKIs, ponatinib does not form a hydrogen bond with T315 and has activity against the T315I mutant form of BCR-ABL1.
Figure 3A: From Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014; 26(3); 430; with permission.
Figure 3B: From O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009; 16(5): 403; with permission.
Of the approved TKIs, imatinib exhibits the broadest spectrum of vulnerabilities and more than 50 different imatinib-resistance KD mutations have been described26,27. Solving the crystal structure of ABL1 in complex with an imatinib analogue was critical for understanding KD mutation-based imatinib resistance. In contrast to expectations imatinib was found to recognize an inactive kinase conformation, with the A-loop in a closed position. Additionally, there was extensive ‘downward’ displacement of the P-loop11. Lastly, imatinib was found to form a hydrogen bond with threonine 315. This binding mode is reflected in the types of KD mutations associated with imatinib resistance28. P-loop mutations are thought to prevent the structural adjustments required for optimal drug binding, the T315I mutant causes a steric clash and A-loop mutations stabilize the kinase in an active conformation from which imatinib is excluded. The degree of resistance conferred by the various KD mutations varies greatly, and some (such as M351T or F311L) remain amenable to dose escalation. In contrast, second-generation TKIs such as dasatinib and nilotinib retain inhibitory activity against the majority of mutants conferring imatinib resistance, with the notable exception of the T315I ‘gatekeeper’ mutation29. Nilotinib was developed from the imatinib scaffold, but has a much improved topological fit, greatly increasing binding affinity. As a result, nilotinib captures many imatinib resistant mutants, although their relative sensitivities to imatinib and nilotinib are similar13,30. Thus nilotinib overcomes resistance through tighter binding to a very similar (inactive) ABL1 conformation. Dasatinib was initially reported to bind to ABL1 with less stringent conformational requirements compared to imatinib, but sophisticated nuclear magnetic resonance studies suggest it is a type I inhibitor12. The dasatinib resistance mutation spectrum is distinct and includes V299 and F317 as hotspots31. However, both nilotinib and dasatinib make a hydrogen bond with T315 and consequently have no activity against T315I. Bosutinib’s resistance mutation spectrum is similar to that of dasatinib, suggesting that type I binding is dominant32. Ponatinib in contrast is a type II inhibitor that binds ABL1 in a conformation that is quite similar to that observed with imatinib, except that no hydrogen bond is formed with T315 (Figure 3B)33. Owing to this and its high target affinity ponatinib exhibits activity against all single BCR-ABL1 mutants at achievable plasma concentrations. In vitro mutagenesis assays developed by us and others fairly accurately predict clinical mutations, validating the fascinating link between structural analysis and clinical observations33. Clinically, the type of BCR-ABL1 mutation informs the selection of salvage therapy and represents a prime example of individualized cancer therapy. It is important to note though that the convenient heat maps displaying the differential activity of the approved TKIs toward the various KD mutants are a guide, but not a dogma (Figure 4). For example achievable plasma concentrations and plasma protein binding are additional variables not captured by in vitro assays of BCR-ABL1 expressing cell lines. Further, correlations are tight only toward the negative side. Thus, the presence of a T315I mutation predicts resistance, but there is no guarantee that a patient with a ‘sensitive’ mutant will respond to a given TKI. Failure to respond to TKI therapy in this setting could be due to alternative BCR-ABL1-dependent mechanisms of resistance (e.g. efflux pumps, see below), or to BCR-ABL1 independent mechanisms.
Figure 4.
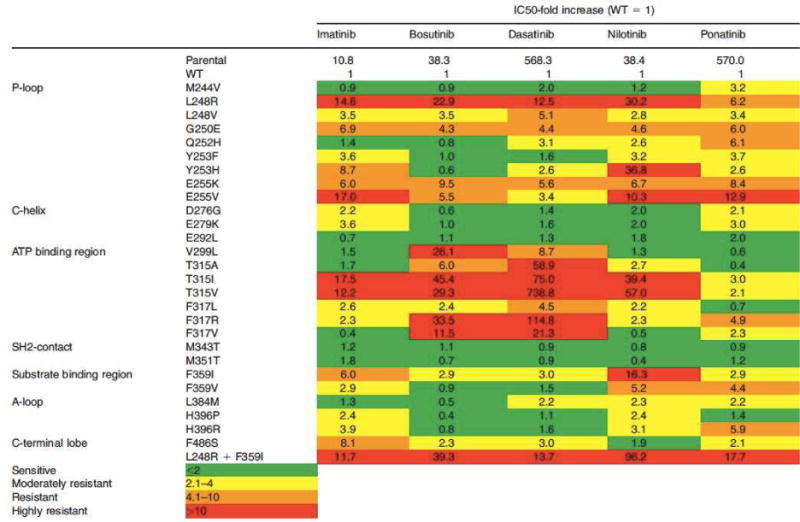
Activity of TKIs against mutant isoforms of BCR-ABL1 in Ba/F3 cells. The relative increase in IC50 value over wild-type BCR-ABL1 is depicted for each TKI against single BCR-ABL1 mutants. Green indicates sensitive mutants, yellow indicates moderate resistance and yellow indicates marked resistance. In patients, TKI efficacy is dependent on other factors, such as oral and cellular bioavailability.
From Eiring AM, Deininger MW. Individualizing kinase-targeted cancer therapy: the paradigm of chronic myeloid leukemia. Genome Biol 2014;15(9):461; with permission.
No single BCR-ABL1 KD mutation has been demonstrated to confer resistance to ponatinib. However, T315I-inclusive compound mutations, defined as a BCR-ABL1 allele with two or more mutations including T315I, have been associated with ponatinib failure21 in advanced phase CML and Philadelphia chromosome-positive (PH+) acute lymphoblastic leukemia (ALL). A recent analysis of CP CML patients in the PACE trial failed to demonstrate that baseline compound mutation status, regardless of T315I inclusion, affects cytogenetic or molecular responses to ponatinib in this cohort34.
Increased BCR-ABL1 expression
Increased BCR-ABL1 expression via BCR-ABL1 gene amplification, Ph duplication and differential regulation of oncogene transcription has been demonstrated in patients, but its relationship to acquired clinical resistance is less certain than in cases of KD mutations. High levels of the BCR-ABL1 oncoprotein are associated with more advanced phase disease, often preceding the development of overt resistance via KD mutations35. Thus, higher levels of BCR-ABL1 may allow for sufficient kinase activity to persist despite the presence of TKIs, enabling leukemia cell survival until a KD mutation is acquired and confers overt resistance. One indication that these relationships are complex is the seemingly paradoxical observation that primary CD34+ CML cells engineered to express high levels of BCR-ABL1 have been reported to exhibit increased sensitivity to imatinib in vitro19,28,36,37.
Drug influx/efflux pumps
OCT-1
Organic-cation transporter-1 (OCT-1) is a cellular influx pump for imatinib that has been demonstrated to influence intracellular drug availability. Low OCT-1 activity imparts BCR-ABL1 dependent imatinib resistance. High OCT-1 activity is predictive of improved MMR rates, event free survival (EFS) and overall survival (OS) in patients treated with imatinib38,39. Patients with low OCT-1 activity and imatinib trough plasma levels <1200ng/mL have inferior outcomes and benefit from imatinib dose intensification40. Imatinib trough levels <1200ng/mL do not necessarily predict inferior outcomes in patients with high OCT-1 activity and these patients are likely to meet molecular milestones on standard-dose imatinib. OCT-1 does not regulate cellular uptake of dasatinib, nilotinib or ponatinib41–43. In the future baseline OCT-1 testing may identify candidates for trough imatinib monitoring and imatinib dose intensification, thereby avoiding unnecessary TKI switching due to perceived imatinib failure, but it is not part of current routine clinical practice. Similarly, although several members of the ATP-binding cassette (ABC) transporter family, including ABCB1 and ABCG2, have been implicated in TKI resistance, testing for polymorphisms and increased expression of ABC transporters is not clinically routine27,37,43–48 49,50.
TKI bioavailability
All of the TKIs used in CML undergo extensive hepatic first-pass metabolism by CYP3A4 and strong inducers of CYP3A4 can contribute to TKI resistance. Patients on TKIs should undergo thorough medication reconciliation to avoid potential drug-drug interactions that can negatively impact TKI efficacy. Common CYP3A4-inducing medications and supplements include dexamethasone, rifampicin, phenobarbital, phenytoin, carbamazepine and St. John’s wort51. Gastric pH-modifying medications such as H2 antagonists and proton pump inhibitors can affect the bioavailability of dasatinib due to the drug’s poor solubility in solutions with a pH >4.0. These patients must be counseled to take antacids 2 hours prior or 2 hours after dasatinib administration to avoid decreases in dasatinib exposure that can occur with their concomitant administration52,53.
BCR-ABL1 INDEPENDENT RESISTANCE
General considerations
Point mutations in BCR-ABL1 are an important mechanism of TKI resistance in CML, but nearly 40% of cases of clinical TKI failure occur in the setting of sustained BCR-ABL1 inhibition54. In this scenario, activation of alternative survival pathways must be responsible for primary or secondary resistance. Conceptually CML cell survival can be mediated through cell-autonomous (leukemia cell intrinsic) mechanisms or through cell-extrinsic microenvironmental factors provided by the bone marrow niche55. It is worth noting that while BCR-ABL1 independent resistance can confer overt resistance in active disease, it is also an important contributor to MRD, likely accounting for leukemia stem cell persistence despite DMR to TKI therapy. Multiple (and counting) signaling pathways have been implicated in BCR-ABL1-independent resistance (Table 1). We have proposed that various upstream pathways may converge on common downstream mediators, offering therapeutic opportunities despite the diversity of upstream signaling56. Moreover it seems that the pathways activated by extrinsic and intrinsic resistance mechanisms overlap. In this frame of thinking, extrinsic resistance may enable survival of leukemogenic cells despite TKI inhibition of BCR-ABL1, until the surviving cells manage to activate the very same pathway through cell-intrinsic mechanisms, leading to overt resistance.
Table 1.
Targets for eradication of LSCs in CML
| Target | References | Trial | Drug(s) tested | Comments |
|---|---|---|---|---|
| Wnt/β-catenin MNK | Zhao et al. Cancer Cell 200772 Lim et al. PNAS 201373 |
+ | PRI-724 | Difficult target |
| Hedgehog | Dierks et al. Cancer Cell 200884 Zhao et al. Nature 2009 |
+ | BMS-833923 LDE225 |
Failed (toxicity) |
| 5-Lipoxygenase | Chen et al. Nat Genet 200985 | + | Zileuton | Currently recruiting |
| BCL6 | Hurtz et al. JExMed 2011 | − | RI-BPI | Small molecule inhibitor in development |
| MYC | Reavie et al. Cancer Cell 201394 Abraham et al. Nature 201695 |
− | – | Difficult target |
| PP2A | Neviani et al. J Clin Invest 2007; 201386 | − | – | Fingolimod approved for MS |
| PPAR-γ | Prost et al. Nature 2015125 | + | Pioglitazone | Ongoing |
| SIRT1 | Bhatia et al. Cancer Cell 2012 | + | Panobinostat Vorinostat |
Tested in refractory CML |
| Rad52 | Cramer-Morales et al. Blood 201387 | − | – | – |
| MEK | Ma et al. Sci Transl Med 201463 Packer et al. Cancer Cell 201164 |
+ | MEK-162 | Ongoing |
| BCL2 family | Goff et al. Cancer Stem Cell 2013 | + | Obatoclax | Tested in advanced hematologic malignancies, including CML-BC |
| Autophagy | Bellodi et al. J Clin Invest 200990 | + | Hydroxychloroquine | Ongoing (CHOICES) |
| PML | Ito et al. Nature 200891 | + | As2O3 | Ongoing |
| JAK2 | Traer et al. Leukemia 201257 Neviani et al. J Clin Invest 201386 |
+ | Ruxolitinib | Ongoing |
| ADAR1 | Jiang et al. PNAS 201393 | − | – | – |
| STAT3 | Eiring et al. Leukemia 201456 | − | – | Difficult target |
| EZH2 | Xie et al.69 Scott et al.68 Cancer Discovery 2016 | − | – | – |
| Heat shock proteins | Peng et al. Blood 200788 | + | STA-9090 | Ongoing |
| Fap1 | Huang et al. Leukemia 201692 | − | – | – |
| BCR-ABL1 | Pinilla-Ibarz et al. Blood 2000115 Bocchia et al. Lancet 2005116 |
+ | Breakpoint peptide vaccines | Suggestion of activity |
| PR1 | Molldrem et al. Nat Med 2000117 Rezvani et al. Haematologica 2011118 |
+ | Peptide vaccines | Negative studies |
| WT1 | Gao et al. Blood 2000119 Dubrovsky et al. Blood 2014120 |
+ | Peptide vaccines Peptide-specific antibody WT transduced autologous T cells |
Ongoing (some) |
| IL1RAP | Järås M et al. PNAS 2010121 | − | Antibody | – |
| IL3R (CD123) | Frolova et al. Br J Haematol 2014122 | − | DT-conjugated antibody (SL-401; SL-501) | – |
STAT3
STAT3 activation has been demonstrated to impart survival cues to leukemic cells via cell-intrinsic and extrinsic mechanisms. Co-culture of TKI-sensitive CML primary cells with HS-5 human bone marrow stromal cells was shown to promote STAT3Y705 phosphorylation and leukemia cell survival through soluble BM-derived factors despite BCR-ABL1 inhibition57,58. Moreover, in the absence of BM-derived factors, BCR-ABL1 independent activation of STAT3 was demonstrated to be a recurring feature of TKI-resistant cell lines and primary CML cells from patients with clinical resistance to multiple TKIs, suggesting that cell-autonomous activation of STAT3 can mediate CML cell survival56. Thus, consistent with the concepts described above, pro-survival cues appear to converge on STAT3 as a crucial distal signal integrator and arbiter of drug resistance. As a result, synthetic lethality approaches designed to inhibit both BCR-ABL1 and pSTAT3Y705 hold therapeutic potential, both in active disease and as a tactic to eliminate MRD.
PI3K/AKT
PI3K signaling is required for the proliferation and growth of CML cells59. Activation of the PI3K/AKT/mTOR pathway has been shown to facilitate primary CML cell survival during imatinib treatment until overt resistance through secondary mutations emerges60. Co-treatment of CML primary cells with nilotinib and the PI3K inhibitor NVP-BEZ235 was shown to inhibit cell growth and increase apoptosis61. Increased cytoplasmic retention of FOXO1, a transcription factor downstream of the PI3K signaling axis, has been reported to contribute to BCR-ABL1 independent resistance in TKI-resistant CML cell lines54. Elevation in FOXO1 levels has also been demonstrated in primary cells from relapsed CML patients lacking BCR-ABL1 KD mutations. TKI-resistant cells appear to be sensitive to combination drug strategies involving BCR-ABL1 TKIs and PI3K inhibitors that facilitate nuclear translocation of FOXO1.
RAF/MEK/ERK
Enhanced MAP kinase signaling has previously been observed in imatinib-treated CD34+ CML progenitor cells62. More recently Ma and colleagues performed a large-scale RNA interference screen that revealed increased RAF/MEK/ERK pathway activity mediated through PRKCH in BCR-ABL1-independent imatinib-resistant CML cell lines and patient samples63. They found that dual treatment with imatinib and the MEK inhibitor trametinib preferentially killed human CML CD34+ cells while sparing normal hematopoietic cells and prolonged survival in their murine models of BCR-ABL1-independent imatinib-resistant CML. In line with this, another study described paradoxical RAS-dependent activation of the RAF/MEK/ERK pathway in nilotinib-treated primary CML cells containing T315I and found that nilotinib synergizes with MEK inhibition to induce synthetic lethality in these cells64. In TKI-sensitive CML cells, MEK activity appears to facilitate BCR-ABL1-mediated oncogene addiction, suggesting that activation of this pathway is critical for leukemia cell survival and a potential target for combination drug inhibition strategies65.
Nucleocytoplasmic transport
More recently, XPO1 and RAN, components of the nucleocytoplasmic transport complex, were identified as genes whose shRNA-mediated knockdown decreased cell proliferation in a BCR-ABL1-independent imatinib-resistant cell line66. Both shRNA-mediated inhibition of RAN and treatment with the XPO1 inhibitor KPT-330 (selinexor) increased the sensitivity of resistant cells to imatinib. KPT-330 has also demonstrated preclinical anti-leukemic activity in mouse models of CML and was observed to decrease leukocytosis and palliate symptoms in a TKI-resistant patient with AP-CML who was provided the drug on a compassionate use basis67.
EZH2
EZH2, a histone methyltransferase that provides the catalytic subunit of polycomb repressive complex 2 (PRC2), has been shown to be overexpressed in CML leukemia stem cells (LSCs). Two recent publications have highlighted the importance of EZH2 misregulation and its association with reprogramming of H3K27me3 targets in LSCs, resulting in LSC protection from apoptosis and TKI resistance68,69. EZH2 inactivation was shown to delay the development of leukemia and prolong survival in mouse models of CML independent of BCR-ABL1 mutational status. In mice with pre-existing gene inactivation of EZH2 through CRISPR/Cas9-mediated gene editing slowed disease progression and extended survival. Combination treatment with nilotinib and EZH2 inhibitors in CML primary cells engrafted into NOD/SCID mice led to a greater reduction of the LSC population compared to nilotinib treatment alone. Normal hematopoietic stem and progenitor cells appear to be spared from EZH2 inhibition, perhaps due to compensation from EZH1, which is expressed at higher levels in normal HSCs compared to LSCs. The selective vulnerability of LSCs to EZH2 inhibition may provide a therapeutic window to eradicate TKI-persistent LSCs with minimal effects on normal hematopoiesis.
Numerous other BCR-ABL1 independent factors have been proposed to contribute to CML LSC persistence and TKI resistance, including activation of SRC family kinases, Wnt-β-catenin, hypoxia-inducible factor 1α, arachidonate 15-lipoxygenase, miR-126, p53, MYC, ADAR1, SIRT1, RAD21 heat shock proteins, PP2A, Fap1, apoptotic regulators, the Hedgehog pathway and the IL-2/CD25 signaling circuit55,70–95. The number of theoretical synthetic lethality approaches involving TKIs and other inhibitors is destined to grow as new resistance mechanisms are unearthed, yet it remains unclear which combinations harbor clinical potential above and beyond TKI monotherapy.
NEW THERAPIES
Tyrosine kinase inhibitors
ABL001
One of the most anticipated new therapies for CML is ABL001, a novel allosteric inhibitor of BCR-ABL1 targeting the myristoyl pocket of the ABL1 kinase. In physiological conditions, the myristoylated N-terminus of ABL1 serves to negatively regulate kinase activity, but is lost upon fusion with BCR in CML. ABL001 was designed to restore this autoregulatory function to the BCR-ABL1 fusion protein, thereby inhibiting oncogenic signaling. Single-agent ABL001 led to tumor regression in mice xenografted with the KCL22 CML cell line, though all tumors eventually recurred. In vivo combination treatment with nilotinib and ABL001 induced complete and sustained regression of disease in mice, with no relapses observed as long as 5 months out from active drug treament96. These encouraging results led to a dose-finding phase I trial of ABL001 monotherapy in CP and AP CML patients with failure of ≥ 2 TKIs due to resistance/intolerance97. Over 50% of patients enrolled had failed ≥ 3 TKIs. Initial results from the trial are promising – 82% of TKI resistant patients in cytogenetic relapse achieved MCyR by 3 months, including 55% who achieved CCyR. Nearly 30% of TKI-resistant patients achieved MMR by 5 months, and clinical activity was pronounced across a range of mutations. A single relapse was attributed to a mutation in the myristoyl pocket97. Overall the drug was well-tolerated, with common grade 3 toxicities including lipase elevation and cytopenias. At the time of last reporting, the maximum tolerated dose had not been reached. Other arms of the Phase I study are assessing the safety and tolerability of ABL001 in combination with imatinib, nilotinib and dasatinib, respectively.
Several other TKIs were previously in development for CML, including bafetinib (BCR-ABL1/Lyn inhibitor), and rebastinib (ABL1/TIE2 inhibitor), but have been sidelined due to poor efficacy in early phase clinical trials98,99. A phase I trial of the intravenous ABL1/Aurora kinase inhibitor danusertib produced modest responses in T315I-positive, TKI-resistant AP/BC CML and Ph+ ALL100. The VEGFR inhibitor axitinib has been found to inhibit BCR-ABL1 mutants with substitutions at positions 315 and 299, but its clinical use is limited by this mutational selectivity101,102. Radotinib, a second-generation oral BCR-ABL1 inhibitor with an almost identical chemical structure as nilotinib, is approved for second-line treatment of CML in South Korea. An ongoing Phase 3 study investigating radotinib versus imatinib in newly diagnosed CML demonstrated superior 12-month CCyR and MMR rates with radotinib 300mg BID (CCyR: 91% vs 76%; MMR: 52% vs 30%)103. Not surprisingly, the in vitro efficacy of radotinib against single BCR-ABL1 mutants appears to be similar to that of nilotinib104.
Drug combinations to eradicate LSCs and eliminate MRD
Patients who have maintained long-term (one to two years minimum) DMR on TKI therapy may be candidates for TKI discontinuation. When treated with single-agent TKI therapy, at best half of newly diagnosed CML patients will eventually be eligible for TKI discontinuation trials, and of these, at most 50–60% will successfully maintain treatment-free remission (TFR) one year following TKI discontinuation105. The finding that a portion of patients are “operationally cured” following TKI treatment is surprising given the wealth of data suggesting CML LSCs are not eradicated by BCR-ABL1 inhibition. It also remains unclear why patients with seemingly identical deep responses segregate in their responses to TKI discontinuation. Recent data has emerged to support the role of immune surveillance by NK and T cells in maintaining successful TFR, implying that alternative biological factors contribute to optimal disease control106. Various TKI discontinuation trials are ongoing, and attempts to clarify the clinical and biologic characteristics predictive of successful TFR are reflected in a trend toward more liberalized patient eligibility criteria and an emphasis on correlative studies (Table 2).
Table 2. Summary of TKI discontinuation studies.
Adapted from Saußele S, Richter J, Hochhaus A, et al. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 2016; 30(8):1641; with permission.
| Trial | Patients reported | Treatment prior to discontinuation | Eligibility for TKI discontinuation by MR | Threshold for restarting TKI | TFR% (median follow-up time) | |
|---|---|---|---|---|---|---|
| Imatinib discontinuation trials | ||||||
| STIM1 | 100 | Imatinib +/− prior IFN | MR5(≥2y) | ≥2 consecutive samples with detectable PCR and a 1-log increase | 39% (55m) | |
| STIM2 | 124 | Imatinib | MR4.5(≥2y) | ≥2 consecutive samples with detectable PCR and a 1-log increase | 46% (2y) | |
| TWISTER | 40 | Imatinib +/− prior IFN | MR4.5(≥2y) | Detectable PCR | 45% (42m) | |
| A-STIM | 80 | Imatinib +/− prior IFN | Undetectable PCR (≥2y) with low level positives occasionally allowed | Loss of MMR | 64% (23m) | |
| ISAV | 112 | Imatinib | Undetectable PCR (18 months) | Loss of MMR | 51.9% at 36 months (21m) | |
| KID | 90 | Imatinib +/− prior IFN | MR4.5(≥2y) | Loss of MMR | 50% (26.6m) | |
| HOVON | 18 | Imatinib | MR4.5(≥2y) | Detectable PCR | 33% (36m) | |
| Imatinib and/or 2G-TKI discontinuation trials | ||||||
| STOP-2G TKI | 52 | Nilotinib or dasatinib | MR4.5(≥2y) | Loss of MMR | 61% (6m); ongoing | |
| ENEST Freedom | 190 | Nilotinib | MR4.5(≥1y) | Loss of MMR | 51.6% (week 48);ongoing | |
| ENESTop | 126 | 2nd-line Nilotinib | MR4.5(≥1y) | Loss of MMR or confirmed loss of MR4 | 57.9% (week 48);ongoing | |
| ENEST Path | 1058 (estimated) | Imatinib followed by Nilotinib | MR4 (≥1–2y) | Loss of MMR or confirmed loss of MR4 | Ongoing | |
| ENEST Goal | 300 (estimated) | Imatinib without MMR followed by Nilotinib | MR4.5(≥1–2y) | Confirmed loss of MR4 | Ongoing | |
| DADI | 63 | Second-line dasatinib | MR4(≥1y) | Loss of MR4 | 49% (6m) | |
| DASFREE | 79(estimated) | Dasatinib | MR4.5(≥1y) | Loss of MMR | Ongoing | |
| CML V (TIGER) | 652(estimated) | Nilotinib vs. Nilotinib + IFN | MR4((≥1y) | Loss of MMK | Ongoing | |
| LAST | 173(estimated) | Imatinib, nilotinib, dasatinib or bosutinib | MR4(≥2y) | Detectable PCR | Ongoing | |
| DESTINY | 168 (estimated) | Imatinib, nilotinib or dasatinib | Patients in MMR or MR4 (≥ly) who can maintain MMR response on half-dose TKI for 12 months | Loss of MMR | Ongoing | |
| EURO-SKI | 200 | Imatinib, nilotinib or dasatinib | MR4((≥1y) | Loss of MMR | 61% (6m); ongoing | |
TKI discontinuation is an evolving goal of CML therapy and has been embraced by patients motivated to come off these chronic medications due to undesirable side effects, which, in some cases, can be quite serious (i.e. pulmonary hypertension on dasatinib or arterial occlusive events on nilotinib). The reality that the majority of CML patients will never attain TFR with current therapies has led to efforts to combine TKIs with other drugs in hopes of eliminating TKI-persistent LSCs and the reservoir of cells responsible for MRD.
TKIs plus immune therapies
Prior to imatinib, interferon-α-(IFN) based therapy was standard of care for CML. Anecdotal evidence suggests that IFN preferentially targets leukemic stem cells in CML, as demonstrated by the fact a small minority of CML patients treated with IFN alone were functionally cured of their disease107. Randomized trials of imatinib and pegylated IFN report improved molecular response rates with combination therapy compared to imatinib alone108,109. With the advent of TKI discontinuation and documentation of successful TFRs, there has been renewed interest in pegylated IFN as an adjunct to TKI therapy in promoting DMR. This had led to early phase trials investigating pegylated IFN in combination with second-generation TKIs. Non-randomized trials of nilotinib or dasatinib in combination with pegylated IFN in newly diagnosed CML patients have reported 12-month MR4.5 rates of 17% and 27–30%, respectively, which compare favorably to the 12-month MR4.5 rates observed in the registration trials of frontline nilotinib (ENESTnd) and dasatinib (DASISION)110–114. A phase 3 randomized trial of IFN in combination with nilotinib is underway in Germany. There remains considerable interest in developing novel immune therapies against a variety of tumor antigens and while early-phase trials investigating peptide vaccines have had mixed results, antibody-based treatments may hold promise115–122.
TKIs plus inhibitors of additional pathways
Despite mounting evidence implicating diverse pathways in BCR-ABL1-independent resistance and LSC persistence, there are a limited number of clinical trials investigating inhibitors of these pathways in combination with TKIs.
Leukemic stem and progenitor cells may be protected in the bone marrow niche via JAK2/STAT5 activation by exogenous growth factors in the setting of BCR-ABL1 inhibition57,123,124. CML CD34+ cells display reduced engraftment when treated ex vivo with the combination of TKI and ruxolitinib (a clinically available JAK2 inhibitor) and transplanted into NSG mice124. The impact of the addition of ruxolitinib to baseline TKI therapy in CML is being studied in a phase 1/2 trial (NCT01751425) and the specific combination of ruxolitinib and nilotinib in CML and Ph+ ALL is being investigated in a separate phase 1/2 study (NCT02253277).
Pioglitazone, an agonist of peroxisome proliferator-activated receptor-γ (PPARγ) belonging to the glitazone family of anti-diabetic drugs, has been found to induce apoptosis in LSCs when used in combination with imatinib, presumably by downregulating STAT5 transcriptional targets, including HIF2α and CITED2125. The addition of pioglitazone to TKI therapy in three CML patients unable to reach CMR after several years of continuous imatinib treatment was associated with sustained MR4.5 in all three patients at 6 months to 1 year following initial pioglitazone exposure. These findings led to phase II trial combining imatinib and pioglitazone in patients with persistent MRD on imatinib. The incidence of PCR-negativity was reported at 57% for the combination group and 27% for a historical cohort receiving imatinib alone. Currently there are several trials investigating pioglitazone in combination with TKIs for CML, including one study (PIO2STOP) attempting to define its use in a second trial of TKI discontinuation for patients who experienced loss of MMR after initial TKI discontinuation.
CONCLUSIONS
Due to improved survival, the prevalence of CML is estimated to exceed 180,000 cases by 2050, thereby establishing CML as the most common form of leukemia in the United States126. While excellent progress has been made through the introduction of targeted molecular therapy over the last two decades, new strategies to eliminate MRD and increase the pool of candidates eligible for trials of TFR are needed. Eliminating TKI resistance and LSC persistence by dual targeting of BCR-ABL1 and alternative pathways appears to be the most promising therapeutic avenue to decrease leukemic disease burden and potentiate “operational cures.” The number of alternative pathways posited to establish synthetic lethality with TKIs is overwhelming, and it will take time and effort to sift through the multiple permutations with rigorous clinical testing. Ultimately though, responses to cancer therapy depend not just on the efficacy of target inhibition, but also on factors such as patient compliance and tolerability of side effects that need to be addressed with a completely different set of tools. It is for these reasons that mechanisms of resistance will always keep pace with therapeutic developments, and we will be contending with them for as long as we continue our fight against cancer.
Synopsis.
Chronic myeloid leukemia (CML) is increasingly viewed as a chronic illness, with most patients expected to have a life expectancy close to that of the general population. Despite the great progress that has been made using BCR-ABL1 tyrosine kinase inhibitors (TKIs), drug resistance via BCR-ABL1-dependent and BCR-ABL1-independent mechanisms continues to be an issue for many patients. BCR-ABL1-dependent resistance is primarily mediated through oncoprotein kinase domain mutations and usually results in overt clinical resistance to TKIs. However, BCR-ABL1-independent resistance, which occurs in the setting of effective BCR-ABL1 inhibition, has become increasingly recognized a major contributor to minimal residual disease (MRD) and efforts to eradicate persistent leukemic stem cells (LSCs) have largely focused on combination therapy with TKIs and drugs targeting these pathways.
Key Points.
Over 25% of CML patients will switch TKIs during their lifetime due to resistance or intolerance. While most cases of clinical resistance are due to kinase domain mutations (BCR-ABL1-dependent resistance), 20–40% of patients exhibit resistance despite effective BCR-ABL1 inhibition (BCR-ABL1-independent resistance).
Ponatinib is the only TKI effective against the T315I BCR-ABL1 mutation. Ponatinib’s activity against this mutant isoform derives from its lack of dependence on forming a critical hydrogen bond with residue T315 for high-affinity binding to BCR-ABL1.
Diverse pathways involving growth factors, epigenetic regulators and apoptotic machinery have been implicated in BCR-ABL1-independent resistance. BCR-ABL1-independent resistance can be classified as cell-extrinsic or cell-intrinsic depending on the relative influence of the microenvironment.
CML leukemic stem cells (LSCs) are resistant to TKI therapy and contribute to minimal residual disease (MRD). Combination strategies to eradicate MRD using TKIs and other drugs are an intense focus of investigation in CML.
A minority of CML patients who achieve sustained deep molecular responses on TKI therapy are able to discontinue treatment without molecular recurrence, entering a state called “treatment-free remission (TFR).” Multiple TKI discontinuation trials are ongoing worldwide and will help determine which patients are most likely to have successful TFR and what biological factors govern maintenance of response.
Acknowledgments
Disclosures:
Michael W. Deininger is supported by the NIH (1R01CA178397-01 and 1R21CA205936-01), V Foundation for Cancer Research, Hope Foundation, and University of Utah seed funding.
Thomas O’Hare is supported by the NIH (1R01CA178397-01 and 1R21CA205936-01), V Foundation for Cancer Research, Hope Foundation, and University of Utah seed funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.SEER Cancer Statistics Factsheets: Chronic Myeloid Leukemia. National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/statfacts/html/cmyl.html. [Google Scholar]
- 2.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 3.Bower H, Bjorkholm M, Dickman PW, et al. Life Expectancy of Patients With Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J Clin Oncol. 2016;34:2851–7. doi: 10.1200/JCO.2015.66.2866. [DOI] [PubMed] [Google Scholar]
- 4.Steegmann JL, Baccarani M, Breccia M, et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016 doi: 10.1038/leu.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872–84. doi: 10.1182/blood-2013-05-501569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Brien S, Radich JP, Abboud CN, et al. Chronic myelogenous leukemia, version 1.2015. J Natl Compr Canc Netw. 2014;12:1590–610. doi: 10.6004/jnccn.2014.0159. [DOI] [PubMed] [Google Scholar]
- 7.Press RD, Galderisi C, Yang R, et al. A half-log increase in BCR-ABL RNA predicts a higher risk of relapse in patients with chronic myeloid leukemia with an imatinib-induced complete cytogenetic response. Clin Cancer Res. 2007;13:6136–43. doi: 10.1158/1078-0432.CCR-07-1112. [DOI] [PubMed] [Google Scholar]
- 8.Hochhaus A. Chronic myelogenous leukemia (CML): resistance to tyrosine kinase inhibitors. Ann Oncol. 2006;17(Suppl 10):x274–9. doi: 10.1093/annonc/mdl273. [DOI] [PubMed] [Google Scholar]
- 9.Treiber DK, Shah NP. Ins and outs of kinase DFG motifs. Chem Biol. 2013;20:745–6. doi: 10.1016/j.chembiol.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 11.Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 12.Vajpai N, Strauss A, Fendrich G, et al. Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib. J Biol Chem. 2008;283:18292–302. doi: 10.1074/jbc.M801337200. [DOI] [PubMed] [Google Scholar]
- 13.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Levinson NM, Boxer SG. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One. 2012;7:e29828. doi: 10.1371/journal.pone.0029828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou T, Commodore L, Huang WS, et al. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem Biol Drug Des. 2011;77:1–11. doi: 10.1111/j.1747-0285.2010.01054.x. [DOI] [PubMed] [Google Scholar]
- 16.Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 17.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 18.Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–83. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 19.Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–6. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 20.von Bubnoff N, Peschel C, Duyster J. Resistance of Philadelphia-chromosome positive leukemia towards the kinase inhibitor imatinib (STI571, Glivec): a targeted oncoprotein strikes back. Leukemia. 2003;17:829–38. doi: 10.1038/sj.leu.2402889. [DOI] [PubMed] [Google Scholar]
- 21.Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26:428–42. doi: 10.1016/j.ccr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soverini S, Colarossi S, Gnani A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12:7374–9. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 23.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–8. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 24.Willis SG, Lange T, Demehri S, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106:2128–37. doi: 10.1182/blood-2005-03-1036. [DOI] [PubMed] [Google Scholar]
- 25.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–9. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 26.O’Hare T, Zabriskie MS, Eiring AM, et al. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513–26. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 27.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 28.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 29.Eide CA, O’Hare T. Chronic myeloid leukemia: advances in understanding disease biology and mechanisms of resistance to tyrosine kinase inhibitors. Curr Hematol Malig Rep. 2015;10:158–66. doi: 10.1007/s11899-015-0248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weisberg E, Manley P, Mestan J, et al. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer. 2006;94:1765–9. doi: 10.1038/sj.bjc.6603170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jabbour E, Hochhaus A, Cortes J, et al. Choosing the best treatment strategy for chronic myeloid leukemia patients resistant to imatinib: weighing the efficacy and safety of individual drugs with BCR-ABL mutations and patient history. Leukemia. 2010;24:6–12. doi: 10.1038/leu.2009.193. [DOI] [PubMed] [Google Scholar]
- 32.Redaelli S, Piazza R, Rostagno R, et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol. 2009;27:469–71. doi: 10.1200/JCO.2008.19.8853. [DOI] [PubMed] [Google Scholar]
- 33.O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deininger MW, Hodgson JG, Shah NP, et al. Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients. Blood. 2016;127:703–712. doi: 10.1182/blood-2015-08-660977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barnes DJ, Palaiologou D, Panousopoulou E, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65:8912–9. doi: 10.1158/0008-5472.CAN-05-0076. [DOI] [PubMed] [Google Scholar]
- 36.Modi H, McDonald T, Chu S, et al. Role of BCR/ABL gene-expression levels in determining the phenotype and imatinib sensitivity of transformed human hematopoietic cells. Blood. 2007;109:5411–21. doi: 10.1182/blood-2006-06-032490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milojkovic D, Apperley J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin Cancer Res. 2009;15:7519–7527. doi: 10.1158/1078-0432.CCR-09-1068. [DOI] [PubMed] [Google Scholar]
- 38.White DL, Dang P, Engler J, et al. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J Clin Oncol. 2010;28:2761–7. doi: 10.1200/JCO.2009.26.5819. [DOI] [PubMed] [Google Scholar]
- 39.White DL, Saunders VA, Dang P, et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood. 2007;110:4064–72. doi: 10.1182/blood-2007-06-093617. [DOI] [PubMed] [Google Scholar]
- 40.White DL, Radich J, Soverini S, et al. Chronic phase chronic myeloid leukemia patients with low OCT-1 activity randomized to high-dose imatinib achieve better responses and have lower failure rates than those randomized to standard-dose imatinib. Haematologica. 2012;97:907–14. doi: 10.3324/haematol.2011.056457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White DL, Saunders VA, Dang P, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- 42.Hiwase DK, Saunders V, Hewett D, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008;14:3881–8. doi: 10.1158/1078-0432.CCR-07-5095. [DOI] [PubMed] [Google Scholar]
- 43.Lu L, Saunders VA, Leclercq TM, et al. Ponatinib is not transported by ABCB1, ABCG2 or OCT-1 in CML cells. Leukemia. 2015;29:1792–4. doi: 10.1038/leu.2015.35. [DOI] [PubMed] [Google Scholar]
- 44.Eadie LN, Hughes TP, White DL. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin Pharmacol Ther. 2014;95:294–306. doi: 10.1038/clpt.2013.208. [DOI] [PubMed] [Google Scholar]
- 45.Sen R, Natarajan K, Bhullar J, et al. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol Cancer Ther. 2012;11:2033–44. doi: 10.1158/1535-7163.MCT-12-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eadie LN, Dang P, Saunders VA, et al. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia. 2016 doi: 10.1038/leu.2016.179. [DOI] [PubMed] [Google Scholar]
- 47.Giannoudis A, Davies A, Harris RJ, et al. The clinical significance of ABCC3 as an imatinib transporter in chronic myeloid leukaemia. Leukemia. 2014;28:1360–3. doi: 10.1038/leu.2014.38. [DOI] [PubMed] [Google Scholar]
- 48.Agrawal M, Hanfstein B, Erben P, et al. MDR1 expression predicts outcome of Ph+ chronic phase CML patients on second-line nilotinib therapy after imatinib failure. Leukemia. 2014;28:1478–85. doi: 10.1038/leu.2014.6. [DOI] [PubMed] [Google Scholar]
- 49.Ni LN, Li JY, Miao KR, et al. Multidrug resistance gene (MDR1) polymorphisms correlate with imatinib response in chronic myeloid leukemia. Med Oncol. 2011;28:265–9. doi: 10.1007/s12032-010-9456-9. [DOI] [PubMed] [Google Scholar]
- 50.Dulucq S, Bouchet S, Turcq B, et al. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2008;112:2024–7. doi: 10.1182/blood-2008-03-147744. [DOI] [PubMed] [Google Scholar]
- 51.Haouala A, Widmer N, Duchosal MA, et al. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2011;117:e75–87. doi: 10.1182/blood-2010-07-294330. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi N, Miura M, Niioka T, et al. Influence of H2-receptor antagonists and proton pump inhibitors on dasatinib pharmacokinetics in Japanese leukemia patients. Cancer Chemother Pharmacol. 2012;69:999–1004. doi: 10.1007/s00280-011-1797-3. [DOI] [PubMed] [Google Scholar]
- 53.Eley T, Luo FR, Agrawal S, et al. Phase I study of the effect of gastric acid pH modulators on the bioavailability of oral dasatinib in healthy subjects. J Clin Pharmacol. 2009;49:700–9. doi: 10.1177/0091270009333854. [DOI] [PubMed] [Google Scholar]
- 54.Wagle M, Eiring AM, Wongchenko M, et al. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia. 2016;30:1493–501. doi: 10.1038/leu.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eiring AM, Khorashad JS, Anderson DJ, et al. beta-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. 2015;29:2328–37. doi: 10.1038/leu.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eiring AM, Kraft IL, Page BD, et al. STAT3 as a mediator of BCR-ABL1-independent resistance in chronic myeloid leukemia. Leuk Suppl. 2014;3:S5–6. doi: 10.1038/leusup.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Traer E, MacKenzie R, Snead J, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26:1140–3. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bewry NN, Nair RR, Emmons MF, et al. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7:3169–75. doi: 10.1158/1535-7163.MCT-08-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skorski T, Kanakaraj P, Nieborowska-Skorska M, et al. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726–36. [PubMed] [Google Scholar]
- 60.Burchert A, Wang Y, Cai D, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19:1774–82. doi: 10.1038/sj.leu.2403898. [DOI] [PubMed] [Google Scholar]
- 61.Okabe S, Tauchi T, Tanaka Y, et al. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with nilotinib against BCR-ABL-positive leukemia cells involves the ABL kinase domain mutation. Cancer Biol Ther. 2014;15:207–15. doi: 10.4161/cbt.26725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chu S, Holtz M, Gupta M, et al. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004;103:3167–74. doi: 10.1182/blood-2003-04-1271. [DOI] [PubMed] [Google Scholar]
- 63.Ma L, Shan Y, Bai R, et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6:252ra121. doi: 10.1126/scitranslmed.3009073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Packer LM, Rana S, Hayward R, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–27. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asmussen J, Lasater EA, Tajon C, et al. MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction. Cancer Discov. 2014;4:200–15. doi: 10.1158/2159-8290.CD-13-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khorashad JS, Eiring AM, Mason CC, et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood. 2015;125:1772–81. doi: 10.1182/blood-2014-08-588855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker CJ, Oaks JJ, Santhanam R, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122:3034–44. doi: 10.1182/blood-2013-04-495374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scott MT, Korfi K, Saffrey P, et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-16-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie H, Peng C, Huang J, et al. Chronic myelogenous leukemia initiating cells require Polycomb group protein EZH2. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang B, Li M, McDonald T, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood. 2013;121:1824–38. doi: 10.1182/blood-2012-02-412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ge X, Wang X. Role of Wnt canonical pathway in hematological malignancies. J Hematol Oncol. 2010;3:33. doi: 10.1186/1756-8722-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–41. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lim S, Saw TY, Zhang M, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A. 2013;110:E2298–307. doi: 10.1073/pnas.1301838110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu J, Meng F, Lu H, et al. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood. 2008;111:3821–9. doi: 10.1182/blood-2007-08-109330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu J, Meng F, Kong LY, et al. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. J Natl Cancer Inst. 2008;100:926–39. doi: 10.1093/jnci/djn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Donato NJ, Wu JY, Stapley J, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–8. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- 77.O’Hare T, Eide CA, Deininger MW. Persistent LYN signaling in imatinib-resistant, BCR-ABL-independent chronic myelogenous leukemia. J Natl Cancer Inst. 2008;100:908–9. doi: 10.1093/jnci/djn204. [DOI] [PubMed] [Google Scholar]
- 78.Ng KP, Manjeri A, Lee KL, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood. 2014;123:3316–26. doi: 10.1182/blood-2013-07-511907. [DOI] [PubMed] [Google Scholar]
- 79.Irvine DA, Zhang B, Kinstrie R, et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci Rep. 2016;6:25476. doi: 10.1038/srep25476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu L, Yu J, Chen R, et al. Dual inhibition of Bcr-Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib-resistant CML cells. Clin Cancer Res. 2015;21:833–43. doi: 10.1158/1078-0432.CCR-13-3317. [DOI] [PubMed] [Google Scholar]
- 81.Kobayashi CI, Takubo K, Kobayashi H, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123:2540–9. doi: 10.1182/blood-2013-07-517847. [DOI] [PubMed] [Google Scholar]
- 82.Chen Y, Peng C, Abraham SA, et al. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J Clin Invest. 2014;124:3847–62. doi: 10.1172/JCI66129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang B, Li L, Chen C-C, et al. Knockdown (KD) of Mir-126 Expression Enhances Tyrosine Kinase Inhibitor (TKI)-Mediated Targeting of Chronic Myelogenous Leukemia (CML) Stem Cells. Blood. 2015;126:51–51. [Google Scholar]
- 84.Dierks C, Beigi R, Guo GR, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–49. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 85.Chen Y, Hu Y, Zhang H, et al. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783–92. doi: 10.1038/ng.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neviani P, Harb JG, Oaks JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123:4144–57. doi: 10.1172/JCI68951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013;122:1293–304. doi: 10.1182/blood-2013-05-501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peng C, Brain J, Hu Y, et al. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood. 2007;110:678–85. doi: 10.1182/blood-2006-10-054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li L, Wang L, Li L, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–81. doi: 10.1016/j.ccr.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–23. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ito K, Bernardi R, Morotti A, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang W, Luan CH, Hjort EE, et al. The role of Fas-associated phosphatase 1 in leukemia stem cell persistence during tyrosine kinase inhibitor treatment of chronic myeloid leukemia. Leukemia. 2016;30:1502–9. doi: 10.1038/leu.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang Q, Crews LA, Barrett CL, et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013;110:1041–6. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reavie L, Buckley SM, Loizou E, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–75. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abraham SA, Hopcroft LE, Carrick E, et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature. 2016;534:341–6. doi: 10.1038/nature18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wylie A, Schoepfer J, Berellini G, et al. ABL001, a Potent Allosteric Inhibitor of BCR-ABL, Prevents Emergence of Resistant Disease When Administered in Combination with Nilotinib in an <em>in Vivo</em> Murine Model of Chronic Myeloid Leukemia. Blood. 2014;124:398–398. [Google Scholar]
- 97.Ottmann OG, Alimena G, DeAngelo DJ, et al. ABL001, a Potent, Allosteric Inhibitor of BCR-ABL, Exhibits Safety and Promising Single-Agent Activity in a Phase I Study of Patients with CML with Failure of Prior TKI Therapy. Blood. 2015;126:138–138. [Google Scholar]
- 98.Smith BD, Hood MM, Kaufman MD, et al. Abstract B78: Rebastinib, a small molecule TIE2 kinase inhibitor, prevents primary tumor growth and lung metastasis in the PyMT breast cancer model. Cancer Research. 2013;73:B78–B78. [Google Scholar]
- 99.Santos FP, Kantarjian H, Cortes J, et al. Bafetinib, a dual Bcr-Abl/Lyn tyrosine kinase inhibitor for the potential treatment of leukemia. Curr Opin Investig Drugs. 2010;11:1450–65. [PubMed] [Google Scholar]
- 100.Borthakur G, Dombret H, Schafhausen P, et al. A phase I study of danusertib (PHA-739358) in adult patients with accelerated or blastic phase chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant or intolerant to imatinib and/or other second generation c-ABL therapy. Haematologica. 2015;100:898–904. doi: 10.3324/haematol.2014.115279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zabriskie MS, Eide CA, Yan D, et al. Extreme mutational selectivity of axitinib limits its potential use as a targeted therapeutic for BCR-ABL1-positive leukemia. Leukemia. 2016;30:1418–21. doi: 10.1038/leu.2015.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pemovska T, Johnson E, Kontro M, et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature. 2015;519:102–5. doi: 10.1038/nature14119. [DOI] [PubMed] [Google Scholar]
- 103.Kwak J-Y, Kim H, Kim JA, et al. Efficacy and Safety of Radotinib Compared with Imatinib in Newly Diagnosed Chronic Phase Chronic Myeloid Leukemia Patients: 12 Months Result of Phase 3 Clinical Trial. Blood. 2015;126:476–476. [Google Scholar]
- 104.Zabriskie MS, Vellore NA, Gantz KC, et al. Radotinib is an effective inhibitor of native and kinase domain-mutant BCR-ABL1. Leukemia. 2015;29:1939–42. doi: 10.1038/leu.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhalla S, Tremblay D, Mascarenhas J. Discontinuing Tyrosine Kinase Inhibitor Therapy in Chronic Myelogenous Leukemia: Current Understanding and Future Directions. Clin Lymphoma Myeloma Leuk. 2016;16:488–494. doi: 10.1016/j.clml.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 106.Saussele S, Richter J, Hochhaus A, et al. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia. 2016 doi: 10.1038/leu.2016.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Talpaz M, Hehlmann R, Quintas-Cardama A, et al. Re-emergence of interferon-alpha in the treatment of chronic myeloid leukemia. Leukemia. 2013;27:803–12. doi: 10.1038/leu.2012.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Preudhomme C, Guilhot J, Nicolini FE, et al. Imatinib plus Peginterferon Alfa-2a in Chronic Myeloid Leukemia. New England Journal of Medicine. 2010;363:2511–2521. doi: 10.1056/NEJMoa1004095. [DOI] [PubMed] [Google Scholar]
- 109.Simonsson B, Gedde-Dahl T, Markevärn B, et al. Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood. 2011;118:3228–3235. doi: 10.1182/blood-2011-02-336685. [DOI] [PubMed] [Google Scholar]
- 110.Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044–54. doi: 10.1038/leu.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hjorth-Hansen H, Stentoft J, Richter J, et al. Safety and efficacy of the combination of pegylated interferon-alpha2b and dasatinib in newly diagnosed chronic-phase chronic myeloid leukemia patients. Leukemia. 2016;30:1853–60. doi: 10.1038/leu.2016.121. [DOI] [PubMed] [Google Scholar]
- 112.Nicolini FE, Etienne G, Dubruille V, et al. Nilotinib and peginterferon alfa-2a for newly diagnosed chronic-phase chronic myeloid leukaemia (NiloPeg): a multicentre, non-randomised, open-label phase 2 study. Lancet Haematol. 2015;2:e37–46. doi: 10.1016/S2352-3026(14)00027-1. [DOI] [PubMed] [Google Scholar]
- 113.ROY L, Chomel J-C, Guilhot J, et al. Combination of Dasatinib and Peg-Interferon Alpha 2b in Chronic Phase Chronic Myeloid Leukemia (CP-CML) First Line: Preliminary Results of a Phase II Trial, from the French Intergroup of CML (Fi-LMC) Blood. 2015;126:134–134. [Google Scholar]
- 114.Cortes J, Saglio G, Baccarani M, et al. Final Study Results of the Phase 3 Dasatinib Versus Imatinib in Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Trial (DASISION, CA180-056) Blood. 2014;124:152–152. [Google Scholar]
- 115.Pinilla-Ibarz J, Cathcart K, Korontsvit T, et al. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood. 2000;95:1781–7. [PubMed] [Google Scholar]
- 116.Bocchia M, Gentili S, Abruzzese E, et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. Lancet. 2005;365:657–62. doi: 10.1016/S0140-6736(05)17945-8. [DOI] [PubMed] [Google Scholar]
- 117.Molldrem JJ, Lee PP, Wang C, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med. 2000;6:1018–23. doi: 10.1038/79526. [DOI] [PubMed] [Google Scholar]
- 118.Rezvani K, Yong AS, Mielke S, et al. Repeated PR1 and WT1 peptide vaccination in Montanide-adjuvant fails to induce sustained high-avidity, epitope-specific CD8+ T cells in myeloid malignancies. Haematologica. 2011;96:432–40. doi: 10.3324/haematol.2010.031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gao L, Bellantuono I, Elsasser A, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95:2198–203. [PubMed] [Google Scholar]
- 120.Dubrovsky L, Pankov D, Brea EJ, et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood. 2014;123:3296–304. doi: 10.1182/blood-2014-01-549022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Järås M, Johnels P, Hansen N, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci U S A. 2010;107:16280–5. doi: 10.1073/pnas.1004408107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Frolova O, Benito J, Brooks C, et al. SL-401 and SL-501, targeted therapeutics directed at the interleukin-3 receptor, inhibit the growth of leukaemic cells and stem cells in advanced phase chronic myeloid leukaemia. Br J Haematol. 2014;166:862–74. doi: 10.1111/bjh.12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lin H, Chen M, Rothe K, et al. Selective JAK2/ABL dual inhibition therapy effectively eliminates TKI-insensitive CML stem/progenitor cells. Oncotarget. 2014;5:8637–50. doi: 10.18632/oncotarget.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gallipoli P, Cook A, Rhodes S, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124:1492–501. doi: 10.1182/blood-2013-12-545640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Prost S, Relouzat F, Spentchian M, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature. 2015;525:380–3. doi: 10.1038/nature15248. [DOI] [PubMed] [Google Scholar]
- 126.Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118:3123–7. doi: 10.1002/cncr.26679. [DOI] [PMC free article] [PubMed] [Google Scholar]