

Abstract
Cancer growth and proliferation rely on intracellular iron availability. We studied the effects of Deferiprone (DFP), a chelator of intracellular iron, on three prostate cancer cell lines: murine, metastatic TRAMP-C2; murine, non-metastatic Myc-CaP; and human, non-metastatic 22rv1. The effects of DFP were evaluated at different cellular levels: cell culture proliferation and migration; metabolism of live cells (time-course multi-nuclear magnetic resonance spectroscopy cell perfusion studies, with 1-13C-glucose; and Seahorse extracellular flux analysis); and expression (Western Blot) and activity of mitochondrial aconitase (m-Acon), an iron-dependent enzyme. The half and 90% inhibitory concentration (IC50 and IC90, respectively) of DFP for the three cell lines after 48 h incubation ranged within 51–67 μM and 81–186 μM, respectively. Exposure to 100 μM DFP led to: (i) Significant inhibition of cell migration after different exposure times, ranging from 12 h (TRAMP-C2) to 48 h (22rv1), in agreement with the respective cell doubling times; (ii) Significantly decreased glucose consumption and glucose-driven TCA cycle activity in metastatic TRAMP-C2 cells, during the first 10 h of exposure, and impaired cellular bioenergetics and membrane phospholipid turnover after 23 h exposure, consistent with a cytostatic effect of DFP. At this time point, all cell lines studied showed (iii) significant decreases in mitochondrial functional parameters associated with oxygen consumption rate, and (iv) both significantly lower m-Acon expression and activity. Our results indicate the potential of DFP to inhibit prostate cancer proliferation at clinically relevant doses and plasma concentrations.
Keywords: Prostate cancer, Cell metabolism, Deferiprone, inhibition of proliferation, Aconitase
Graphical abstract
Deferiprone (DFP), an intracellular iron-chelator, impaired mitochondrial metabolic activity, leading to inhibition of cell growth (IC50, IC90) and migration in different prostate cancer cell lines: murine, metastatic TRAMP-C2; murine, non-metastatic Myc-CaP; and human, non-metastatic 22rv1. Increased, decreased, and unchanged major metabolic effects in response to DFP are depicted by green, pink, and blue markings, respectively. Our results indicate the potential of DFP to inhibit prostate cancer growth at clinically relevant doses.
INTRODUCTION
Iron is known to have both beneficial and deleterious effects in tumor cells, i.e. while potentially toxic to cells and tissues, it is also essential for cancer cell growth and proliferation (1). Although iron has been suggested as a potential chemotherapeutic target (2), most available iron-chelating agents target extracellular iron, leading to systemic iron deficiency and toxicity. Thus, no cancer treatment options targeting intracellular iron are clinically available yet.
Deferiprone (DFP) is an orally administered iron-chelator, used clinically (3). Although mostly used clinically for the treatment of thalassemia (4), Friedreich ataxia (5), and kidney disease (6), DFP has also been shown to inhibit proliferation of skin fibroblasts and frataxin-depleted neuroblastoma-derived cells in vitro (7,8). Unlike other chelating agents, DFP readily enters cells and reaches the major intracellular sites of iron accumulation (8). Specifically, DFP has been shown to remove iron from the mitochondria and impair the activity of mitochondrial aconitase (m-Acon) (7). This enzyme, which catalyzes the two-step isomerization of citrate to isocitrate in the tricarboxylic acid (TCA) cycle, has a unique [Fe4S4]2+ cluster with a labile iron atom that must be replaced occasionally, and is therefore particularly sensitive to cellular iron levels – when these become depleted, the [Fe3S4]+ cluster cannot be regenerated and the enzyme becomes inactive (9).
Normal prostate peripheral tissue has low mitochondrial aconitase (m-Acon) activity, as shown in the rat prostate (10). This has been associated with zinc-induced inhibition of m-Acon in the peripheral prostate epithelial cells of rat and pig (10,11). Activation of m-Acon is an early biochemical change during prostate cancer development and has been associated with a down-regulation of zinc transporters (12). This leads to a shift from citrate-producing to a citrate-oxidizing malignant phenotype, which has been extensively observed in different human prostate cancer cell lines (10). Thus, in the clinical setting, high levels of citrate are typically observed in normal prostate epithelia and is reduced in prostate cancer tissue (13–15), particularly in high-grade tumors (16). These findings, together with the ability of DFP to affect intracellular iron levels and its good clinical profile, make this drug a potential candidate for prostate cancer treatment.
We studied the effects of DFP on proliferation, migration, metabolism, and m-Acon expression in three prostate cancer cell lines. Specifically: murine TRAMP-C2, which can progress to androgen-independent metastatic disease (17); murine Myc-CaP, non-metastatic (18); and human 22rv1, a castration-resistant variant of the parental androgen-dependent CWR22 xenograft (19) that is non-metastatic in mice (20).
EXPERIMENTAL
Cell lines
Three cell lines with androgen receptor expression were used in this work: TRAMP-C2, Myc-CaP, and 22rv1. The TRAMP-C2 cell line was derived from the transgenic adenocarcinoma of the mouse prostate model (TRAMP) (17), and metastasizes. The non-metastatic Myc-CaP cell line was derived from a c-myc transgenic mouse with prostate cancer (18). The 22rv1 cell line was derived from a human prostatic carcinoma xenograft (CWR22), serially propagated in mice after castration-induced regression and relapse of the parental cell line (19). TRAMP-C2 cells were kindly provided by Dr. Sumit Subudhi (Dr. James Allison’s laboratory at MSKCC; originated from ATCC Cat. No. CRL-2731) in 2012 and grown in Dulbecco’s Modified Eagle’s (DME) medium, containing 25 mM glucose, 4 mM glutamine, 1% penicillin/streptomycin (Gibco BRL, USA), 5% fetal bovine serum (FBS, Sigma-Aldrich, St. Louis, MO, USA) and 5% Nu-serum IV (BD Scientific, USA), 5 μg/mL human insulin (Gibco BRL, USA), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, and 10 nM dihydrotestosterone (Steraloids Newport, RI, USA). Myc-CaP and 22rv1 cells were obtained from Dr. Michael Evans (Dr. Charles Sawyer’s laboratory at MSKCC; also available from the American Type Culture Collection (ATCC, Manassas, VA, USA) – catalog numbers CRL-3255 and CRL-2505, respectively) in 2011. Myc-CaP cells were grown in DME medium (5.6 mM glucose, 4 mM glutamine, and 1 mM pyruvate), supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco BRL, USA); 22rv1 cells were grown in Roswell Park Memorial Institute (RPMI) culture medium supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco BRL, USA). All cell lines were grown in 5% CO2 / 95% air (resulting in 20% O2) at 37 °C in a humidified chamber, split every two (or three) days and used up to passage ten. All cell lines were tested by short tandem repeat (STR) profiling in March 2016 at the DNA Diagnostics Center (DDC Medical, Fairfield, OH); 22rv1 cells were authenticated by comparison to the ATCC STR profile database for human cell lines (100% match to CRL-2505). All compounds were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise specified.
Cell proliferation studies
Cells were cultured in 12-well plates, with initial seeding densities of 3.8 × 104 cells (TRAMP-C2 and Myc-CaP) or 5.0 × 104 cells (22rv1). All cells were allowed to attach for 24 h. After that, the number of viable cells was determined in three control wells (Cellsi). In the remaining wells, the medium was changed to culture medium with different concentrations of DFP (139 g/mol): 0, 16, 30, 66, 100, 120, 300, and 660 μM for each cell line. Triplicate wells were used for each DFP concentration. After a 48 h or 96 h incubation period, the cells (Cellsf) were counted with the Trypan Blue exclusion assay using a LUNA™ Automated Cell Counter (Logos Biosystems, Inc., Annandale, VA). The cell doubling times were calculated according to the formula (Eq. 1):
| (Eq.1) |
Cellsi and Cellsf indicate the initial and final number of cells, respectively. The half maximal inhibitory concentration (IC50) and 90% inhibitory concentration (IC90) of DFP were determined from normalized log-lin curves of cell proliferation, using a 3-parameter fit. The normalization consisted in the following formula (Eq. 2):
| (Eq.2) |
Where T and C represent the number of viable cells: pretreated at time 0 (T0); untreated control at time t (C); treated at time t (T). The fitting equations used to determine IC50 and IC90, between Bottom and Top plateaus in the Y axis (100%, untreated control), were as follows: for IC50, a 3-parameter fit of Bottom, HillSlope, and Log(IC50) (Eq. 3).
| (Eq.3) |
The IC90 was then determined based on the HillSlope and the constant F (percentage between Top and Bottom plateau: 50 for IC50 and 10 for IC90), according to the formula (Eq. 4):
| (Eq.4) |
Five independent experiments were carried out for the 48 h incubation period with DFP for each cell line, each from duplicate samples; and three for 96 h incubation (22rv1 cells only), each from triplicate samples.
Cell migration studies
To measure cell migration we used the scratch (wound healing) assay (21). Each cell line was seeded in 6 well plates (1.5 × 106 cells per well) and grown to confluent monolayers. At that point, an ~ 500-μm-wide strip was cut across the well with a standard 200-μl pipette tip (21). The cut monolayers in each well were washed twice with Phosphate Buffered Saline (PBS), to remove remaining cellular debris. Cell migration (the wound-healing process) was monitored at different time points post-scratch (0 to 30 h for TRAMP-C2 and Myc-CaP; 0 to 192 h for 22rv1), using a Nikon Eclipse TS100 inverted microscope and an Insight Mono 2Mp microscope camera without IR filter (Spot Imaging™, Diagnostic Instruments, Sterling HTS, MI). Data were analyzed with TScratch (Developed by the Koumoutsakos group, Computational Science & Engineering Laboratory, ETH Zürich, Switzerland; http://cse-lab.ethz.ch/software/) as the mean percentage of cell-free area at each time point normalized to the area of the initial wound (22). Three independent experiments were carried out for each cell line, each from duplicate samples.
Cell perfusion magnetic resonance (MR) studies
TRAMP-C2 cells were studied in our MR-compatible cell perfusion system (23). For each study, 3.0 ×106 cells were initially seeded on polystyrene microcarrier beads of 125–212 μm diameter (PP104-1521, SoloHill, Ann Arbor, MI, USA), and cultured for two days on bacteriological Petri dishes (08-757-28, Thermo Fisher Scientific, Pittsburgh PA, USA), with daily medium change until reaching ≥70% confluence (23). At that point, the number of cells growing on beads was estimated by counting the nuclei of a representative sample (Petri dish), using the citric acid/crystal violet method, as before (23). A total of 1.3±0.1 ×107 cells (mean±SE, n=7) were counted; the remaining cells growing on the microcarrier beads (9.5±0.9 ×107 (mean±SE, n=7)) were loaded into the perfusion system. The latter consisted of a custom-made, 10 mm MR screw-cap tube (Wilmad-Labglass, Vineland, NJ, USA) where culture medium and oxygen were continuously re-circulated, and the temperature controlled (37 °C) by a heating water jacket and the control unit at the MR spectrometer (23). The studies were carried out on a 500 MHz AVANCE III Bruker scanner equipped with a 10 mm broad-band probe for 1H and 31P/13C acquisitions (Bruker BioSpin, Billerica, MA, USA). During the experiments, the cells were continuously perfused for 32 h with DME media: after setup and the first 1.5 h of each experiment, the perfusion medium was replaced with 250 ml of medium containing 98–99% 1-13C-glucose (CLM-420-0, Cambridge Isotope Labs, Billerica, MA, USA; and 297046, ISOTEC®, Sigma-Aldrich, Miamisburg, OH, USA), for the remaining 30.5 h of the experiment. Three independent experiments were performed in regular medium conditions and four additional experiments in medium containing 100 μM DFP (~IC90). Several 31P MR spectra and 1H-decoupled, Nuclear Overhauser Effect-enhanced 13C MR spectra were acquired repeatedly, using in both cases a pulse-acquire sequence with a 45° flip angle. Phosphorous (31P) MR spectra were acquired with 20 kHz sweep width, 8 k points, 1.2 s relaxation time, and 1280 averages (26.5 min total acquisition time); 13C MR spectra with 30 kHz sweep width, 16 k points, 1.0 s relaxation time, and 303 averages (8.5 min total acquisition time). Spectral analyses of peak areas were carried out in the time domain using AMARES in jMRUI v4.0 (24) (Supplementary Fig. S1). For each experiment, metabolite peaks areas calculated from 13C MR data were normalized for initial cell number using the average initial areas of β-NTP (β-NTPinit) – the latter obtained from 31P MR spectra at the beginning of the experiment (1 hour) before change to culture medium containing 1-13C-glucose with or without DFP.
Extracellular flux analysis
Cells were seeded on 96-well XF96 polystyrene cell culture microplates (Seahorse Bioscience, Billerica, MA), with the following seeding densities per well: TRAMP-C2 and Myc-CaP, 1.0 ×104; 22rv1, 3.0 ×104. The cells were allowed to attach for 18 h in regular cell culture conditions. The medium was then changed in each well, incubating half of the wells with culture medium containing 100 μM DFP and the other half again with regular medium for 24 h. At that point, the medium in each well was changed to unbuffered XF Assay medium (102365-100, Seahorse Bioscience, Billerica, MA), with the same glutamine, glucose and DFP concentrations as the respective 24 h-incubation media, and the plates kept for 1 h in a CO2-free atmosphere. The cells were then studied in an Extracellular Flux XF96e Analyzer (Seahorse Bioscience, Billerica, MA, USA), measuring in each well the basal extracellular acidification rate (ECAR), and time-course oxygen consumption rates (OCR) in response to sequential injections of: oligomycin (inhibition of ATP synthase); Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; H+ ionophore, uncoupler); and antimycin A plus rotenone (inhibition of mitochondrial complexes I and III, respectively), disabling the electron transport completely in the respiratory chain (Sigma-Aldrich, St. Louis, MO, USA) (25). For each well, the collected time-course data were normalized to its total protein content. The latter were determined at the end of the experiment from the cell lysates (RIPA buffer: Thermo Scientific, Rockford, IL, USA) using the bicinchoninic acid protein assay (BCA kit: Thermo Scientific Pierce, Rockford, IL, USA). Mitochondrial functional parameters (basal respiration, ATP production, maximal respiration, spare respiratory capacity, non-mitochondrial respiration, and proton leak) were calculated from OCR curves, normalized to the number of cells in each well (estimated from the already determined total protein content), as previously reported (23). Two independent experiments were carried out for each cell line, each one measuring an average of 16 wells per experimental condition.
Expression of m-Acon
TRAMP-C2, MyCaP and 22rv1 cells were seeded in T75 flasks (TRAMP-C2, 7.5×105; Myc-CaP and 22rv1, 1.0 ×106) and left to attach for 24 h, in standard conditions (as described in the “cell lines” sub-section). The medium was changed and cells were incubated with 100 μM DFP or in standard medium, for an additional 24 h period, after which they were used for Western Blot studies. Cell pellets from 80% confluent TRAMP-C2, Myc-CaP and 22rv1 cells were collected, kept frozen at −80 °C for up to 2 weeks, and then lysed with RIPA Buffer and protease inhibitors cocktail (Halt Protease Inhibitor Cocktail 1:100; Thermo Scientific, Rockford, IL, USA). The protein concentrations were determined with the BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, IL, USA). For each cell line, equivalent amounts of protein (20 mg) were separated by electrophoresis using a NuPAGE 4–12% Bis-Tris gradient gel (Invitrogen, Life Tech. Corporation, Grand Island, NY, USA) and transferred to an Immun-Blot PVDF membrane (BioRad, Hercules, CA, USA) as before (26). Membranes were blocked in 5% milk in Tris-buffered saline (TBS) with Tween-20 buffer and were immunoblotted with anti-m-Acon antibody (#6922; Cell Signaling Technology, Danvers, MA, USA) at a 1:1000 dilution. Bound primary antibodies were visualized with appropriate horseradish peroxidase–conjugated secondary antibodies (1:2000) using enhanced chemiluminescence reagent (Western Lightning-ECL, PerkinElmer, Waltham, MA, USA). Immunoblots were stripped using Restore Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL, USA) and re-probed with anti-β-Actin antibody (Cell Signaling, Beverly, MA, USA) at a 1:1000 dilution. The m-Acon and β-Actin bands in the membranes were then scanned and quantified by pixel analyses using ImageJ (NIH open source software, USA: http://rsb.info.nih.gov/ij/). Three independent experiments were carried out for each cell line.
Activity of m-Acon
For m-Acon activity measurements, cells were seeded in T225 flasks (TRAMP-C2, 3.5×106; Myc-CaP and 22rv1, 1.0 ×107) and left to attach for 24 h, in standard conditions (as described in the “cell lines” sub-section). The medium was changed and cells were incubated with 100 mM DFP or in standard medium, for an additional 24 h period. The total activity of mitochondrial aconitase (m-Acon) was assessed in TRAMP-C2, Myc-CaP, and 22rv1 cells, using a specific enzyme activity microplate assay kit (ab109712, Abcam, Cambridge, MA, USA). The protocol consisted of two steps, as described by the manufacturer: (i) isolating the mitochondria from fresh cell lysates; (ii) measuring the activity of m-Acon, as an increase in absorbance at 240 nm associated with the formation of cis-aconitate (mmol/min). Activity was determined from the slope between two time point measurements (10 and 30 min of incubation time, based on previous experiments showing a strong linear dependence: Supplementary Fig. S2) and normalized to total cellular protein content. Three independent experiments were performed for each cell line, with triplicate measurements for each experimental condition, to evaluate the effect of 24 h exposure to DFP.
Statistical analyses
Statistical analyses were carried out with Graph Pad Prism v6.01 (Graph Pad Software, La Jolla, CA, USA). DFP inhibitory concentrations were compared between cell lines in each experimental condition, using 1-way ANOVA with Tukey’s correction for multiple comparisons; IC50 and IC90 were assessed independently. The effect of DFP treatment in each cell line was evaluated independently for each parameter measured, using 2-way ANOVA with Sidak’s multiple comparisons test, to account for either the cell passage / experiment number (doubling times, ECAR and OCR parameters, and m-Acon activity) or the incubation periods (migration). Finally, the treatment effects of DFP assessed by MR cell perfusion and Western Blot expression, were analyzed independently for each cell line with a Student’s t-Test: paired, when comparing the same cells in consecutive stages during the same study to the initial reference value (MR cell perfusion studies), or cells from the same batch in treatment and control groups (Western Blot studies); or unpaired, when comparing different experiments (MR cell perfusion studies). * Indicates statistical significance (p < 0.05) and error bars indicate the standard error (SE) of the means, unless indicated otherwise.
RESULTS
DFP inhibits prostate cancer cell proliferation
DFP had a significant inhibitory effect on proliferation in all cell lines studied after 48 h exposure (Fig. 1A). Under regular growth conditions (0 μM DFP) for 48 h, the doubling times of TRAMP-C2, Myc-CaP, and 22rv1 cells were (average ± SD): 12.1±0.3 h, 12.0±0.6 h, and 26.8±3.7 h, respectively (Fig. 1A). After 48 h, TRAMP-C2 and Myc-CaP cells in control wells (0 μM DFP) would become confluent, which for Myc-CaP cells led to detachment from the microplate surface. Due to their longer doubling time, we additionally incubated 22rv1 cells with DFP for 96 h. All cell lines and treatment times tested showed a significant inhibition of cell proliferation after incubation with all DFP concentrations tested above 30 μM (Fig. 1A). Log-lin curves of base count-corrected, normalized cell numbers over DFP concentrations were calculated for each cell line and treatment time, showing a cytostatic effect of DFP (Fig. 1B) and resulting in IC50 and IC90 values of about 50 and 100 μM, respectively (average ± SD): 48 h TRAMP-C2, 50.7±1.0 and 81.1±4.7 μM; 48 h Myc-CaP, 55.1±2.5 and 94.2±11.5 μM; 48 h 22rv1, 67.2±5.7 and 185.9±72.3 μM; 96 h 22rv1, 44.4±4.9 and 110.6±30.1 μM (Fig. 1C). Thus, for further studies we selected 100 μM DFP as the average minimum concentration that generates a maximum inhibition of proliferation in the three cell lines tested – a concentration also attainable clinically in plasma (27).
Figure 1.
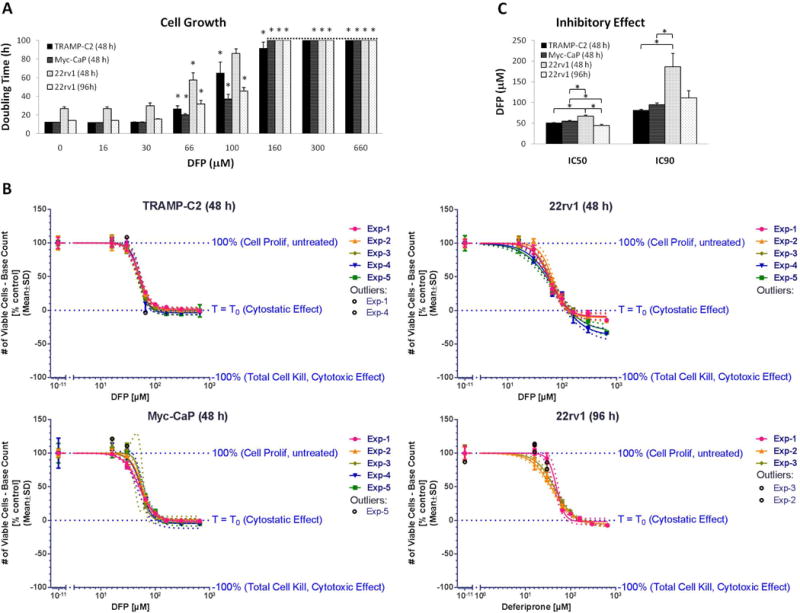
Inhibition of prostate cancer cell proliferation after incubation with different concentrations of DFP. TRAMP-C2, Myc-CaP and 22rv1 cells were exposed to DFP for 48 h; 22rv1 cells were additionally exposed for 96 h because of their longer doubling times. A, Doubling times based on the number of viable cells at the time of medium change (base count, average ± SD): 48 h TRAMP-C2 group, (4.5±1.4) ×104; 48 h Myc-CaP group, (7.4±1.0) ×104; 48 h 22rv1 group, (4.8±0.8) ×104; 96 h 22rv1 group, (4.6±0.5) ×104. * mark significant differences in cell doubling time compared to untreated control cells for each cell line. B, Normalized log-lin curves (n=5 for 48 h exposure and n = 3 for 96 h exposure of 22Rv1) for each cell line, indicating the thresholds for untreated (Top), cytostatic effect (Bottom), and cytotoxic effect (inferior limit). Markers depict the experimental data, overlaid with the curve fit (solid line) and the 95% confidence bands (dotted lines). For each independent experiment, data were averaged from triplicate wells for each condition. C, IC50 and IC90 average values determined for each cell line and incubation time, from the respective log-lin curves, depicted in B.
DFP inhibits prostate cancer cell migration
Besides its inhibitory effect on cell proliferation, DFP also inhibited cell migration, as measured by the Scratch Assay (Fig. 2). We noticed significant differences in cell migration starting at different time points for each cell line, ranging from 12 h in TRAMP-C2 cell to 48 h in 22rv1 cells; Myc-CaP cells showed a significant difference after 30 h incubation. These differences should be related in part to the different basal cell doubling times, which are considerably longer for 22rv1 cells (~27 h) than for TRAMP-C2 or Myc-CaP cells (~12 h).
Figure 2.
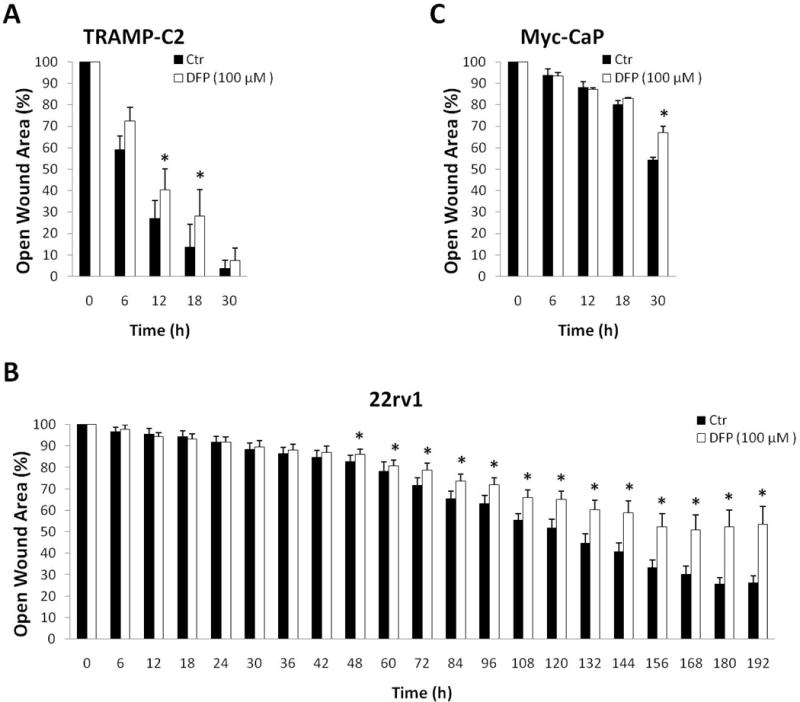
Inhibition of cell migration due to DFP (100 μM) exposure. Scratch assays for: A, TRAMP-C2; B, Myc-CaP; C, 22rv1.
DFP induces early changes in the metabolism of TRAMP-C2 cancer cells
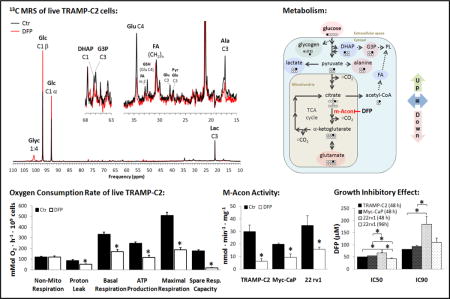
We used our MR-compatible cell perfusion system to determine the effects of DFP on prostate cancer cell metabolism during the first 32 h of exposure. Thus, TRAMP-C2 cells were studied with and without 100 μM DFP in the circulating perfusion medium. During that time, dynamic 31P-MRS monitoring (Fig. 3A) showed that cells exposed to DFP internalize significantly less Pi from the medium after 17 h exposure (Fig. 3B); at this time point, no apparent differences in β-NTP (which is mostly ATP (28,29)) were detected between control and treated cells. No significant differences between control and DFP-treated cells were detectable throughout the entire experiment for intra- and extra-cellular pH (Supplementary Fig. S3). However, after 23 h exposure to the drug, tendencies were clearly noted for lower ATP (β-NTP) and NAD(P)+(H) levels (Fig. 3B) in treated cells. At the same time, differences in choline metabolism became significant, as shown by the increase in the glycerophosphocholine-to-phosphocholine ratio (GPC/PC) in treated cells, while GPC/PC was constant during the experiment for untreated cells (Fig. 3B). These observations are consistent with inhibition of cell proliferation in DFP-treated cells; whereas the over time stable intra- and extra-cellular pH, as well as increasing phospholipid precursor signal with a steady GPC/PC and energy (NTPs) metabolites in the control cells, indicate healthy cell growth and proliferation without undue cellular stress due to lack of nutrients or perfusion medium acidification (Fig. 3).
Figure 3.
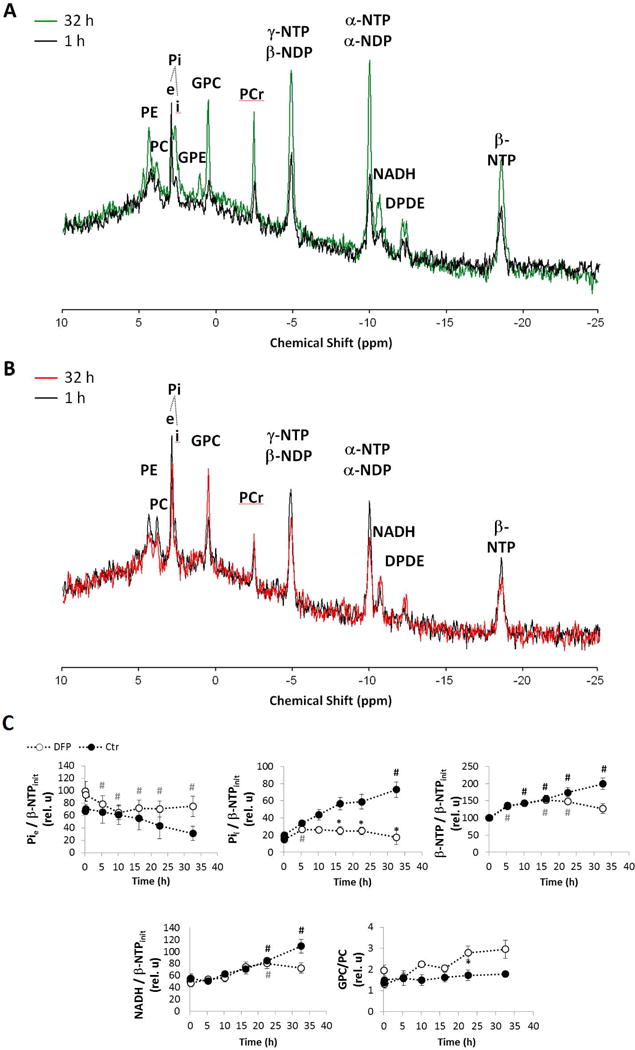
Time-course effect of DFP (100 μM) on live TRAMP-C2 cell metabolism during MR perfusion experiments, as detected by 31P MRS. A-B, Representative 31P MR spectral profiles after 1 h (black) and at 32 h of cell perfusion for untreated control cells (green, A) and DFP-treated cells (red, B) – major peak assignments displayed: phosphoethanolamine (PE); phosphocholine (PC); extracellular (e) and intracellular (i) inorganic phosphate (Pi); glycerophosphoethanolamine (GPE); glycerophosphocholine (GPC); phosphocreatine (PCr); α, β and γ nucleoside-diphosphate (NDP) and -triphosphate (NTP); total nicotinamide adenine dinucleotides (NADH, NAD+, NADPH, NADP+); and diphosphodiesters (DPDE). C, Average time-course changes of Pie; and Pii, β-NTP, NADH, normalized to initial value of β-NTP (β-NTPinit), and GPC-to-PC ratio; experiments carried out in regular medium (filled circles) and in the presence of 100 μM DFP (open circles). # (black for Ctr and grey for DFP-treated cells), significantly different compared to the initial level, for each cell line (paired t-Test); * significantly different values between Ctr and DFP-treated cells at the same time point (unpaired t-Test).
Parallel to 31P MRS, dynamic 13C MRS monitoring of TRAMP-C2 cells perfused with 1-13C-glucose (Fig. 4A) showed marked changes in glucose metabolism(Fig. 4B). Specifically, DFP treatment led to a 6-fold decrease in 1-13C-glucose uptake rate from the extracellular medium after 5 h perfusion (Fig. 4C). At the same time, cellular incorporation of glucose (1-13C-Glc) into glycogen (1-13C-Glyc) was significantly higher in the presence of DFP. Along the glycolytic pathway, no significant changes were detected at any time in 1-13C-glucose incorporation into dihydroxyacetone phosphate (1-13C-DHAP), although the incorporation rate into glycerol-3-phosphate (3-13C-G3P) decreased after 22 h exposure to DFP. Moreover, DFP did not induce any changes in the glycolytic synthesis rate of lactate (3-13C-Lac), but less 1-13C-glucose was used for de novo synthesis of alanine (3-13C-Ala) after 11 h exposure. It is noted that the unchanged rate of conversion of 1-13C glucose into labeled lactate indicates that the cells maintain glycolysis despite the diminishing glucose supply over time. As far as incorporation of 13C labeling from glucose into the mitochondria, specific changes were noted in the TCA (tricarboxylic acid) cycle, downstream of citrate. Thus, DFP essentially blocked the synthesis of glutamate (4-13C-Glu) within the first hours of exposure, according to the kinetic plots (Fig. 4B), reaching significance after 7 h (Fig. 4C). These observations suggest an inhibition of m-Acon, and therefore restriction of the glucose-driven TCA cycle (Fig. 4D). The low labeling of Glu-C2 and Glu-C3 in the untreated cells, relative to Glu-C4 (Supplementary Fig. S4), also indicate that most of the 1-13C-glucose labeling enters the TCA cycle via pyruvate dehydrogenase, and not pyruvate carboxylase: while the latter would only generate Glu-C2 labeling in the first turn of the TCA cycle (not observed), the former leads to Glu-C4 labeling in the first turn (most prevalent), and additional labeling of Glu-C2 and Glu-C3 in the second turn. Moreover, no changes were detected in the rate of incorporation of acetate (2-13C-acetate, derived from 13C-labeled citrate exported to the cytosol) into fatty acids ((CH2)-13C-FA) but the synthesis rate of G3P decreased.
Figure 4.
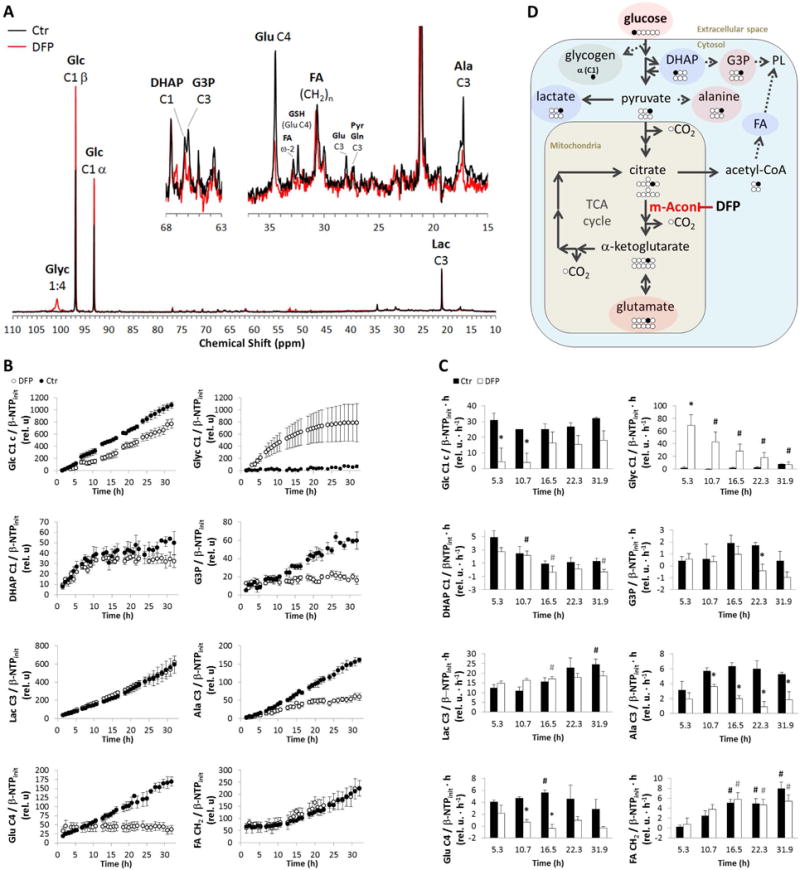
Time-course effect of DFP (100 μM) on live TRAMP-C2 cell metabolism during perfusion experiments, as detected by 13C MRS. Position of carbon labeled by 13C from 1-13C-labeled glucose (Glc C1) depicted by number following C. A, Representative 13C-MR spectral profiles after 24 h of experiment time for untreated control (Ctr, black) and DFP-treated (DFP, red) TRAMP-C2 cells – major peak assignments displayed: glycogen (Glyc); glucose (Glc); dihydroxyacetone phosphate (DHAP); glycerol-3-phosphate (G3P); glutamate (Glu); fatty acids (FA); glutathione (GSH); pyruvate (Pyr), glutamine (Gln); alanine (Ala); and lactate (Lac). B, Average time course of 1-13C-Glc consumption and synthesis of 1-13C-Glyc, 1-13C-DHAP, 3-13C-G3P, 3-13C-Lac, 3-13C-Ala, 4-13C-Glu, and (CH2)-13C-FA, normalized to initial value of β-NTP (β-NTPinit). C, Average metabolite synthesis (consumption) rates at different time intervals. D, Biochemical model illustrating the inhibitory effect of DFP on m-Acon and the major metabolic changes detected in response to DFP exposure: increase (green); decrease (red); no change (blue). PL, phospholipids. # (black for Ctr and grey for DFP-treated cells), significantly different compared to the initial level, for each cell line (paired t-Test); * significantly different values between Ctr and DFP-treated cells at the same time point (unpaired t-Test).
DFP decreases oxygen consumption rate (OCR) and mitochondrial function in prostate cancer cells
To further investigate the effects of DFP on prostate cancer cells, we carried out extracellular flux analysis on TRAMP-C2, Myc-CaP, and 22rv1 cells. Our preliminary studies with TRAMP-C2 cells suggested that OCR significantly decreased with increasing DFP concentration and incubation periods (Supplementary Fig. S5). When studying the three cell lines in parallel (Fig. 5), we observed that DFP (100 μM) did not induce significant changes in basal ECAR in 22rv1 cells, while increasing this parameter by 40–48% in TRAMP-C2 and Myc-CaP cells (Fig. 5A). Based on the time-course OCR curves (Fig. 5B), DFP induced significant reductions in both basal respiration and maximal respiration in all cell lines tested (Fig. 5C): a stronger effect was noticed in TRAMP-C2 cells (−49% and −63%, respectively), while basal respiration was only reduced by −14% in 22rv1 cells. TRAMP-C2 and Myc-CaP cells also showed significant reductions in both mitochondrial ATP production (−53% and −32%, respectively) and proton leak (−38% and −42%, respectively), whereas spare respiratory capacity significantly decreased in TRAMP-C2 and 22rv1 cells (−90% and −237%, respectively).
Figure 5.
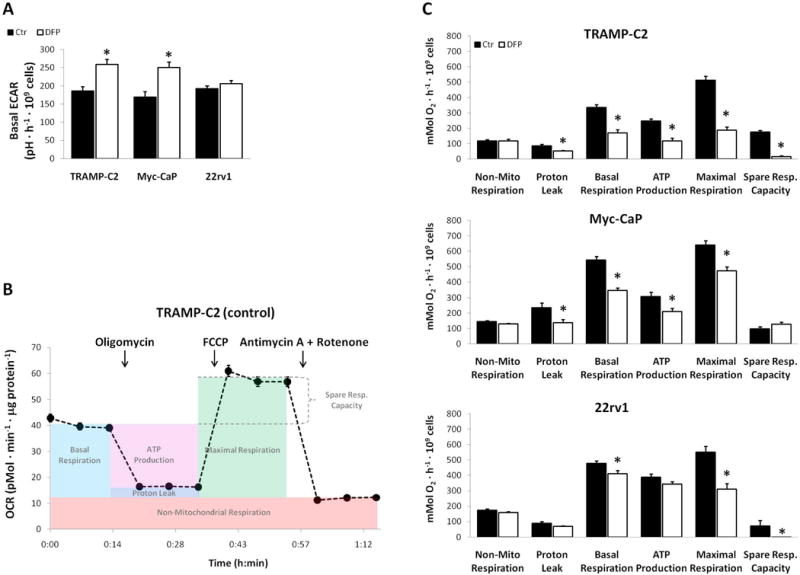
Extracellular flux analysis in TRAMP-C2, Myc-CaP and 22rv1 cells incubated with 100 μM DFP for 24 h. A, Basal ECAR measurements for treated (DFP) and untreated (Ctr) groups, averaged over 15 min experiment time. B, Average OCR time-course measurements in TRAMP-C2 cells (single, control experiment) during sequential injections of different drugs, and ranges for calculating the functional parameter, as described by the manufacturer. C, Mitochondrial functional parameters derived from OCR time-course curves, for treated (DFP) and untreated (Ctr) groups.
DFP reduces the expression and activity of m-Acon in prostate cancer cells
The expression of m-Acon was investigated by Western Blot in TRAMP-C2, Myc-CaP, and 22rv1 cells, after 24 h exposure to 100 μM DFP (Fig. 6A). Deferiprone significantly downregulated the expression of m-Acon in all cell lines, up to −55% in Myc-CaP cells (Fig. 6B). In the same experimental conditions (24 h exposure to 100 μM DPF), the activity of m-Acon was also significantly reduced in all cell lines, by 2-fold in Myc-CaP and 22 rv1 cells and up to −79% in TRAMP-C2 cells (Fig. 6C).
Figure 6.
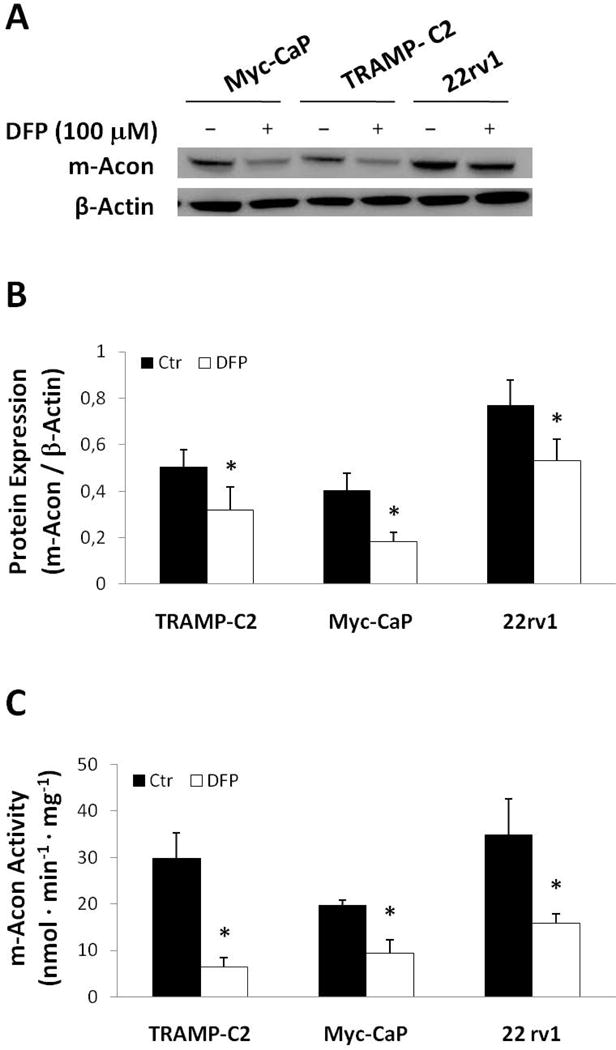
Effect of DFP (100 μM) on m-Acon expression in TRAMP-C2, Myc-CaP and 22rv1 cells after a 24 h incubation period. A, Representative Western blot membrane showing expression bands for m-Acon (top) and β-Actin (bottom) for each cell line and condition. B, Quantification of m-Acon expression by Western Blot normalized to β-Actin for each treated (DFP 100 μM) and untreated (Ctr) cell line (n=3 each). C, Effect of DFP (100 μM) on m-Acon activity in each cell line. Activity normalized to total cell protein content for each cell line. * significant difference between Ctr and DFP-treated cells for each cell line (paired t-Test).
DISCUSSION
We have investigated the effects of DFP on cell proliferation, migration, and metabolism in three prostate cancer cell lines: murine TRAMP-C2 (metastatic) and Myc-CaP (non-metastatic), as well as human 22rv1 (non-metastatic). After a 48 h incubation period, the IC50 of DFP was in the 51–67 μM range for all 3 cell lines; with full cytostatic effect at ~100 μM (IC90 in the 81–186 μM range). Deferiprone (100 μM) inhibited cell migration at different time points according to doubling times, with significant effects detected between 12 h (TRAMP-C2) and 48 h (22rv1). Time-course cell perfusion MR studies using TRAMP-C2 cells showed early metabolic changes during the first 24 h of exposure to 100 μM DFP, indicating altered glucose and phospholipid metabolism associated with mitochondrial impairment. The latter was supported in all cell lines by extracellular flux analysis, showing decreased mitochondrial functional parameters based on oxygen consumption, such as basal and maximal respiration. Our results also indicate significant inhibition of m-Acon by DFP, which was confirmed in all cell lines by Western Blot expression and enzymatic activity assays, and consistent with reduced TCA cycle activity (Fig. 4D) and reduced oxygen consumption rates (Fig. 5C).
The earliest metabolic changes in response to DFP exposure (100 μM) were dynamically monitored during cell perfusion in the more aggressive prostate cancer cell line, TRAMP-C2. Our cell perfusion studies showed a significant decrease in (1-13C labeled) glucose uptake from the extracellular medium, and its higher incorporation into glycogen was detected during the first 5 h. After 11 h of incubation with DFP, no significant changes were detected in dihydroxyacetone phosphate or lactate synthesis rates, although other metabolites branching from the glycolytic pathway were significantly decreased (alanine and glycerol-3-phosphate), and glutamate synthesis through the TCA cycle had essentially been blocked. The latter observation could (1) explain the simultaneous lower synthesis rate of 3-13C-alanine, as a consequence of reduced supply of glutamate for the transamination of pyruvate, (2) is in agreement with inhibition of m-Acon detected in all cell lines (Western Blot and activity assays), and (3) matches the decrease in mitochondrial functional parameters in the complete cell line panel (extracellular flux analysis). While reducing their mitochondrial metabolism under stress (iron chelation leading to cytostatic effect), TRAMP-C2 retain their ability to synthesize fatty acids from glucose at a stable rate, in agreement with the “lipogenic nature” of prostate cancer (30). However, the higher GPC/PC ratio indicated less aggressive growth and proliferation after 23 h, consistent with studies on human prostate cancer tissues (31), and consistent with the associated lower levels of glucose-derived glycerol-3-phosphate, as this metabolite is essential for de novo synthesis of phospholipids.
The metabolic data obtained from TRAMP-C2 MR cell perfusion studies, including the differences in ATP and total NAD (NAD+, NADH, NADP+, NADPH) detected after 23 h of incubation with DFP, are in good agreement with the decreased cell proliferation detected in all cell lines. More specifically, viable DFP treated cells with normal ATP levels (β-NTP) internalize significantly less Pi than controls after 10 h of incubation while presenting no significant changes in their total NAD levels throughout the entire study (Supplementary Fig. S6). These changes are also compatible with the extracellular flux analysis results for the complete cell line panel studied, showing a decrease in mitochondrial functional parameters related to oxidative phosphorylation and TCA cycle activity. Accordingly, cells should become more dependent on glycolysis to keep their redox balance (NAD+ recycling). This is suggested both by the early extracellular acidification rates measured in 2D cell culture microplates (total lactate exported increased in the first 15 min in TRAMP-C2 and Myc-CaP cell lines treated with DFP, while remaining unchanged in 22rv1 cells) and the long term glucose-derived lactate synthesis rates determined in 3D growing, perfused cells (unchanged after 32 h in treated TRAMP-C2 cells). These metabolite changes strengthen the importance of glycolysis in prostate cancer metabolism (32), as a major provider of biosynthetic precursors and bioenergetics.
We acknowledge that extending the cell perfusion studies of intermediary 1-13C-glucose metabolism to additional prostate cancer cell lines could help elucidating the consistency of our findings across different phenotypes of the disease. However, this was not compatible with the long experimental times required for such studies and our limited access to the MR system. We also acknowledge, and consider it likely, that DFP may target other intra-cellular, iron-containing enzymes besides m-Acon, such as respiratory chain complexes and ribonucleotide reductase, and may impact other cellular processes leading to cell inhibition of cell proliferation. For example, the iron-chelator L-mimosine has been shown to downregulate m-Acon, fatty acid synthase and lactate dehydrogenase A expression in prostate cells through HIF-1α stabilization (33), demonstrating the potential multi-targeted effects of iron. While we expect m-Acon to be particularly susceptible to the early effects of intracellular iron chelation by DFP (due to its unique structure with a labile iron), further studies – beyond the scope of the current study – are necessary to characterize other potential biological effects of intracellular iron-chelation in prostate cancer by DFP, including and not limited to testing additional prostate cancer cell lines with distinct glycolytic profiles (34).
Based on the current clinical dosage of DFP in thalassemia (75 mg/kg/day (4)) and the serum levels in patients (80–450 μM / 3 g dose (27)), our studies indicate the potential of DFP to inhibit prostate cancer growth and proliferation at clinically relevant doses. Importantly, patients have been treated for thalassemia for periods exceeding 15 years without significant effects limiting quality of life (Alan R Cohen MD, University of Pennsylvania and Children’s Hospital of Philadelphia, personal communication, 2013). While the majority of these patients have been in an iron overloaded state due to thalassemia, other studies (5,6,35) have also investigated patients with non-iron overload disease, at DFP doses ranging from 20–30 mg/kg/day to 75mg/kg/day, wherein it was well tolerated. Thus, further studies with DFP, alone and in combination with chemotherapeutics, shall elucidate to a greater extent its intracellular iron-chelation effects in prostate cancer and other tumors, and investigate its efficacy in vivo in animal models to evaluate potential clinical translation. The latter could be relatively quick, since dose, toxicity and schedule in patients are known.
Supplementary Material
Acknowledgments
The authors thank Dr. George Sukenick for granting access to the NMR Core Facility at MSKCC, and Dr. Jaya Satagopan for helpful advice on statistical analyses. We acknowledge support from the following grants: R01 CA163980 (NCI, RGB), P30 CA008748 (NCI, Cancer Center Support – Core Grant), W81XWH-13-1-0386 (DOD, JAK), P50-CA092629 – Prostate SPORE (NCI, Koutcher PI of Imaging Core).
ABBREVIATIONS
- Ala
alanine
- Ctr
control
- DFP
Deferiprone
- DHAP
dihydroxyacetone phosphate
- DME
Dulbecco’s Modified Eagle’s
- ECAR
extracellular acidification rate
- FA
fatty acids
- FBS
fetal bovine serum
- FCCP
Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- G3P
glycerol-3-phosphate
- Glc
glucose
- Gln
glutamine
- GSH
glutathione
- Glu
glutamate
- Glyc
glycogen
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IC
inhibitory concentration
- Lac
lactate
- m-Acon
mitochondrial aconitase
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- Pyr
pyruvate
- RPMI
Roswell Park Memorial Institute
- STR
short tandem repeat
- TCA
tricarboxylic acid
- TRAMP
transgenic adenocarcinoma of the mouse prostate model
References
- 1.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kovacevic Z, Kalinowski DS, Lovejoy DB, Yu Y, Suryo Rahmanto Y, Sharpe PC, Bernhardt PV, Richardson DR. The medicinal chemistry of novel iron chelators for the treatment of cancer. Curr Top Med Chem. 2011;11:483–499. doi: 10.2174/156802611794785190. [DOI] [PubMed] [Google Scholar]
- 3.Piga A, Roggero S, Salussolia I, Massano D, Serra M, Longo F. Deferiprone. Ann N Y Acad Sci. 2010;1202:75–78. doi: 10.1111/j.1749-6632.2010.05586.x. [DOI] [PubMed] [Google Scholar]
- 4.Cohen AR, Galanello R, Piga A, De Sanctis V, Tricta F. Safety and effectiveness of long-term therapy with the oral iron chelator deferiprone. Blood. 2003;102:1583–1587. doi: 10.1182/blood-2002-10-3280. [DOI] [PubMed] [Google Scholar]
- 5.Boddaert N, Le Quan Sang KH, Rotig A, Leroy-Willig A, Gallet S, Brunelle F, Sidi D, Thalabard JC, Munnich A, Cabantchik ZI. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–408. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- 6.Rajapurkar MM, Hegde U, Bhattacharya A, Alam MG, Shah SV. Effect of deferiprone, an oral iron chelator, in diabetic and non-diabetic glomerular disease. Toxicology mechanisms and methods. 2013;23:5–10. doi: 10.3109/15376516.2012.730558. [DOI] [PubMed] [Google Scholar]
- 7.Goncalves S, Paupe V, Dassa EP, Rustin P. Deferiprone targets aconitase: implication for Friedreich’s ataxia treatment. BMC Neurol. 2008;8:20. doi: 10.1186/1471-2377-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glickstein H, El RB, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106:3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 9.Beinert H, Kennedy MC. Aconitase, a two-faced protein: enzyme and iron regulatory factor. FASEB J. 1993;7:1442–1449. doi: 10.1096/fasebj.7.15.8262329. [DOI] [PubMed] [Google Scholar]
- 10.Costello LC, Franklin RB. Aconitase activity, citrate oxidation, and zinc inhibition in rat ventral prostate. Enzyme. 1981;26:281–287. doi: 10.1159/000459195. [DOI] [PubMed] [Google Scholar]
- 11.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem. 1997;272:28875–28881. doi: 10.1074/jbc.272.46.28875. [DOI] [PubMed] [Google Scholar]
- 12.Singh KK, Desouki MM, Franklin RB, Costello LC. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol Cancer. 2006;5:14. doi: 10.1186/1476-4598-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurhanewicz J, Vigneron DB. Advances in MR spectroscopy of the prostate. Magn Reson Imaging Clin N Am. 2008;16:697–710. ix–x. doi: 10.1016/j.mric.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Martin ML, Adrados M, Ortega MP, Fernandez Gonzalez I, Lopez-Larrubia P, Viano J, Garcia-Segura JM. Quantitative (1) H MR spectroscopic imaging of the prostate gland using LCModel and a dedicated basis-set: correlation with histologic findings. Magn Reson Med. 2011;65:329–339. doi: 10.1002/mrm.22631. [DOI] [PubMed] [Google Scholar]
- 15.Bertilsson H, Tessem MB, Flatberg A, Viset T, Gribbestad I, Angelsen A, Halgunset J. Changes in gene transcription underlying the aberrant citrate and choline metabolism in human prostate cancer samples. Clin Cancer Res. 2012;18:3261–3269. doi: 10.1158/1078-0432.CCR-11-2929. [DOI] [PubMed] [Google Scholar]
- 16.Zakian KL, Sircar K, Hricak H, Chen HN, Shukla-Dave A, Eberhardt S, Muruganandham M, Ebora L, Kattan MW, Reuter VE, Scardino PT, Koutcher JA. Correlation of proton MR spectroscopic imaging with gleason score based on step-section pathologic analysis after radical prostatectomy. Radiology. 2005;234:804–814. doi: 10.1148/radiol.2343040363. [DOI] [PubMed] [Google Scholar]
- 17.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- 18.Watson PA, Ellwood-Yen K, King JC, Wongvipat J, Lebeau MM, Sawyers CL. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65:11565–11571. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 19.Sramkoski RM, Pretlow TG, 2nd, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999;35:403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 20.Kovar JL, Johnson MA, Volcheck WM, Chen J, Simpson MA. Hyaluronidase expression induces prostate tumor metastasis in an orthotopic mouse model. Am J Pathol. 2006;169:1415–1426. doi: 10.2353/ajpath.2006.060324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 22.Geback T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 23.Simoes RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, Sukenick G, Locasale JW, Blasberg RG, Koutcher JA, Ackerstaff E. Metabolic plasticity of metastatic breast cancer cells: adaptation to changes in the microenvironment. Neoplasia. 2015;17:671–684. doi: 10.1016/j.neo.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefan D, Di Cesare F, Andrasescu A, Popa E, Lazariev A, Vescovo E, Strbak O, Williams S, Starcuk Z, Cabanas M, van Ormondt D, Graveron-Demilly D. Quantitation of magnetic resonance spectroscopy signals: the jMRUI software package. Meas Sci Technol. 2009;20 [Google Scholar]
- 25.Invernizzi F, D’Amato I, Jensen PB, Ravaglia S, Zeviani M, Tiranti V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion. 2012;12:328–335. doi: 10.1016/j.mito.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serganova I, Rizwan A, Ni X, Thakur SB, Vider J, Russell J, Blasberg R, Koutcher JA. Metabolic imaging: a link between lactate dehydrogenase A, lactate, and tumor phenotype. Clin Cancer Res. 2011;17:6250–6261. doi: 10.1158/1078-0432.CCR-11-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kontoghiorghes GJ, Goddard JG, Bartlett AN, Sheppard L. Pharmacokinetic studies in humans with the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Clin Pharmacol Ther. 1990;48:255–261. doi: 10.1038/clpt.1990.147. [DOI] [PubMed] [Google Scholar]
- 28.Allavena C, Guerquin-Kern JL, Lhoste JM. Follow-up by 31P NMR spectroscopy of the energy metabolism of malignant tumor in rats during treatment. Radiother Oncol. 1991;21:48–52. doi: 10.1016/0167-8140(91)90340-m. [DOI] [PubMed] [Google Scholar]
- 29.Evanochko WT, Sakai TT, Ng TC, Krishna NR, Kim HD, Zeidler RB, Ghanta VK, Brockman RW, Schiffer LM, Braunschweiger PG, et al. NMR study of in vivo RIF-1 tumors. Analysis of perchloric acid extracts and identification of 1H, 31P and 13C resonances. Biochim Biophys Acta. 1984;805:104–116. doi: 10.1016/0167-4889(84)90042-9. [DOI] [PubMed] [Google Scholar]
- 30.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012;98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson MG, Keshari KR, Tabatabai ZL, Simko JP, Shinohara K, Carroll PR, Zektzer AS, Kurhanewicz J. Quantification of choline- and ethanolamine-containing metabolites in human prostate tissues using 1H HR-MAS total correlation spectroscopy. Magn Reson Med. 2008;60:33–40. doi: 10.1002/mrm.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, Zamboni N, Schulze A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012;2:328–343. doi: 10.1158/2159-8290.CD-11-0234. [DOI] [PubMed] [Google Scholar]
- 33.Tsui KH, Chung LC, Wang SW, Feng TH, Chang PL, Juang HH. Hypoxia upregulates the gene expression of mitochondrial aconitase in prostate carcinoma cells. J Mol Endocrinol. 2013;51:131–141. doi: 10.1530/JME-13-0090. [DOI] [PubMed] [Google Scholar]
- 34.Vaz CV, Alves MG, Marques R, Moreira PI, Oliveira PF, Maia CJ, Socorro S. Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int J Biochem Cell Biol. 2012;44:2077–2084. doi: 10.1016/j.biocel.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Mariani R, Arosio C, Pelucchi S, Grisoli M, Piga A, Trombini P, Piperno A. Iron chelation therapy in aceruloplasminaemia: study of a patient with a novel missense mutation. Gut. 2004;53:756–758. doi: 10.1136/gut.2003.030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.