

Abstract
Human islet amyloid polypeptide (hIAPP), also known as amylin, is a 37 residue peptide hormone that is stored and co-secreted with insulin. hIAPP plays a pivotal role in type 2 diabetes and is the major component of amyloid deposits found in the pancreas of patients afflicted with the disease. The self-assembly of hIAPP and the formation of amyloid is linked to the death of insulin producing β-cells. Recent findings suggest that soluble hIAPP oligomers are the cytotoxic species responsible for β-cell loss whereas amyloid fibrils themselves may indeed be innocuous. Potential avenues of therapeutic intervention include the development of compounds that prevent hIAPP self-assembly as well as those that reduce or eliminate lag time and rapidly accelerate the formation of amyloid fibrils. Both of these approaches minimize temporal exposure to soluble cytotoxic hIAPP oligomers. Toward this end our laboratory has pursued an electrostatic repulsion approach to the development of potential inhibitors and modulators of hIAPP self-assembly. Peptide conjugates were constructed in which benzene carboxylic acids of varying charge were employed as electrostatic disrupting elements and appended to the N-terminal of the hIAPP22–29 (NFGAILSS) self-recognition sequence. The self-assembly kinetics of conjugates were characterized by turbidity measurements and the structure of aggregates probed by Raman and CD spectroscopy while the morphology was assessed using transmission electron microscopy. Several benzene carboxylic acid peptide conjugates failed to self-assemble and some were found to inhibit the aggregation of full-length amylin while others served to enhance the rate of amyloid formation and/or increase the yield of amyloid produced. Studies reveal that the geometric display of free carboxylates on the benzene ring of the conjugates plays an important role in the activity of conjugates. In addition, a number of free benzene carboxylic acids were found to modulate amylin self-assembly on their own. The results of these investigations confirm the viability of the electrostatic repulsion approach to the modulation of amyloid formation and may aid the design and development of potential therapeutic agents.
Graphical abstract
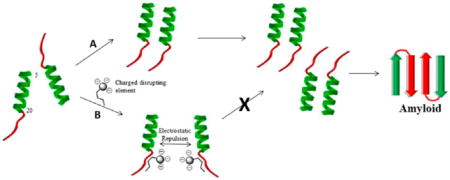
INTRODUCTION
Human islet amyloid polypeptide (hIAPP) is a 37 residue hormone that is co-stored and co-secreted with insulin in pancreatic β-cells. The polypeptide functions in maintaining glucose homeostasis and appetite satiation.1,2 hIAPP also plays a pivotal role in the development and progression of adult onset diabetes (type 2 diabetes or diabetes mellitus).3–7 Type 2 diabetes is characterized by a loss of β-cell mass which is correlated with the appearance of amyloid deposits in the pancreas. Indeed, post-mortem analysis has revealed that approximately 95% of type 2 diabetes patients have pancreatic amyloid deposits.8,9 The depletion of insulin producing β-cells ultimately leads to the hyperglycemia associated with type 2 diabetes and is a major contributor to the pathology of the disease.
Human islet amyloid polypeptide is the major protein component of amyloid fibrils in type 2 diabetes. In these insoluble fibers, the polypeptide exists in a structure known as the cross β motif in which extended polypeptide chains run perpendicular to the long axis of the fibril in a parallel β-sheet conformation. While initial investigations regarded hIAPP derived amyloid deposits as being culpable for the polypeptide’s cytotoxicity, more recent findings illustrate that soluble hIAPP oligomers are responsible for inducing β-cell death.10 A significant and growing body of evidence indicates that the cytotoxic effects of hIAPP stems from the polypeptides ability to induce membrane damage.11,12 In keeping with this, negatively charged phospholipids have been shown to promote and enhance the aggregation of hIAPP while dye leakage studies have confirmed the ability of hIAPP to disrupt model membranes.13–15 It is hypothesized that the phospholipid surface may serve to increase the local concentration of hIAPP thereby promoting interactions between hIAPP molecules or inducing conformational changes that drive fibril assembly. The effect of anionic phospholipids on amylin aggregation is further supported by the observation that heparin can also promote hIAPP aggregation.16,17
The membrane disrupting ability of hIAPP has been mapped to the N-terminal region of the polypeptide. Residues 1–19 are responsible for facilitating the interaction of hIAPP with phospholipids and model membranes.18 This observation is consistent with the fact that all positively charged amino acids in the hIAPP sequence reside in the 1–19 region.
The mechanism of membrane disruption by hIAPP is believed to occur through a two stage process and displays biphasic kinetics.19,20 The first stage is characterized by the formation of pores in the membrane followed by oligomer growth on the membrane surface leading to further membrane damage. Loss of ion homeostasis, in particular Ca2+, along with the activation of various stress signaling pathways culminate with cell death.
Other mechanisms of hIAPP cytotoxicity have also been proposed including oxidative and ER stress as well as the activation of death receptors and apoptotic signaling pathways.20 In reality, it is very likely that multiple mechanisms of hIAPP toxicity are at work in vivo including the disruption of cell and organelle membranes. What is consistent between these various cytotoxic mechanisms is that they appear to be initiated by exposure to soluble oligomers of hIAPP.
Possible therapeutic approaches to the treatment of type 2 diabetes include circumventing the buildup of cytotoxic soluble oligomers responsible for β-cell death. This can be achieved in one of two ways. The first approach is the development of novel chemical entities that can inhibit amylin aggregation. A second approach is to develop compounds capable of hastening the formation of insoluble amyloid fibrils. By rapidly driving the formation of amyloid, the temporal buildup of soluble oligomers can be avoided. Such an approach may avert the damage done by cytotoxic soluble oligomers by minimizing exposure times.
Small peptide fragments derived from Aβ and hIAPP have been shown to serve as inhibitors of amyloid formation. The self-recognition ability of these peptides directs them to corresponding regions of the target protein where they can exert their inhibitory effects. For example, the peptide KLVFF is known to inhibit Aβ aggregation.21 Scorcchi et al. have identified a number of peptide amylin aggregation inhibitors derived from the hIAPP sequence.22 Porat and co-workers discovered that replacement of Phe in the hIAPP22–29 sequence (NFGAILSS) resulted in a peptide that failed to self-assemble like the native sequence.23,24 The NYGAILSS peptide was also found to inhibit amyloid formation by full-length hIAPP.24 Likewise, Nilsson et al. identified a nonaggregating peptide based on the hIAPP20–29 sequence containing a Phe-23 to Trp substitution which inhibits amylin self-assembly.25 More recently, our laboratory identified several nonaggregating peptides that are effective inhibitors of amylin aggregation.26 These peptides are believed to bind to the 22–29 region of full-length hIAPP and enforce local secondary structure in an otherwise flexible region of full-length hIAPP and thereby hindering formation of the characteristic U-shaped monomers of amyloid fibrils.
The use of self-recognition element (SRE) peptide sequences derived from amyloidogenic proteins has been exploited to deliver bulky groups and/or secondary structural elements to specific sites within the target protein to prevent aggregation. For example, Findies and co-workers appended cholic acid to the LVFF sequence of Aβ to produce a peptide conjugate that was able to inhibit self-assembly of Aβ due to steric repulsion.27 Likewise, peptide sequences containing β-sheet breaking residue such as Pro and 2-amino isobutyric acid (Aib) have also led to compounds capable of inhibiting amyloid formation.28–30
Charge repulsion has been employed to disrupt and modulate amyloid formation. Murphy et al. synthesized and evaluated peptide constructs based on the Aβ 16–20 recognition sequence that contained charged amino acid residues appended to the N- or C-terminal as potential Aβ amyloid inhibitors.31,32 The peptides KLVFFKKKK and KLVFFEEEE were found to alter the kinetics of Aβ aggregation by enhancing amyloid formation and providing protection against Aβ cellular toxicity. Thus, changing the kinetic pathway of fibril formation and driving oligomers to the state of insoluble deposits minimizes the buildup of soluble oligomers and their membrane damaging cytotoxic effects.
We were interested in applying the approach of Murphy et al. toward the modulation and/or inhibition of amylin self-assembly. However, the increased molecular mass associated with appending several amino acid residues to the N- or C-terminal of a recognition sequence was of some concern as we wished to keep the potential inhibitor as compact as possible. A smaller molecule would more readily facilitate the future development of peptide mimetic compounds. On the basis of this, we contemplated appending more “charge dense” moieties on the terminal region of a peptide self-recognition element. Toward this end we chose to conjugate various benzene carboxylic acids to the N-terminal of the hIAPP22–29 recognition sequence. The benzene carboxylic acids vary in charge and should serve as potential disrupting elements of amylin aggregation. We now report the amyloidogenic propensity and biophysical characteristics of these novel peptide conjugates and describe how they affect the self-assembly of the full-length amylin.
RESULTS AND DISCUSSION
Conjugate Design and Synthesis
Peptide conjugates were designed to prevent self-assembly through a charge repulsion mechanism. We chose the 22–29 region of hIAPP as the SRE because this peptide fragment is well studied and characterized. The native hIAPP22–29 sequence forms aggregates on its own while specific point mutations at Phe-23 have led to the acquisition of nonaggregating inhibitors of amylin self-assembly.24–26 NFGAILSS has also has been used to seed full-length amylin to drive fibril production.22
For charged disrupting elements we chose inexpensive, commercially available benzene carboxylic acid derivatives that contain varying numbers of carboxyl groups (Figure 1). These include benzene-1,4-dicarboxylic acid (terephthalic acid) (1), benzene-1,3,5-tricarboxylic acid (trimesic acid) (2), benzene-1,2,4,5-tetracarboxylic acid, (pyromellitic acid) (5), benzene-1,2,3,4,5,6-hexacarboxylic acid (mellitic acid) (6), and 5-sulfosalicylic acid (7). Benzene-1,2,4-tricarboxylic-1,2-anhydride-4-chloride (3) and benzene-1,2,4-tricarboxylic acid anhydride (4) were employed to prepare isomeric versions of the trimesic acid (2) containing conjugate that varied in the display of carboxylates on the aromatic ring.
Figure 1.
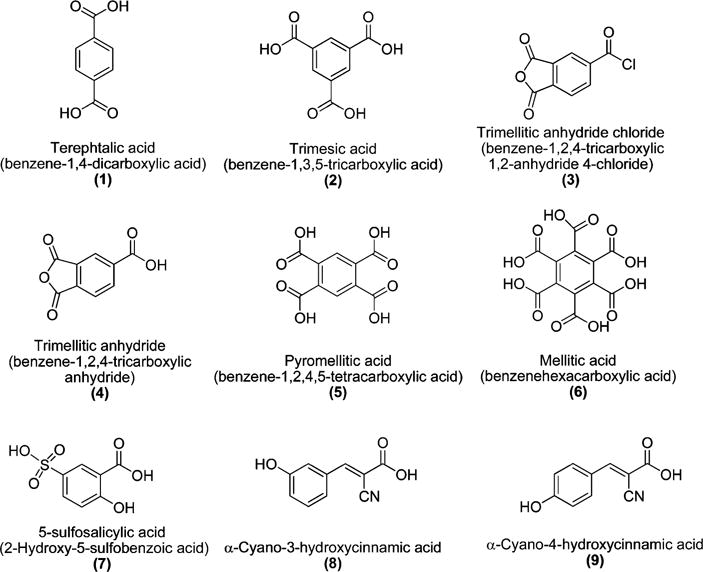
Structures of benzene carboxylic and cinnamic acid derivatives employed to prepare peptide conjugates.
The peptide conjugates synthesized as potential inhibitors of amylin aggregation are illustrated in Figure 2. Conjugates are identified by the prefix “C” followed by the number of the corresponding free benzene carboxylic acid from which they are derived. For ease of synthesis, benzene carboxylic acids were conjugated to the N-terminal of the hIAPP22–29 SRE through an amide bond linkage. After conjugation, each benzene carboxylic acid moiety has n − 1 (where n = the total number of carboxyls) carboxyl groups available to function as charged disrupting elements. At physiological pH, each carboxyl group should be ionized and provide the benzoic acid moieties with net negative charges ranging from −1 to −5. Intense charge repulsion between the N-termini of adjacent peptide strands should prevent self-association to form the characteristic parallel β-sheet structure described for NFGAILSS.26 The ability of the conjugates to adopt an alternative antiparallel β-sheet structure should also be hindered as this arrangement would bring the negatively charged free C-terminal of one peptide strand into proximity with the highly negatively charged benzene carboxylic acid moiety of an adjacent strand. Systematic variation in the number of carboxyl groups on the benzene carboxylic acids should facilitate the ability to discern the minimum charge requirements necessary to inhibit conjugate self-assembly.
Figure 2.
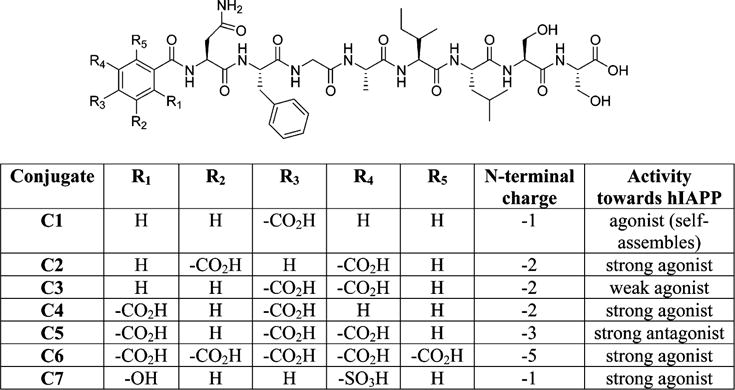
Structure and activity of benzene carboxylic acid peptide conjugates.
While conjugates containing terephthalic (1) and sulfosalicylic (7) acid moieties both possess an N-terminal with a single negative charge at pH 7.2, it was contemplated that the sulfonic acid may lead to some enhanced interactions with full-length amylin. This is due to the observation that amyloid binding compounds such as Congo red, phenolsulfothalien, and acid fuchsin contain sulfonic acid moieties.33–35 In addition, sulfonated molecules such as 3-amino-1-propanesulfonic acid and 1,3-propanedisulfonic acid have been shown to be effective inhibitors of Aβ and serum amyloid A aggregation, respectively.36,37
Benzene-1,2,4-tricarboxylic 1,2-anhydride 4-chloride (3) and benzene-1,2,4-tricarboxylic anhydride (4) and were employed to prepare isomers of C2 in an effort to elucidate if the substitution pattern of carboxylates on the aromatic ring influences the amyloidogenic propensity of individual conjugates and their interaction with full-length amylin. C3, with a 3,4 substitution pattern, was synthesized by preferential reaction of the N-terminal amino group of NFGAILSS peptidyl resin with the acid chloride functionality of benzene-1,2,4-tricarboxylic 1,2-anhydride 4-chloride.
Reaction of the peptidyl resin with benzene-1,2,4-tricarboxylic acid anhydride (4) yielded a single isomer as the major product (C4). Equal attack of the amine at the C-1 and C-2 carbonyls of the anhydride moiety was initially anticipated to provide two discrete products. However, resonance in benzene-1,2,4-tricarboxylic acid anhydride indicates that the C-1 carbonyl should be the most electrophilic and therefore the primary site of attack. On the basis of this, we rationalized that conjugate C4 contains a 1,3 carboxylic acid substitution pattern. The isomeric conjugates C2, C3, and C4 displayed individual reverse phase analytical HPLC retention times of 31.01, 32.09, and 30.76 min, respectively, each with a molecular mass of 999.53 in agreement with the calculated value of 999.42. Co-injection of C2, C3, and C4 reveals three closely eluting peaks confirming these compounds are distinct isomeric conjugates (Figure S1).
Peptide conjugates C8 and C9, which are derived from α-cyano-3-hydroxycinnamic acid and α-cyano-4-hydroxycinnamic, respectively (Figure 1), were also investigated as molecules potentially capable of influencing amylin aggregation. α-Cyano-4-hydroxycinnamic acid readily self-associates and is well-known for its use as a matrix in MALDI-TOF mass spectrometry.38 However, its isomer, α-cyano-3-hydroxycinnamic acid, does not self-associate, which has excluded its use as a matrix material. On the basis of this, we rationalized that it might be possible to exploit the different self-association properties of these isomeric hydroxycinnamic acids to prevent peptide aggregation by appending α-cyano-3-hydroxycinnamic acid to the N-terminal of the hIAPP22–29 SRE. Hence, we anticipated that the 3- and 4-hydroxycinnamic acid peptide conjugates C8 and C9 might display significantly different amyloidogenic propensities. Finally, the charged amyloid disrupting peptide, EEEENFGAILS (P10), was also prepared to compare and contrast the behavior of the charged peptide conjugates.
Peptides and conjugates were synthesized manually on Fmoc-Ser(OtBu)-Wang resin using a standard Fmoc solid phase synthesis protocol. Low substitution resin was chosen to minimize potential cross-linking between adjacent peptide chains during conjugation of the benzene carboxylic acids. With the exceptions of compounds C3 and C4, carboxylic acids were conjugated to the N-terminal of the peptidyl resin using HBTU, HOBT, and DIEA in DMF as described in the Experimental Section. The use of DIEA proved to be critical for coupling of higher substituted benzene carboxylic acids as their corresponding N-methylmorpholine salts were insoluble in DMF at the concentrations employed. Conjugates were purified by reverse phase HPLC and their structures confirmed by MALDI-TOF mass spectrometry.
Self-Assembly of hIAPP22–29 Conjugates
Turbidity (light scattering) was employed to determine the amyloidogenic propensity of individual peptide conjugates. The use of turbidity is a well-established method for monitoring the self-assembly of short peptide sequences such as NFGAILSS. The results of turbidity measurements for the conjugates, P10, and NFGAILSS are presented in Figure 3. From the data it can be seen that conjugates C1, C8, and C9 containing the terephthalic acid and cyano-hydroxycinnamic acid moieties readily form aggregates which then begin to settle out of solution. Conjugates C2–C7 fail to aggregate, as does the amyloid disrupting peptide EEEENFGAILSS (P10). Interestingly, the hydroxycinnamic acid conjugates are both capable of undergoing self-assembly. This behavior does not reflect the different propensities of the individual free acids to self-associate. Although both conjugates form amyloid, the α-cyano-4-hydroxycinnamic acid derivative appears to produce a higher amount of total amyloid than the 3-hydroxy isomer.
Figure 3.
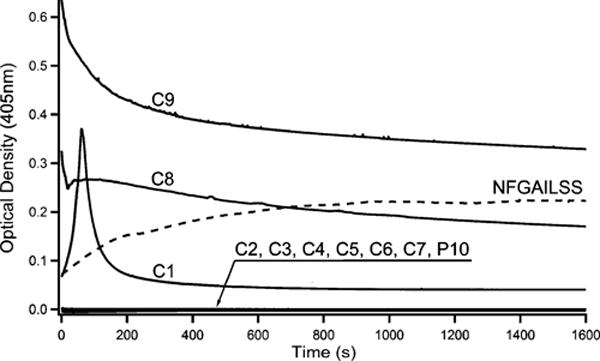
Changes in optical density at 405 nm monitored over time for 1 mM solutions of conjugates C1–C9 and P10 in tris-HCl buffer (pH 7.2). NFGAILSS turbidity plot is included for comparison and was taken from ref 39.
From the turbidity data it cannot be definitively stated what the minimum charge requirements are to prevent peptide self-assembly, although it does appear that an N-terminal charge greater than −1 is needed. However, there is still ambiguity. For example, conjugates C1 and C7 which contain the terephthalic and sulfosalycylic acid moieties, respectively, display drastically different amyloidogenic propensities despite both compounds having a single negative charge associated with the benzene carboxylic acid functionality. The finding that the terephthalic acid derivative self-assembles while the sulfosalycyclic acid compound does not implies that the sulfonic acid group provides additional interactions other than charge that effectively inhibit peptide aggregation. It is possible that the more polar sulfo group, in addition to the phenolic hydroxyl, may serve to confer enhanced water solubility on C7 making it less prone to aggregation. While it cannot definitively be stated what the minimum charge requirements are to inhibit self-assembly of the conjugates, it is clear that charge does play a role as more highly charged conjugates fail to self-assemble.
TEM analysis confirms the findings of turbidity measurements that compounds C2–C7 as well as P10 do not self-assemble. No evidence of fibrils was observed in aged samples of these conjugates upon microscopic examination. In contrast, TEM images of C1, C8, and C9 reveal a dense field of amyloid fibrils (Figure 4).
Figure 4.
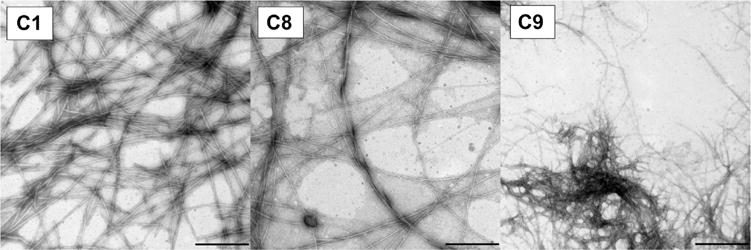
TEM of aggregated conjugates C1, C8, and C9 from turbidity samples. The bar on each panel defines a 500 nm distance.
Raman and IR spectroscopic analyses of the aggregating conjugates indicate that these samples exhibit similar characteristics as the hIAAP22–29 peptide mutants studied by our group and others.24,26,39,40 Regardless of the net ionic charge on the N-terminal, the aggregates yielded a similar parallel β-sheet configuration. Our group has previously established two particular Raman markers that interrogate the configuration of the aggregated structures, the Amide I mode at ca. 1674 cm−1 and some ring modes at ca. 1200–1210 cm−1.26 Aggregates exhibiting parallel β-sheet configuration display a single Amide I mode at 1674 cm−1 and a doublet peak in the vicinity of the 1200 cm−1 region. The latter indicating π–π interaction of the rings stacked upon one another. If the aggregates on the other hand, form an antiparallel configuration the Amide I mode at 1674 cm−1 plus a shoulder at around 1689 cm−1 is observed while the ring mode displays a single peak at approximately 1214 cm−1.
A close inspection of Table 1 indicates C1, C8, and C9 take on a parallel β-sheet configuration as aggregates form. Conjugates C1, C8, and C9 all present a single Amide I peak and two-peak ring modes that are ca. 10 cm−1 apart. The net ionic charge on the N-terminal group does not influence the resulting configuration.
Table 1.
Raman and IR Peaks Observed from Aggregates of C1, C8, and C9 along with the Apparent β-Sheet Orientationa
| Raman Shift (cm−1)
|
IR Shift (cm−1)
|
|||
|---|---|---|---|---|
| conjugate | ring mode | amide I mode | amide I mode |
β-sheet orientation |
| C1 | 1204 | 1665 | 1631 | Parallel |
| 1211 | – | – | ||
| C8 | 1202 | 1672 | 1631 | Parallel |
| 1215 | – | – | ||
| C9 | 1204 | 1664 | 1631 | Parallel |
| 1217 | – | – | ||
Raman and IR spectra on Figures S2 and S3, respectively.
Effects of Benzene Carboxylic Acid Conjugates and Free Acids on Amylin (hIAPP1–37) Self-Assembly
The self-assembly of hIAPP1–37 is a nucleation dependent event. The kinetics of amyloid formation is characterized by a sigmoidal curve with three distinct regions representing different phases of the self-assembly process. The first phase, known as the lag phase, is where hIAPP1–37 monomers associate to form soluble oligomers. Once a critical number of oligomers have assembled, nucleation occurs triggering an exponential polymerization of amyloid fibrils. This rapid polymerization is defined as the growth phase. The final phase of amyloid formation occurs when equilibrium is reached between soluble hIAPP1–37 aggregates and insoluble fibers and is exemplified by a plateau in the kinetic profile. This final stage of amyloid formation is the saturation phase. Agonists or antagonists of hIAPP1–37 may alter one or more of the kinetic phases of amyloid assembly.
Previous work from our laboratory, as well as others, has demonstrated that nonaggregating peptides derived from SREs can serve as inhibitors of amylin aggregation. On the basis of this, we examined the effects of C2–C7 on hIAPP1–37 fibrillization. Thioflavin T fluorescence data for amylin aggregation in the absence and presence of conjugates are presented in Figure 5 (see also Figure S4 for the effect of an aggregating conjugate, C1, on amylin self-assembly). From the data it is clear that the conjugates have a profound effect on the self-assembly kinetics of full-length amylin. There does not appear to be a direct correlation between the magnitude of the charge associated with the benzene carboxylic acid moiety and the amyloid disrupting ability of the peptide conjugates. Conjugates C2, C3, C4, C6, and C7 are all agonists of amylin self-assembly with distinctly different characteristics. The characterization of C2–C4 facilitates insight as to how the geometric display of free carboxylates affects the activity of these compounds.
Figure 5.
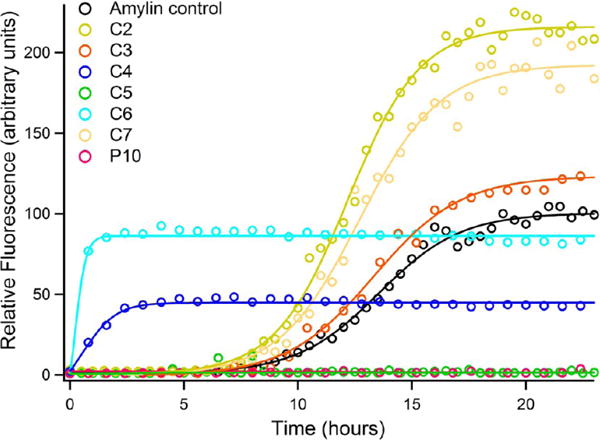
Thioflavin T fluorescence emission plot corresponding to β-sheet formation of amylin samples upon incubation with non-aggregating peptide constructs.
Conjugates C2–C4 all possess benzene carboxylic acid moieties with a net charge of −2; yet, these compounds exhibit different properties with regard to modulating the amyloidogenic propensity of hIAPP1–37. Conjugate C2 exhibits a 2,4 substitution pattern of carboxylates whereas C3 and C4 possess 3,4 and 1,3 displays of carboxyl groups, respectively. Both C2 and C3 have little effect on the lag time of amylin self-assembly. However, C2 significantly increases the total yield of amyloid produced to over twice that of the amylin control. This observation suggests that C2 is capable of shifting the equilibrium between soluble and insoluble hIAPP1–37 aggregates. In contrast, C3 exhibits a significantly slower growth rate than C2 and produces much less total amyloid. The overall quantity of amyloid produced in the presence of C3 is less than C2 but slightly higher than the amylin control.
The influence of conjugate C4 on the self-assembly of full-length amylin is markedly different than that of C2 and C3. Conjugate C4 virtually eliminates the lag time associated with hIAPP1–37 self-assembly. Full-length amylin begins to aggregate almost immediately in the presence of C4, but the overall yield of aggregates is much less than the amylin control. Indeed, C4 produced the lowest amount of amyloid of all the conjugates that enhance amylin self-assembly. However, the rate of fibril growth does not appear to be significantly affected by C4.
The above observations reveal that the presentation of carboxylate groups plays an important role in determining how conjugates C2–C4 interact with full-length amylin and exert their agonistic effects. While thorough SAR studies are needed to parse out the exact structural requirements for this agonistic behavior it is clear that it is not the magnitude but the spatial arrangement of charge that is key to the activity of these compounds.
Conjugates C6 and C7 are also agonists of amylin self-assembly. C6 exhibits carboxylates at positions 1 through 5 and has a charge of −5 associated with the benzene carboxylic acid, whereas C7 contains the sulfosalicylic acid moiety with a charge of −1. Conjugate C6 is a strong agonist of amyloid formation. In the presence of C6, lag time is all but abolished, the fibrillation rate is increased, and the total amount of amyloid produced is similar to that of the amylin control. In juxtaposition to C6, conjugate C7 does not have as dramatic an effect on lag time but significantly increases the total yield of amyloid to approximately twice that of the control. C7 also induces an increase in the rate of fibril growth, although not to the same degree as C6.
Interestingly the influence of C2 and C7 on amylin aggregation is almost completely opposite to that of C4 and C6. Both C4 and C6 eliminate lag time but do not produce high yields of amyloid, whereas C2 and C7 have a minimal effect on lag time but produce significantly larger quantities of amyloid fibers. TEM data confirms the presence of fibrils formed by hIAPP1–37 incubated with conjugates C2, C3, C4, C6, and C7 and corroborates the findings of ThT kinetic assays (Figure 6). The TEM data for EEEENFGAILSS (P10) reveals aggregate formation that was perhaps only minimally registered in the ThT assay. Indeed if the fluorescence intensity axis of the ThT plot is expanded (Figure S5), a slight increase in signal is observed reflecting the formation of a small amount of aggregates. Despite this observation P10 is still an effective inhibitor of amylin at the concentration utilized in the assay.
Figure 6.

TEM of amylin aggregates formed upon incubation with peptide constructs. Image A is amylin alone. The bar on each panel defines a 500 nm distance.
In sharp contrast to C2, C3, C4, C6, and C7, conjugate C5, which is derived from benzene-1,2,4,5-tetracarboxylic acid, is a potent antagonist of amylin self-assembly. C5 completely abolishes the self-assembly of hIAPP1–37 (Figures 5 and 6). The charge associated with the benzene carboxylic acid in C5 is −3. CD spectra taken of full-length hIAPP incubated with C5 indicates that the conjugate is able to trap or stabilize the polypeptide in a unique conformation that is unable to transition to the β-sheet structure (Figure 7). The CD spectra appear to only change in intensity over the time course of the experiment. Transmission electron microscopy verifies the absence of any amyloid fibrils when hIAPP1–37 is incubated in the presence of C5 and corroborates CD and ThT data (Figures 5 and 6). It is also particularly significant that C5 inhibits amylin aggregation more effectively as the designed disrupting peptide P10 while being lower in molecular mass, size, and charge. Likewise, conjugate C5 is able to inhibit amyloid formation without the use of a large bulky group appended to the N-terminal as in the case of the Aβ inhibitor cholyl-KLVFF.
Figure 7.
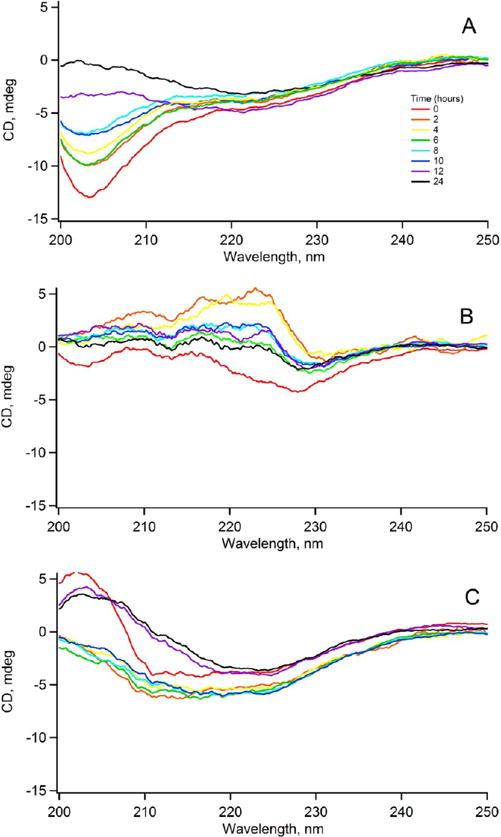
Time course of amylin self-assembly in the absence or presence of peptide constructs as monitored by CD. (A) amylin alone; (B) amylin incubated with C5; and (C) amylin with P10.
The ability of P10 to inhibit amylin self-assembly is also strikingly different from what has been observed with similar peptides designed to disrupt Aβ aggregation. For example, the peptides KLVFFEEEE and KLVFFKKKK were found to serve as agonists of Aβ aggregation. Neither was able to prevent Aβ self-assembly, although both peptides protected cells against the toxic effects of amyloid. There is a substantial body of evidence that indicates the toxicity of amyloid, regardless of the source protein, can be attributed to the membrane disrupting capabilities of soluble oligomers and not amyloid fibrils themselves. Peptides such as KLVFFEEEE and KLVFFKKKK presumably exert their protective effects by rapidly driving the formation of insoluble amyloid fibrils, thereby preventing the buildup of soluble oligomers.
We next investigated the ability of the various benzene carboxylic acids to inhibit amylin self-assembly on their own. Figure 8 illustrates the results of these studies. The data demonstrates that benzene-1,2,3,4,5,6-carboxylic acid (6) and benzene-1,2,4,5-tetracarboxylic acid (5) strongly induce amylin aggregation. Benzene-1,2,3,4,5,6-carboxylic acid (6) eliminates the lag phase and increases the rate of fibril growth while producing similar amounts of amyloid as the control. Benzene-1,2,4,5-tetracarboxylic acid (5) reduces lag time, increases the rate of fibrillization, and approximately doubles the amount of amyloid fibrils produced at equilibrium. Benzene-1,3,5-tricarboxylic acid (2) is a significant inhibitor of amyloid formation whereas 5-sulfosalycylic (7), terephthalic (1), and benzoic acid (C6H5COOH, (11)) all exhibit only minor effects on amylin self-assembly. TEM reveals the presence of amyloid fibrils in the samples of amylin containing the various benzene carboxylic acids (Figure 9).
Figure 8.
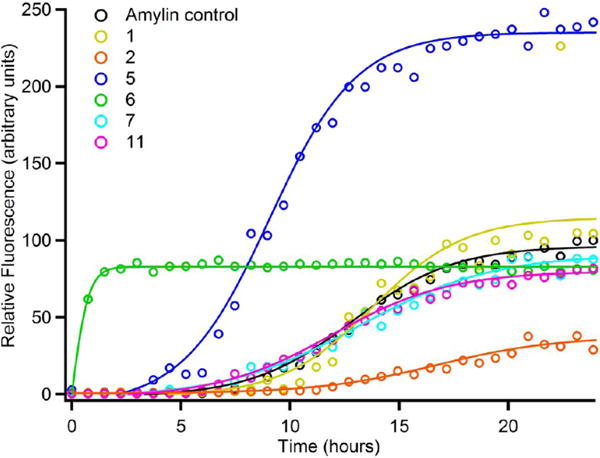
Fluorescence emission plot of thioflavin-T upon incubation of amylin with different benzene carboxylic acid derivatives; terephthalic acid (1); 1,3,5-tricarboxylic acid (2); 1,2,4,5-tetracarboxylic acid (5); mellitic acid (6); sulfosalicylic acid (7); and benzoic acid (11).
Figure 9.
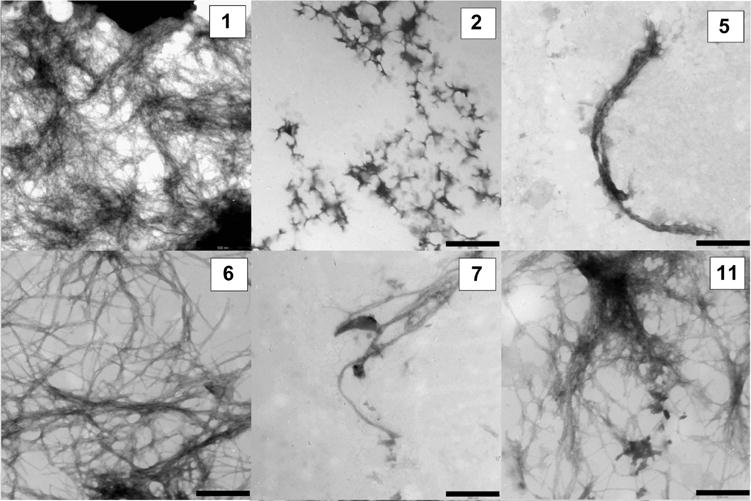
TEM of aggregates formed from mixtures of 40 μM free carboxylic acids and 4 μM amylin in Tris buffer pH 7.21. All samples were incubated for 24 h before imaging. The bar on each panel defines a 500 nm distance.
The observation that the more highly charged benzene carboxylic acids such as mellitic acid (6) and benzene-1,2,4,5-tetracarboxylic acid (5) serve as agonists of hIAPP1–37 self-association is consistent with previous observations that negatively charged phospholipids catalyze amylin aggregation. The highly charged benzene carboxylic acids may serve as a nucleation site for amylin aggregation just as the negatively charged surface of a phospholipid bilayer does. Studies have shown that the membrane disrupting ability of full-length hIAPP is indeed due to self-association and fibril growth on the membrane surface. Hamilton et al. have recently reported the synthesis and characterization of negatively charged, oligoamide α-helix mimetics displaying carboxylates equivalent to the i, i +3/4, and i+7 positions that demonstrate agonist activity toward amylin self-assembly.41 Likewise, Hassanpour et al. have developed doubly charged carboxylic acid derivatives of 2,5-diaryl-substituted thiophene helical mimetics that reduced the lag phase and dramatically increased the total amount of amyloid produced by hIAPP1–37.42 These compounds are believed to work through a mechanism involving interactions between negatively charged carboxylates and positively charged residues located in the N-terminal portion of amylin. The mode of action of negatively charged helical mimetics as well as the influence of phospholipids are consistent with the effects of the free mellitic and benzene-1,2,4,5-carboxylic acids on the amyloidogenic propensity of amylin. However, it should be noted that helical mimetics are more likely to optimally present their carboxylate groups to engage in and exploit specific interactions between the positively charged residues in amylin due to their conformational and spatial constraints. For example, terephthalic acid which contains two negatively charged carboxyl groups has little effect on amylin aggregation, whereas thiophene helical mimetics presenting two carboxylates have been reported to significantly alter the amyloidogenic propensity of amylin.42 The interaction of 5 and 6 with amylin may only occur through general nonspecific electrostatic interactions.
Comparison of the free benzene carboxylic acids with the conjugates reveals some significant findings. Conjugate C5 which contains the 1,2,4,5-benzene tetracarboxylic acid moiety serves to totally abolish amyloid formation, whereas the free acid is a strong agonist of amylin aggregation. This observation implies that there are two distinctly different mechanisms of action involved with the conjugate and the free acid and suggests that the peptide SRE plays an important role in how this conjugate functions.
It is perhaps plausible to make the argument that C5 should be more aptly compared to free 1,3,5-benzene carboxylic (2) acid, as the benzene carboxylic acid moieties of both these compounds possess a net −3 charge. While benzene-1,3,5-tricarboxylic acid (2) is an effective inhibitor of amylin self-assembly it is not as efficacious as C5. Furthermore, the corresponding conjugate that actually contains the benzene-1,3,5-tricarboxylic acid moiety (C2) is an agonist of amylin self-assembly. Conjugates C2 and C7 also act in a manner contrary to the free benzene carboxylic acids from which they are derived. Terephthalic and 5-sulfosalicylic acid exhibit little effect on amylin aggregation; yet, the corresponding conjugates dramatically enhance the amyloidogenic propensity of hIAPP1–37 (Figures 5 and S5). These observations again support an important role for the peptide SRE in the mechanism of action of the conjugates.
It is intriguing to speculate how C5 and peptide P10 exert their inhibitory effect on full-length amylin. The 5–20 region of monomeric hIAPP1–37 transiently exists in a helical conformation. Wiltzius et al. have proposed that, during amylin self-assembly, helical intermediates associate to bring the amyloidogenic C-terminal of monomers into proximity thereby driving intermolecular β-sheet formation to produce the characteristic U-shaped monomers of amyloid fibrils.43 This process is believed to be mediated through amylin dimers which orient themselves in an antiparallel fashion. A plausible mechanism for the inhibition of amyloid by C5 and peptide P10 would entail binding of the SRE to monomeric amylin at the 22–29 region. Charge repulsion between amylin-peptide complexes could prevent the initial dimerization of helical intermediates necessary to drive amyloid formation (Figure 10).
Figure 10.
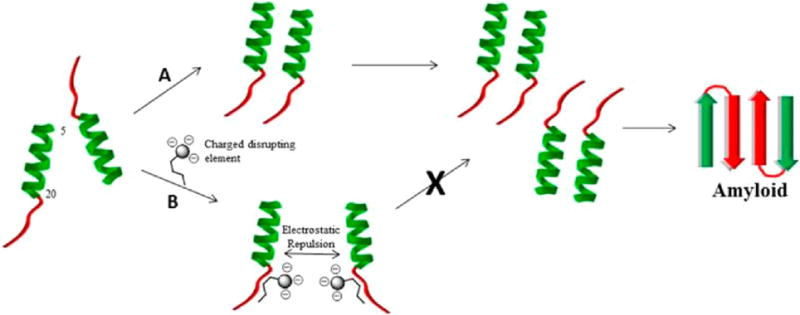
(A) Proposed mechanism of amyloid formation based on the model of Wilzius et al. illustrating the formation of two layers of cross-β structure (top layer is shown in color, bottom in gray). The 5–20 region of amylin monomers transiently form helical intermediates that assemble into dimers. Sets of dimers orient themselves in a head to tail fashion, which brings amyloidogenic C-terminal sequences together and drives β-sheet formation to produce the characteristic U-shaped monomers associated with the cross-β architecture of amyloid fibrils. The cross-β motif is depicted from above with the long axis of the fibril coming out of the plane of the page. (B) Potential mechanism of amyloid inhibition by C5 and possibly P10. The SRE binds to the 22–29 region of amylin monomers. Electrostatic repulsion between amylin-conjugate complexes prevents the dimerization of amylin monomers blocking further progression toward the formation of amyloid deposits.
Alternative models of amyloid formation suggest that helical intermediates may actually be off-pathway species in the amyloidogenic process while others implicate β-hairpins as being key on-pathway species.44,45 In the context of these models it may be that C5 and P10 somehow stabilize off-pathway helical intermediates thereby preventing amyloid formation. In the β-hairpin model, it is proposed that hIAPP monomers interconvert between α-helix and β-hairpin conformations. Constructs C5 and P10 may somehow prevent this key transition. This is consistent with CD data that suggests C5 traps hIAPP1–37 in a unique conformation (Figure 7). Clearly, thorough mechanistic investigations are needed to address exactly how the peptide conjugates are exerting their specific effects on amylin.
The inhibitory action of C5 is likely to be more complex than that of P10 with the geometric arrangement of carboxylates playing a role. The fact that conjugates of lesser and higher charges than that of C5 serve as agonists of amylin self-assembly indicates that charge of the conjugate alone is not sufficient for its inhibitory activity.
The N-terminal region of amylin contains the positively charged residues Lys-1 and Arg-11. Depending on the pH of the medium, His-18 may also exhibit a positive charge. It is a possibility that these conjugates are somehow interacting with Lys-1, Arg-11, or His-18 in a fashion that inhibits or enhances the kinetics of fibril formation. Interactions with His-18 may be of particular importance as the protonation state and presence of this residue has been shown to play a significant role in amylin self-assembly.46
In conjugate C6, the SRE may not serve any critical purpose as its behavior resembles that of the free benzene carboxylic acid. This demonstrates that the behavior of C6 is most likely governed by the benzene carboxylic acid moiety with the SRE exerting little to no effect. The highly charged, amphipathic nature of 6 may allow it to replicate the behavior of phospholipids or detergent micelles in solution with the negatively charged benzene carboxylic acid moieties resembling lipid “head” groups and the relatively hydrophobic peptide chain mimicking lipid “tails”. It may be possible that fibril growth can occur on the negatively charged surface of micelles derived from C6. Turbidity data demonstrating that C6 does not form insoluble amyloid does not mean the compound fails to undergo self-association. Indeed, C6 may simply form soluble oligomers that promote amylin nucleation or micelles as previously stated.
To probe the possible effect of micelle formation on aggregation propensity, we measured the turbidity of 1 mM C6 dissolved in tris-HCl buffer (pH 7.21) in the presence of sodium dodecyl sulfate (SDS) at concentrations below and above the SDS critical micelle concentration (CMC) of 8.2 mM. After incubating C6 in 2 mM SDS for 15 min, no significant increase in turbidity signal was observed, nor were any fibrils detected. On the other hand, C6 aggregates immediately formed when they were incubated in 10 mM SDS. Fibrils can be clearly observed soon after mixing, and turbidity signal measured at 405 nm is significant. This observation is consistent with the data collected by Sureshbabu et al.,47 demonstrating that in the presence of micelles, aggregation propensity of C6 is enhanced but only at detergent concentrations above CMC. Therefore, at the conditions of our ThT assay (40 μM conjugate to 4 μM amylin) it is unlikely that the conjugates exist as micelles that would affect the aggregation of amylin. We can also surmise that the CMC for the conjugates must be higher than 40 μM because not all aggregated when a 1 mM sample was incubated in buffer (Figure 3).
The sulfonated conjugate C7 may mimic sulfonated compounds such as heparin and the glycosaminoglycan chains of the heparin sulfate proteoglycan (HSPG) Perlecan known to promote amyloid self-assembly.48,49 However, these findings are at odds with the documented inhibition of amyloid by acid fuschin and other sulfonated small molecules.
Employing electrostatic repulsion to mitigate amyloid formation may be an effective strategy for developing potential therapeutic agents. It has been previously demonstrated that small molecules capable of modulating the net charge of a protein can be used to affect the amyloidgenic potential. Abdolvahabi and co-workers demonstrated that acylation of Lys residues in superoxide dismutase 1 with aspirin increases the net negative charge of the polypeptide and slows the rate of aggregation.50 In a similar fashion, Patil and Alexandrescu have recently applied a charge repulsion approach to inhibiting amylin self-assembly by preparing “charge-loaded” analogs of full-length amylin.51 In this study, strings of Arg or Asp residues were strategically incorporated into the native amylin sequence to produce aggregation inhibitors. These amylin analogs displayed a concentration dependent inhibition of wild type amylin self-assembly. Under the experimental conditions employed the amylin analogs greatly reduced amyloid formation. However, none of the polypeptides was able to completely abolish amylin aggregation even at the highest concentration used (120 μM). The most potent of the charged-based analogs (termed Arg-1) was also effective at protecting MIN6 mouse insulinoma cells from the cytotoxic effects of native amylin. In contrast to full-length charge-based amylin analogs, the significantly smaller conjugate C5 along with peptide P10 here are capable of virtually abolishing amylin self-assembly at a concentration of 40 μM in vitro.
CONCLUSION
The benzene carboxylic acid conjugates described in this study may serve as a template for the future design of amyloid inhibitors and further illustrate the viability of using electrostatic interactions to produce agonist and antagonists of amylin aggregation. These compounds successfully address two viable strategies for circumventing the buildup of soluble cytotoxic oligomers by either rapidly driving the fibrillation process to completion or inhibiting it altogether. The identification of free benzene carboxylic acids that are capable of antagonizing or promoting amylin aggregation may also aid in the development of small molecule amyloid inhibitors. Our laboratory is currently pursuing the optimization of several benzene carboxylic acid conjugates as well as conducting detailed mechanistic studies and cellular assays. The results of these investigations should enhance our knowledge of how to regulate the amyloidogenic process and drive the design and development of more efficient amyloid inhibitors.
EXPERIMENTAL SECTION
Preloaded Fmoc-Ser(tBu)-Wang resin was purchased from EMD Chemicals, Inc. (Gibbstown, NJ). All other reagents for peptide synthesis and Fmoc-protected amino acids were obtained from Advanced ChemTech (Louisville, KY). N,N-Dimethylformamide (DMF), dichloromethane (DCM), and acetonitrile (ACN) were purchased from Pharmco-AAPER (Brookfield, CT). Terephthalic acid was obtained from Acros Organics of Thermo Fisher (Pittsburgh, PA). All other reagents were from Sigma-Aldrich Co. (St. Louis, MO).
Peptide Conjugate Synthesis
The resin-bound peptide NFGAILSS was synthesized manually on Fmoc-Ser(OtBu)-Wang resin (substitution level 0.22 mmol/g). Fmoc removal was accomplished using 20% piperidine (v/v) in DMF for 20 min. Couplings were performed using 3 equiv each of Fmoc-protected amino acid, N,N,N′,N′-Tetramethyl-O-(1 H-benzotriazol-1-yl)uranium hexafluorophosphate (HBTU), 1-Hydroxy-benzotriazole (HOBT), and 9 equiv of N-methylmorpholine (NMM) relative to resin-bound amine. Amino acid couplings and Fmoc removal were monitored by Kaiser ninhydrin test.52 After the coupling of Fmoc-Asn(Trt)-OH, the peptidyl resin was aliquoted, deprotected, and the appropriate benzene carboxylic acid coupled according to the protocol described for the conjugation of benzene-1,2,4,5-tetracarboxylic acid unless otherwise specified.
Cleavage of peptides from the resin with simultaneous removal of all side chain protecting groups was accomplished using a cocktail composed of 95% trifluoroacetic acid, 2.5% water, and 2.5% triisopropylsilane (TFA/H2O/TIPS). After concentrating in vacuo, crude peptides were isolated by precipitation with cold diethyl ether. Crude peptides were dissolved in 50% CH3CN/H2O and filtered through a 0.45 μm PDVF filter before being subjected to chromatography. Peptides were purified by reverse phase HPLC (Varian ProStar, Palo Alto, CA) on a Vydac C18 protein and peptide column (2.2 × 25 cm) using a linear gradient of CH3CN/H2O containing 0.1% TFA. Column effluent was monitored at 218 and 254 nm. Appropriate fractions were pooled and lyophilized. Peptides were characterized by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (Waters Corporation, Milford, MA). Peptide purity was confirmed by reverse phase analytical HPLC on a Vydac C18 protein and peptide column (4.6 × 250 mm).
Conjugation of Benzene-1,2,4,5-tetracarboxylic Acid
5 equiv each of benzene-1,2,4,5-tetracarboxylic acid, HBTU, and HOBT relative to resin bound amine and 9 equiv of DIEA relative to benzene-1,2,4,5-tetracarboxylic acid were employed to activate the benzene carboxylic acid prior to amide bond formation. The molar ratio of reagents used favors the activation of a single carboxyl group by ensuring a 4:1 ratio of carboxylate ion to HBTU/HOBT, thereby minimizing the likelihood of cross-linking between adjacent polymer bound peptide chains. A similar procedure was used for the coupling of the other benzene carboxylic acid derivatives.
Conjugation of Benzene-1,2,4-tricarboxylic 1,2-Anhydride 4-chloride (Trimellitic Anhydride Chloride) and Benzene-1,2,4-tricarboxylic Anhydride (Trimellitic Anhydride)
Benzene-1,2,4-tricarboxylic 1,2-anhydride 4-chloride or benzene-1,2,4-tricarboxylic anhydride (5 equiv relative to resin bound amine) along with 9 equiv of DIEA dissolved in DMF was added to the NFGAILSS peptidyl resin and coupling allowed to proceed.
Turbidity Measurements
Lyophilized peptide conjugates were dissolved in 1,1,1,3,3,3-HFIP or DMSO to prepare concentrated stock solutions. Stock solutions of the NFGAILSS control were prepared by dissolving the peptide in 100% HFIP. Stock solutions were sonicated immediately prior to use. Aliquots of the concentrated stocks were diluted into 10 mM Tris-HCl pH 7.5 to a final peptide concentration of 1 mM for peptide aggregation studies. The final concentration of DMSO ranged from 2% to 4%. Turbidity was measured at 405 nm at room temperature as a function of time on a Jasco V-570 spectrophotometer (Easton, MD).
Thioflavin T (ThT) Fluorescence Assay
To assess the influence of nonaggregating conjugates on the self-assembly of full-length amylin we performed ThT fluorescence assays. For the ThT assay, peptide conjugates were dissolved in 100% 1,1,1,3,3,3-HFIP or DMSO to prepare concentrated stock solutions. Stock solutions were sonicated immediately prior to use. Aliquots of the concentrated stocks were diluted into 10 mM Tris-HCl buffer (pH 7.5) containing ThT to a final ThT and peptide conjugate concentration of 3 and 40 μM, respectively. To this mixture amylin (Bachem, Torrance, CA) was added to a final concentration of 4 μM. The ThT chromophore was excited at 450 nm and the emission monitored at 482 nm as a function of time on a FluoroMax-4 Spectrofluorimeter (Horiba Jobin Yvon Inc., Edison, NJ). Monochromator slits were set to 2.5 and 10 nm, respectively, for excitation and emission. Fluorescence was monitored every 30 min for at least 20 h at 25 °C.
Raman Spectroscopy
All Raman spectra were obtained on a Jasco NRS-3100 confocal dispersive Raman spectrometer equipped with a macro-Raman measurement accessory (East-on, MD). Raman scattering was induced by a 12 mW 488 nm laser and collected on a thermoelectrically cooled CCD detector. The macro-Raman assembly permitted direct measurements of solution in quartz cuvettes. Raman spectra were taken of each peptide dissolved in HFIP, before the aggregation process was initiated, and immediately after aliquots of the concentrated stocks were diluted into 10 mM Tris-HCl. The aggregate forming peptides yielded the Raman spectra of the fibrils formed. Aggregates were collected, layered unto a quartz slide, and spectra measured using the confocal Raman setup. Aggregates layered unto a slide yielded stronger Raman signals free of interfering solvent signals. Samples were prepared under the same experimental conditions as described for the kinetic aggregation assay. Measurements were done at room temperature and solutions were at 1 mM.
Circular Dicroism (CD) Spectroscopy
In conjunction with the ThT assay, changes on the secondary structure of amylin in the presence of the different peptide conjugates were done using CD spectroscopy on a Jasco J-810 spectrometer (Easton, MD). Similar solutions as with the ThT assay in the absence of thioflavin t were prepared with the CD spectra taken every 30 min for 24 h. All CD spectra were subtracted with the CD spectra of a blank containing 40 μM peptide conjugate in 10 mM Tris-HCl buffer (pH 7.5).
Transmission Electron Microscopy (TEM)
Samples were deposited onto 300 mesh copper grids with Formvar carbon film (Electron Microscopy Sciences, Hatfield, PA) and negatively stained with 2% uranyl acetate. After excess staining solution was blotted the images taken on a Ziess Libra 120 Plus 120 kV Electron Microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY) equipped with an Olympus Megaview G2 CCD camera with resolution of 1376 × 1032 pixels with pixel size of 6.45 × 6.45 μm2 for viewing samples at low magnification.
Acknowledgments
This work was supported in part by the Institute of General Medicine of the National Institutes of Health, grant no. 5SC3GM89624-2. We would like to thank Pratikumar Rathod from York College of CUNY for providing MALDI-TOF mass spectrometric analysis.
ABBREVIATIONS
- hIAPP
human islet amyloid polypeptide
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- HOBT
1-hydroxy-benzotriazole
- NMM
N-methylmorpholine
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilane
- HFIP
1,1,1,3,3,3-hexafluoroisopropanol
- ThT
thioflavin T
- MALDI-TOF
matrix-assisted laser desorption ionization time-of-flight
- CD
circular dichroism
- CCD
charge coupled device
- TEM
transmission electron microscopy
- SDS
sodium dodecyl sulfate
- CMC
critical micelle concentration
- HSPG
heparin sulfate proteoglycan
- SRE
self-recognition element
- Aib
2-amino isobutyric acid
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.6b00732.
Analytical data of purified peptide conjugates (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Lutz TA. Control of energy homeostasis by amylin. Cell Mol Life Sci. 2012;69:1947–1965. doi: 10.1007/s00018-011-0905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rushing PA, Hagan MM, Seeley RJ, Lutz TA, Woods SC. Amylin: a novel action in the brain to reduce body weight. Endocrinology. 2000;141:850–853. doi: 10.1210/endo.141.2.7378. [DOI] [PubMed] [Google Scholar]
- 3.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 4.Cao P, Marek P, Noor H, Patsalo V, Tu LH, Abedini A, Raleigh DP. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013;587:1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao P, Abedini A, Raleigh DP. Aggregation of islet amyloid polypeptide: from physical chemistry to cell biology. Curr Opin Struct Biol. 2013;23:82–89. doi: 10.1016/j.sbi.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KH. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akter R, Cao P, Noor H, Ridgway Z, Tu LH, Wang H, Wong AG, Zhang X, Abedini A, Schmidt AM. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J Diabetes Res. 2016;2016:1. doi: 10.1155/2016/2798269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark A, Cooper GJ, Lewis CE, Morris JF, Willis AC, Reid KB, Turner RC. Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet. 1987;330:231–234. doi: 10.1016/s0140-6736(87)90825-7. [DOI] [PubMed] [Google Scholar]
- 9.Kapurniotu A. Amyloidogenicity and cytotoxicity of islet amyloid polypeptide. Biopolymers. 2001;60:438–459. doi: 10.1002/1097-0282(2001)60:6<438::AID-BIP10182>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 10.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel MF. Membrane permeabilization by islet amyloid polypeptide. Chem Phys Lipids. 2009;160:1–10. doi: 10.1016/j.chemphyslip.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Lin CY, Gurlo T, Kayed R, Butler AE, Haataja L, Glabe CG, Butler PC. Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce antitoxic oligomer antibodies does not prevent h-IAPP-induced β-cell apoptosis in h-IAPP transgenic mice. Diabetes. 2007;56:1324–1332. doi: 10.2337/db06-1579. [DOI] [PubMed] [Google Scholar]
- 13.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 14.Nanga RPR, Brender JR, Xu J, Hartman K, Subramanian V, Ramamoorthy A. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J Am Chem Soc. 2009;131:8252–8261. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nanga RPR, Brender JR, Xu J, Veglia G, Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP 1–19 in micelles by NMR spectroscopy. Biochemistry. 2008;47:12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jha S, Patil SM, Gibson J, Nelson CE, Alder NN, Alexandrescu AT. Mechanism of Amylin Fibrillization Enhancement by Heparin. J Biol Chem. 2011;286:22894–22904. doi: 10.1074/jbc.M110.215814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Carufel CA, Nguyen PT, Sahnouni S, Bourgault S. New insights into the roles of sulfated glycosaminoglycans in islet amyloid polypeptide amyloidogenesis and cytotoxicity. Biopolymers. 2013;100:645–655. doi: 10.1002/bip.22243. [DOI] [PubMed] [Google Scholar]
- 18.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J Am Chem Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brender JR, Lee EL, Hartman K, Wong PT, Ramamoorthy A, Steel DG, Gafni A. Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophys J. 2011;100:685–692. doi: 10.1016/j.bpj.2010.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao P, Abedini A, Wang H, Tu LH, Zhang X, Schmidt AM, Raleigh DP. Islet amyloid polypeptide toxicity and membrane interactions. Proc Natl Acad Sci U S A. 2013;110:19279–19284. doi: 10.1073/pnas.1305517110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 22.Scrocchi LA, Chen Y, Waschuk S, Wang F, Cheung S, Darabie AA, McLaurin J, Fraser PE. Design of peptide-based inhibitors of human islet amyloid polypeptide fibrillogenesis. J Mol Biol. 2002;318:697–706. doi: 10.1016/S0022-2836(02)00164-X. [DOI] [PubMed] [Google Scholar]
- 23.Porat Y, Stepensky A, Ding FX, Naider F, Gazit E. Completely different amyloidogenic potential of nearly identical peptide fragments. Biopolymers. 2003;69:161–164. doi: 10.1002/bip.10386. [DOI] [PubMed] [Google Scholar]
- 24.Porat Y, Mazor Y, Efrat S, Gazit E. Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry. 2004;43:14454–14462. doi: 10.1021/bi048582a. [DOI] [PubMed] [Google Scholar]
- 25.Doran TM, Kamens AJ, Byrnes NK, Nilsson BL. Role of amino acid hydrophobicity, aromaticity, and molecular volume on IAPP(20–29) amyloid self-assembly. Proteins: Struct, Funct, Genet. 2012;80:1053–1065. doi: 10.1002/prot.24007. [DOI] [PubMed] [Google Scholar]
- 26.Profit AA, Vedad J, Saleh M, Desamero RZB. Aromaticity and amyloid formation: Effect of π-electron distribution and aryl substituent geometry on the self-assembly of peptides derived from hIAPP22–29. Arch Biochem Biophys. 2015;567:46–58. doi: 10.1016/j.abb.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Findeis MA, Lee JJ, Kelley M, Wakefield J, Zhang MH, Chin J, Kubasek W, Molineaux SM. Characterization of cholyl-leu-val-phe-phe-ala-OH as an inhibitor of amyloid beta-peptide polymerization. Amyloid. 2001;8:231–241. doi: 10.3109/13506120108993819. [DOI] [PubMed] [Google Scholar]
- 28.Poduslo JF, Curran GL, Kumar A, Frangione B, Soto C. β-sheet breaker peptide inhibitor of Alzheimer’s amyloidogenesis with increased blood–brain barrier permeability and resistance to proteolytic degradation in plasma. J Neurobiol. 1999;39:371–382. [PubMed] [Google Scholar]
- 29.Frydman-Marom A, Shaltiel-Karyo R, Moshe S, Gazit E. The generic amyloid formation inhibition effect of a designed small aromatic β-breaking peptide. Amyloid. 2011;18:119–127. doi: 10.3109/13506129.2011.582902. [DOI] [PubMed] [Google Scholar]
- 30.Mason JM, Kokkoni N, Stott K, Doig AJ. Design Strategies for Anti-Amyloid Agents. Curr Opin Struct Biol. 2003;13:526–532. doi: 10.1016/s0959-440x(03)00100-3. [DOI] [PubMed] [Google Scholar]
- 31.Pallitto MM, Ghanta J, Heinzelman P, Kiessling LL, Murphy RM. Recognition Sequence Design for Peptidyl Modulators of β-Amyloid Aggregation and Toxicity. Biochemistry. 1999;38:3570–3578. doi: 10.1021/bi982119e. [DOI] [PubMed] [Google Scholar]
- 32.Lowe TL, Strzelec A, Kiessling LL, Murphy RM. Structure-Function Relationships for Inhibitors of β-Amyloid Toxicity Containing the Recognition Sequence KLVFF. Biochemistry. 2001;40:7882–7889. doi: 10.1021/bi002734u. [DOI] [PubMed] [Google Scholar]
- 33.Meng F, Abedini A, Plesner A, Middleton CT, Potter KJ, Zanni MT, Verchere BC, Raleigh DP. The Sulfated Triphenyl Methane Derivative Acid Fuchsin Is a Potent Inhibitor of Amyloid Formation by Human Islet Amyloid Polypeptide and Protects against theToxic Effects of Amyloid Formation. J Mol Biol. 2010;400:555–566. doi: 10.1016/j.jmb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy M, Porat Y, Bacharach E, Shalev DE, Gazit E. Phenolsulfonphthalein, but not phenolphthalein, inhibits amyloid fibril formation: Implications for the modulation of amyloid self-assembly. Biochemistry. 2008;47:5896–5904. doi: 10.1021/bi800043d. [DOI] [PubMed] [Google Scholar]
- 35.Aitken JF, Loomes KM, Konarkowska B, Cooper GJS. Suppression by polycyclic compounds of the conversion of human amylin into insoluble amyloid. Biochem J. 2003;374:779–784. doi: 10.1042/BJ20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, Lacombe D, Kong X, Aman A, Laurin J, et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28:537–547. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Rumjon A, Coats T, Javaid MW. Review of eprodisate for the treatment of renal disease in AA amyloidosis. Int J Nephrol Renovasc Dis. 2012;5:37–43. doi: 10.2147/IJNRD.S19165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beavis RC, Chaudhary T, Chait BT. α-Cyano-4-hydroxycinnamic acid as a matrix for matrix assisted laser desorption mass spectrometry. Org Mass Spectrom. 1992;27:156–158. [Google Scholar]
- 39.Profit AA, Felsen V, Chinwong J, Mojica ERE, Desamero RZB. Evidence of π-stacking interactions in the self-assembly of hIAPP22–29. Proteins: Struct, Funct, Genet. 2013;81:690–703. doi: 10.1002/prot.24229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 41.Saraogi I, Hebda JA, Becerril J, Estroff LA, Miranker AD, Hamilton AD. Synthetic α-helix mimetics as agonists and antagonists of islet amyloid polypeptide aggregation. Angew Chem, Int Ed. 2010;49:736–739. doi: 10.1002/anie.200901694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hassanpour A, De Carufel CA, Bourgault S, Forgione P. Synthesis of 2,5-Diaryl-Substituted Thiophenes as Helical Mimetics: Towards the Modulation of Islet Amyloid Polypeptide (IAPP) Amyloid Fibril Formation and Cytotoxicity. Chem - Eur J. 2014;20:2522. doi: 10.1002/chem.201303928. [DOI] [PubMed] [Google Scholar]
- 43.Wiltzius JJW, Sievers SA, Sawaya MR, Eisenberg D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009;18:1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Carufel CA, Quittot N, Nguyen PT, Bourgault S. Delineating the Role of Helical Intermediates in Natively Unfolded Polypeptide Amyloid Assembly and Cytotoxicity. Angew Chem, Int Ed. 2015;54:14383–14387. doi: 10.1002/anie.201507092. [DOI] [PubMed] [Google Scholar]
- 45.Dupuis NF, Wu C, Shea JE, Bowers MT. The Amyloid Formation Mechanism in Human IAPP: Dimers Have β-Strand Monomer_Monomer Interfaces. J Am Chem Soc. 2011;133:7240–7243. doi: 10.1021/ja1081537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abedini A, Raleigh DP. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry. 2005;44:16284–16291. doi: 10.1021/bi051432v. [DOI] [PubMed] [Google Scholar]
- 47.Sureshbabu N, Kirubagaran R, Jayakumar R. Surfactant-induced conformational transition of amyloid β-peptide. Eur Biophys J. 2009;38:355–367. doi: 10.1007/s00249-008-0379-8. [DOI] [PubMed] [Google Scholar]
- 48.Meng F, Abedini A, Song B, Raleigh DP. Amyloid formation by pro-islet amyloid polypeptide processing intermediates:examination of the role of protein heparan sulfate interactions and implications for islet amyloid formation in type 2 diabetes. Biochemistry. 2007;46:12091–12099. doi: 10.1021/bi7004834. [DOI] [PubMed] [Google Scholar]
- 49.Jha S, Patil SM, Gibson J, Nelson CE, Alder NN, Alexandrescu AT. Mechanism of amylin fibrillization enhancement by heparin. J Biol Chem. 2011;286:22894–22904. doi: 10.1074/jbc.M110.215814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdolvahabi A, Shi Y, Rhodes NR, Cook NP, Martí AA, Shaw BF. Arresting Amyloid with Coulomb’s Law: Acetylation of ALS-Linked SOD1 by Aspirin Impedes Aggregation. Biophys J. 2015;108:1199–1212. doi: 10.1016/j.bpj.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patil SM, Alexandrescu AT. Charge-Based Inhibitors of Amylin Fibrillization and Toxicity. J Diabetes Res. 2015;2015:1. doi: 10.1155/2015/946037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]