

Abstract
The CXCR4 chemokine and Sonic Hedgehog (SHH) morphogen pathways are well-validated therapeutic targets in cancer, including medulloblastoma. However, single-agent treatments with SHH or CXCR4 antagonists have not proven efficacious in clinical trials to date. Here, we discovered that dual inhibition of the SHH and CXCR4 pathways in a murine model of SHH-subtype medulloblastoma exerts potent antitumor effects. This therapeutic synergy resulted in the suppression of tumor-propagating cell function and correlated with increased histone H3 lysine 27 trimethylation within the promoters of stem cell genes, resulting in their decreased expression. These results demonstrate that CXCR4 contributes to the epigenetic regulation of a tumor-propagating cell phenotype. Moreover, they provide a mechanistic rationale to evaluate the combination of SHH and CXCR4 inhibitors in clinical trials for the treatment of medulloblastoma, as well as other cancers driven by SHH that coexpress high levels of CXCR4.
Introduction
Medulloblastoma is the most common pediatric malignant brain tumor, and despite decades of research and clinical trials, overall survival rates remain below 70% (1, 2). Poor outcomes are especially prevalent within select subsets of the disease that are distinguished by their signature gene expression profiles and chromosomal aberrations. The identification of oncogenic mutations and transcriptional programs that drive tumor growth within discrete medulloblastoma subtypes has led to the application of targeted therapeutics (3, 4). One such targeted therapeutic, GDC-0449 (vismodegib/Erivedge, Genentech), has been approved for the treatment of basal cell carcinoma and is currently in clinical trials for medulloblastoma of the Sonic Hedgehog (SHH) subtype (5, 6). GDC-0449 inhibits Smoothened (Smo), an activating proteininthe SHH signaling pathway. Although early results with GDC-0449 showed promise in treating medulloblastoma patients, the response was typified by initial regression, followed by rapid relapse and patient death (7). Moreover, patients with basal cell carcinoma who received GDC-0449 treatment experienced a range of toxicities that limited dose and diminished patient compliance (8).
In some cases, relapse in both medulloblastoma and basal cell carcinoma patients resulted from Smo mutations that reduced its affinity for GDC-0449 (9, 10). In other cases, genetic alterations in downstream components of the SHH pathway rendered tumor cell growth independent of Smo activity (11, 12). Still, in other cases, no Smo or SHH pathway component mutations were identified, and the basis for resistance remains undefined (6, 13). Identifying additional targets to mitigate the risk of GDC-0449 resistance and recurrence and reducing toxicity of SHH pathway inhibition are required.
The SHH subtype of medulloblastoma (SHH-MB) derives from postnatal cerebellar granule neuron precursor cells (GNP), and many insights about medulloblastoma have stemmed from the study of normal cerebellar development (14). Maximal GNP proliferation requires coactivation of the SHH and the CXCR4 chemokine pathways (15). Together, these pathways also synergize to promote maximal medulloblastoma growth, and targeting CXCR4 alone with continuous infusion of specific inhibitors (AMD3100, AMD3465) was effective in preclinical studies of medulloblastoma and other brain cancers (16, 17). Although short-term treatment with AMD3100 (plerixafor) is safe and efficacious in combination with GCSF for bone marrow stem cell mobilization (18), continuous infusion of AMD3100 for 10 days in healthy HIV-positive individuals was associated with significant toxicities (19, 20). Current clinical trials evaluating AMD3100 in patients with newly diagnosed or recurrent glioblastoma are evaluating the safety and efficacy of daily subcutaneous injection (NCI2012-00149) or 2 weeks of continuous intravenous infusion (NCI2013-02012). Here, we sought to determine whether combined CXCR4 and SHH antagonism can be utilized to circumvent GDC-0449 resistance and sensitize medulloblastoma to intermittent CXCR4 antagonism, which may be better tolerated.
Materials and Methods
Chemicals were obtained from Sigma-Aldrich unless otherwise noted.
Animal studies
Animals were used in accordance with an established Animal Studies Protocol approved by The Washington University School of Medicine Animal Studies Committee, ensuring adherence to all federal regulations for the humane care and use of animals in research projects. Both male and female mice were utilized in all studies; no significant effect of sex was observed.
Cerebellar granule neuron preparation
Postnatal day 6 (P6) or adult C57Bl/6J mice (The Jackson Laboratory) mice were euthanized and brains were removed. GNPs were isolated as described previously (17).
SmoA1 tumor tissue processing
SmoA1 tumor cells were harvested from tumor-bearing ND2: SmoA1 (The Jackson Laboratory), as described previously (21). Cells were either used immediately for xenotransplantation or cryopreserved in 90% FBS/10% DMSO.
Xenotransplantation
Flank implants
SmoA1 tumor cells were implanted into the flanks of C57Bl/6J or NCRNU nude (Taconic) mice as in ref. 21.
Intracranial implants
SmoA1 tumor cells (1 ×105) in 5 μL DMEM/F12 were implanted into the cerebellum of NCRNU nude mice at 1 mm lateral and 1 mm posterior from the lambda and 2 mm below the dura (17).
Tumor treatment in vivo
Tumor-bearing mice were separated into four groups based on tumor size, to ensure equivalent average tumor volume between groups. For flank xenograft treatments, each mouse received either AMD3100 (16 μg/g in PBS), cyclopamine (2 mg/mL in HPBCD), or vehicle by subcutaneous injection and GCD-0449 (75 μg/g in DMSO/PBS) or vehicle by oral gavage once per day for 3 days, followed by at least 5 days of no drug administration. This was repeated twice more for a total of 9 treatment days over a 19-day period, and tumors were harvested 24 hours after the last drug administration. Tumor size was measured in three dimensions daily using digital calipers, and tumor volume was calculated as the product of these orthogonal measures. For intracerebellar xenograft treatments, mice received a single 3-day cycle of treatment.
Xenograft tissue processing
Flank tumor processing
Flank tumors were resected, and cells were flash frozen for protein extraction (17), RNA extraction, and chromatin immunoprecipitation. Pieces of tumors were postfixed overnight in cold Prefer (Anatech), cryoprotected in 30% sucrose overnight, and embedded in paraffin for immunohistology. Live cells were diluted for extreme limiting dilution assay (ELDA) and subtransplantation into naïve animals (5,000,000 or 5,000 cells in Matrigel).
Intracranial tumor processing
Brains were removed, processed, and paraffin embedded (17).
ELDA
ELDA assays for clonogenic (stem cell) frequency were performed precisely as in refs. 22, 23.
RNA purification, cDNA synthesis, and qPCR
RNA was harvested from tissue or dissociated cells using TRIzol (Invitrogen), and cDNA was prepared for PCR using standard methods (22). Real-time qPCR was performed using gene-specific primers (Supplementary Table S1) and either Power Sybr Green Master Mix Reagent (Life Technologies) or iTaq Sybr Green (Bio-Rad), with either a MiniOpticon Real-Time PCR machine or CFX Connect Real-Time PCR machine (Bio-Rad). Data were analyzed according to the ΜCt method (2−ΔΔt), where ΔCt is the difference between the gene of interest and GAPDH control Ct value, and data were normalized to the average of all vehicle-treated samples as in refs. 21, 22.
IHC
Immunohistochemical and immunofluorescence analyses for bromodeoxyuridine (BrdUrd; 1:50, Abcam), Ki67 (1:500, Thermo Fisher), cleaved caspase-3 (1:200, Cell Signaling Technology), Nmyc (1:100, Santa Cruz Biotechnology), Gli2 (1:200, Abcam), Bmi1 (1:20, R&D Systems), Sox2 (1:200, Abcam), and H&E staining were performed exactly as described previously (24).
Smo/Sufu amplification and Sanger sequencing
RNA was harvested from GNPs from C57Bl/6J mice (the parent strain of the SmoA1 mice) or from treated SmoA1 tumor cells, and cDNA was synthesized as above. The coding region of the SmoA1 gene encompassing the reported GDC-0449 resistance mutation (D477H, amplified region spanned from leucine380-asparagine562) was amplified using primers specific to the cDNA (Supplementary Table S1) to produce a 550-bp band, gel purified, and sequenced using internal primers (Supplementary Table S1; Genewiz). Similary, the Sufu gene was amplified using specific sequencing primers to cover the 1375 bp cDNA (listed in Supplementary Table S1) and sequenced in both directions.
Flow cytometry and cell sorting
At least 1 ×106 cells were stained with PE-conjugated anti-SSEA1 (CD15, BD Biosciences 560142) and allophycocyanin–conjugated anti-CXCR4 (BD Biosciences 558644) or their respective isotype controls. Cells were analyzed using a FACS-Caliber flow cytometer (BD Biosciences), and data were analyzed using FlowJo (FlowJo, LLC). Negative gating was set according to isotype-stained controls and adjusted for each independent tumor sample. FACS into CXCR4hi/CXCR4lo populations was done using a BD FACSAria II or a Sony Synergy cell sorter after staining with allophycocyanin–anti-CXCR4. Analysis of the CXCR4hi subpopulation was limited to those cells with greater than the 50th percentile surface CXCR4 expression in the CXCR4hi peak. Analysis of the CXCR4lo subpopulation was limited to those cells with less than the 50th percentile surface CXCR4 expression in the CXCR4lo peak. Live cells were identified by exclusion of Sytox Green dye (Life Technologies).
Western blotting
Protein from tumor tissue or cells (30–100 μg/lane) was processed for Western blotting as described previously (21, 22). Antibodies included phospho-Akt (Thr308, 1:300, Cell Signaling Technology), phospho-Akt (Ser473, 1:300, Cell Signaling Technology), pan Akt (Cell Signaling Technology, 1:500), CXCR4 (1:1,000, Leinco Technologies), and actin (1:2,000, Sigma).
Ultra-low input native chromatin immunoprecipitation
Sorted cells were processed for chromatin immunoprecipitation (ChIP) as described previously (18), using magnetic beads (Invitrogen) preloaded with anti-histone H3 trimethylated K4 (Abcam), anti-histone H3 trimethylated K27 (Millipore), or rabbit IgG (Millipore) diluted in ChIP buffer (20 mmol/L Tris-HCl pH 8.0, 2 mmol/L EDTA, 150 mmol/L NaCL, and 0.1% Triton X-100). After standard processing, DNA was amplified using qPCR and primers specific to the promoter region of stem cell genes (Supplementary Table S1). Promoter histone methylation was calculated by normalizing to input DNA signal (pre-cleared lysate control) after subtraction of background signal (rabbit IgG control pulldown).
Statistical analysis
For promoter methylation marks and stem cell gene expression analyses, two sample t tests compared the logarithm of transformed expression for each gene in CXCR4hi versus CXCR4lo cell subpopulations. Linear models were fit on the logarithm-transformed expression with incorporation of gene effect, CXCR4 cell subpopulation effect, and their interaction effect. Quantile–quantile plots of the resulting residuals were generated to confirm conformation to normality. The omnibus F test was conducted to test the null hypothesis that gene expression or histone methylation differed significantly between CXCR4 high and low cells in any of the genes under investigation. All tests were two-sided, and significance was claimed at a 5% level.
Other statistical analyses utilized t tests, ANOVA, or Fisher exact test as appropriate and as indicated in the figure legends.
Results
Multiple lines of evidence indicate that CXCR4 is overexpressed in medulloblastoma and represents an important therapeutic target (16, 17, 21). The mechanism by which CXCR4 promotes medulloblastoma growth remains incompletely described. To evaluate whether the well-characterized SmoA1 mouse model of medulloblastoma would be appropriate for mechanistic studies of CXCR4’s protumorigenic functions, we measured the expression of CXCR4 in spontaneous SmoA1 medulloblastoma tumor samples. The majority of cerebellar development in both mouse and human brains occurs postnatally, with a peak in cerebellar GNP cell proliferation on P6 in murine models (15, 25). Although CXCR4 expression in P6 cerebellum is significantly higher than the expression in adult tissue, SmoA1 tumor tissue contained the highest CXCR4 mRNA levels (Fig. 1A), consistent with our previous reports using human medulloblastoma and cerebellar tissue (21).
Figure 1.
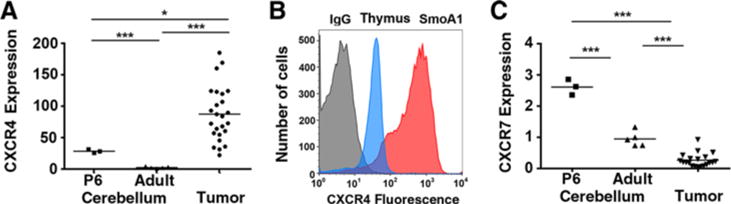
SmoA1 tumor cells express high levels of surface-localized CXCR4. A, qPCR measured CXCR4 expression in RNA derived from P6 cerebellum (n = 3), 2-month-old adult mice (n = 5), and spontaneous medulloblastomas from SmoA1 mice (n = 25). *, P < 0.05; ***, P < 0.001, unpaired t test. B, Surface CXCR4 was measured by flow cytometry. Representative histograms are shown. Gray histogram, control IgG staining (SmoA1 medulloblastoma cells); blue histogram, thymocytes; red histogram, SmoA1 medulloblastoma cells. C, CXCR7 gene expression from mouse brain and tumor tissue was analyzed as in A. ***, P < 0.001, unpaired t test.
As CXCR4 must be expressed on the cell surface for an active response to its ligand, we next investigated its surface expression in dissociated tumor cells using flow cytometry. In primary medulloblastoma cells, we found that CXCR4 was highly localized to the cell surface at levels that exceeded those found for thymocytes, a cell type known to exhibit high levels of surface CXCR4 expression (Fig. 1B). Similar to our studies of human medulloblastoma, the high surface and gene expression of CXCR4 in tumor cells occurred in the absence of expression of the alternate CXCL12 receptor, CXCR7, which functions as a negative regulator of CXCR4 signaling (Fig. 1C; refs. 21, 26). The tumor phenotype of high levels of CXCR4 expression, in the absence of CXCR7, is in stark contrast to the developing cerebellum, where P6 granule precursor cells express high levels of both CXCR4 and CXCR7.
Comparison between the SmoA1 and thymocyte flow cytometry profiles analysis highlights a bimodal distribution of surface CXCR4 in SmoA1 cells. Although the thymocyte peak is sharp and narrow, the SmoA1 profile has a substantial shoulder at lower levels of CXCR4 expression (Fig. 1B). CXCR4 is highly expressed in neural stem cells (27), and we hypothesized that surface CXCR4 expression might correlate with stem cell marker expression and function. We found that the expression of CD15, an established surface carbohydrate antigenic marker of murine medulloblastoma stem cells (28, 29), was positively correlated with surface CXCR4 expression (Fig. 2A). There was no measureable difference in total CXCR4 expression between the CXCR4hi and CXCR4lo subpopulations, suggesting that surface CXCR4 localization might be mechanistically related to tumor-propagating cell (TPC) function (Fig. 2A, inset). To test this hypothesis, we physically separated the CXCR4hi and CXCR4lo subpopulations from primary tumors and directly measured their in vitro clonogenic or stem-like cell potential using ELDA. Examination of the CXCR4hi and CXCR4lo subpopulations by ELDA indicated that clonogenic activity was enriched in the CXCRhi compared with the CXCRlo subpopulation (3% vs. 0.8%; Fig. 2B), confirming that CXCR4 surface localization, not total CXCR4 expression, was correlated with clonogenic function.
Figure 2.
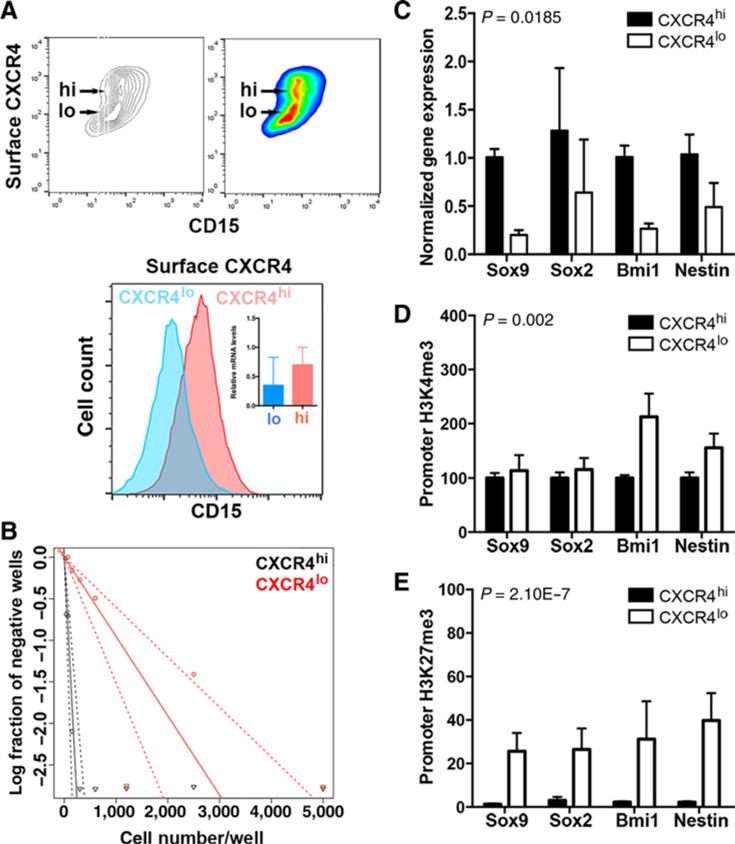
Levels of surface-localized CXCR4 correlate with a stem-like cell phenotype. A, Secondary flank SmoA1 tumor tissue was dissociated into a single-cell suspension and stained for surface CXCR4 protein and CD15 expression using flow cytometry. Top, noncolored and colored contour maps of results. y-axis, surface CXCR4 signal; x-axis, CD15 signal. The CXCR4hi and CXCR4lo subpopulations are indicated. Bottom, CXCR4hi and CXCR4lo subpopulations were computationally separated and analyzed for their CD15 expression. Inset, qPCR for total CXCR4 expression (n = 3). B–E, Single-cell suspensions of tumor homogenate were physically separated using FACS into CXCR4hi and CXCR4lo subpopulations and analyzed as follows. B, Cells were plated in an in vitro ELDA. A representative ELDA plot is shown; clonogenic cell frequency was calculated using raw data, and the analysis tool is available at http://bioinf.wehi.edu.au/software/elda/. C, Stem cell gene expression was measured by qPCR. D and E, Chromatin was precipitated from FACS-sorted CXCR4hi and CXCR4lo cells with antibody directed against histone H3 trimethylated K4 (D) or histone H3 trimethylated K27 (E), followed by qPCR analysis for the indicated promoter regions. P values (see Materials and Methods) were calculated for comparisons between the two subpopulations.
In medulloblastoma, as in other cancers, the clonogenic function of TPCs is dependent upon a transcriptional program and correlates with the expression of stem cell markers (30). We compared the expression of these markers in CXCR4hi and CXCR4lo subpopulations in primary tumors and found greater expression of Sox9, Sox2, Bmi1, and nestin in the CXCR4hi subpopulation, consistent with their greater fraction of clonogenic cells (Fig. 2C, P = 0.0185 for the difference between CXCR4hi and CXCR4lo subpopulations). The stem cell versus differentiated cell transcriptional program is regulated through characteristic histone modifications and transcription factor activity. Promoters for genes associated with differentiation are enriched for both the activation mark, trimethylation of histone H3 lysine 4 (H3K4me3), and the repressive mark, H3K27me3 (31). In contrast, stem cell gene promoters are only enriched in H3K4me3 marks. We examined these histone modifications in the promoters of the stem cell genes and found there was 2- and 1.5-fold greater H3K4me3 at the Bmi1 and nestin promoters in the CXCR4lo compared with the CXCR4hi population, respectively (Fig. 2D). In contrast, the suppressive H3K27me3 mark was approximately 30- to 40-fold greater in the promoters of each of the stem cell genes in the CXCR4lo compared with the CXCR4hi subpopulation (Fig. 2E, P = 2.1E–7 for the difference between CXCR4hi and CXCR4lo subpopulations). Thus, the levels of CXCR4 surface expression are correlated with epigenetic regulation of stem cell gene expression and a functional stem cell state.
These findings suggested that the reported antitumor activity of CXCR4 antagonists might result from a direct effect on tumor cells to abrogate their TPC activity. To study this, we undertook a staged evaluation of combined SHH and CXCR4 antagonism. In pilot experiments, we opted for flank implantation of primary isolates derived from multiple SmoA1 tumors into immunocompromised mice to facilitate continuous measurement of growth and reisolation of tumor cells for posttreatment studies. First, we tested whether intermittent treatment with the CXCR4 antagonist AMD3100 and SHH antagonist cyclopamine, either alone or in combination, would exhibit significant antitumor effects. Neither drug alone was effective at blocking tumor growth (Supplementary Fig. S1A). In contrast, four cycles of 3-day on/5-day off combined AMD3100 and cyclopamine were highly effective and blocked tumor growth. To enhance the clinical relevance of these studies, we performed a second pilot study in nude mice using GDC-0449 instead of cyclopamine. Tumors were established from an independent cohort of SmoA1 primary tumors. Although the cell number was identical in both experiments (1 × 106/injection) and initial tumor volumes were comparable between the experiments (experiment 1, 584 ± 116 mm3; experiment 2, 901 ± 302 mm3), the rate of growth in xenografts derived from the second set of tumors was slower, and fold growth was lower compared with the first trial (compare fold growth in Supplementary Fig. S1A and S1B). In this second pilot experiment, we limited our analysis to measure differences between AMD3100 and GDC-0449 alone or in combination. Although SmoA1 mutation (Smo W539L) results in relative resistance to GDC-0449 (32), 3 days of treatment produced an immediate reduction in tumor growth compared with treatment with AMD3100 alone. However, with each successive treatment, the depth of the response lessened, and tumors rapidly regrew when treatment was stopped (Supplementary Fig. S1B). Again, combined SHH and CXCR4 antagonism was synergistic and yielded superior inhibition of tumor growth over the experimental period. Together, these studies in nude mice indicate that the combination of CXCR4 and SHH antagonism has superior therapeutic effects associated with suppression of tumor regrowth even after treatment cessation.
To further enhance the clinical relevance of these studies, we treated larger cohorts of tumor-bearing wild-type immunocompetent C57Bl/6J mice. Starting tumor volumes were again comparable with those in the pilot experiments (840 ± 71 mm3). As expected, vehicle-treated tumors grew at a rapid and continuous rate (Fig. 3A). Consistent with the findings in nude mice, intermittent AMD3100 did not slow tumor growth, indicating that successful single-agent application of CXCR4 antagonists in the clinic may require continuous exposure.
Figure 3.
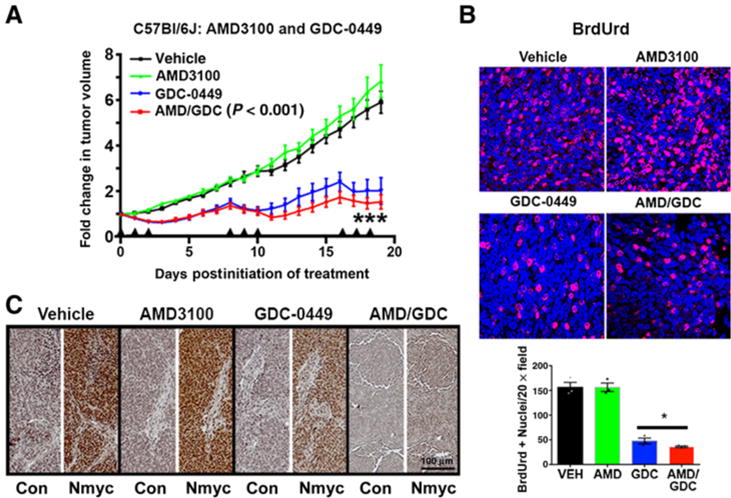
Combined Smo and CXCR4 antagonism is more efficacious than Smo antagonism alone against SmoA1 medulloblastoma. A, Primary SmoA1 tumor cell isolates (n = 5 tumors) were used to generate five cohorts of 20 to 25 immunocompetent C57Bl/6J mice bearing subcutaneous tumor implants. Mice from each cohort were separated into four treatment groups yielding final treatment numbers of vehicle (VEH; n = 19), AMD3100 (n = 21), GDC-0449 (n = 21), and AMD3100/GDC-0449 (n = 23). Mice were treated with the indicated drug for 3 consecutive days (arrowheads), followed by 5 days of no treatment. This dosage schedule was repeated for a total of three treatment periods prior to tumor harvest. Tumors were measured daily, blinded to treatment group, using digital calipers in three dimensions (width, height, and length), and all measurements were normalized to the tumor size at day 0 (pretreatment). ***, P < 0.001 comparing GDC-0449–treated tumors and AMD3100/GDC-0449–treated tumors, two-way ANOVA. B, Representative images of BrdUrd labeling (red) in each of the treatment groups. Nuclei were counterstained with DAPI. BrdUrd-positive and total nuclei were counted in sections from three separate mice from each treatment group. Quantification of the mean and SEM of the numbers of positive nuclei per high powered fields are shown. Both GDC-0449 and AMD3100/GDC-0449 treatments significantly reduced the numbers of BrdUrd-positive nuclei. The combined treatment suppressed BrdUrd incorporation more than GDC alone (*, P < 0.05, as determined by two-tailed t test). C, Representative images of Nmyc or no primary control (Con) immunostaining from each of the treatment groups. Scale bar, 100 μm.
Seventy-two hours of GDC-0449 treatment was adequate to promote transient tumor regression to approximately 50% of pretreatment volumes (vehicle 125.3% ± 5.7 vs. GDC 64.0 ± 6.4, P < 0.0001). In the absence of continued treatment, the tumors consistently rebounded to their pretreatment sizes. Combined AMD3100 and GDC-0449 treatment of tumor-bearing C57Bl/6J mice was superior to treatment with GDC-0449 alone, suggesting that the mechanistic advantage of combined therapy is independent of both immune activity and other strain-dependent differences between nude and C57Bl/6J mice.
We evaluated the basis for the antitumor synergy between AMD3100 and GDC-0449 with measures of proliferation in tumor tissue. Four hours prior to euthanasia, mice were injected with BrdUrd to label actively proliferating cells. Blinded quantification of BrdUrd positivity indicated that AMD3100 had no effect on proliferation, but that GDC-0449 alone significantly reduced it (Fig. 3B). Moreover, the combination of AMD3100 and GDC-0449 more potently blocked proliferation than either drug alone. We next examined the expression of Nmyc by immunohistochemical analysis of tumor tissue. Nmyc is known to be regulated by SHH and to mediate its effects on proliferation (33). AMD3100 had no effect compared with vehicle controls (Fig. 3C), but consistent with its effects on BrdUrd incorporation, GDC-0449 reduced Nmyc expression. Combined AMD3100 and GDC-0449 exhibited the greatest effect on Nmyc expression, which appeared to be completely abrogated by the combined treatment.
We further assessed the potential for antitumor effects through increased apoptosis and examined the abundance of cleaved caspase-3 (CC3). AMD3100 treatment alone had an apparent effect on apoptosis and mildly elevated the fraction of CC3-positive cells compared with controls (Supplementary Fig. S1C). GDC-0449 alone or in combination with AMD3100 had no effect on CC3 positivity. Together, these data indicate that combined AMD3100 and GDC-0449 block tumor growth primarily by inhibiting proliferation.
Finally, we sought to confirm the antitumor effects of combined GDC-0449 and AMD3100 in orthotopic intracerebellar xenografts. Xenografts were established in nude mice and allowed to grow for 8 weeks prior to randomization to the four treatment groups. Mice were treated for 3 days, and 24 hours after the last treatment, brains were processed for IHC. Tumors grew in a highly infiltrative fashion in the cerebellum (Fig. 4A). Proliferation rates (Ki67+ nuclei/total nuclei) were high (22 ± 5.8%) in vehicle-treated tumors as assessed by Ki67 IHC (Fig. 4B). Proliferation rates were reduced in all other treatment groups, with the greatest reduction occurring in the cotreatment group (AMD, 2.8 ± 2.8%; GDC-0449, 1.45 ± 0.2%; AMD/GDC, 0.97 ± 0.2%). Again, reductions in proliferation correlated with decreases in Nmyc expression (Fig. 4C). Together, these data confirm the synergistic effects of combined CXCR4 and SHH antagonism in an orthotopic model of medulloblastoma.
Figure 4.
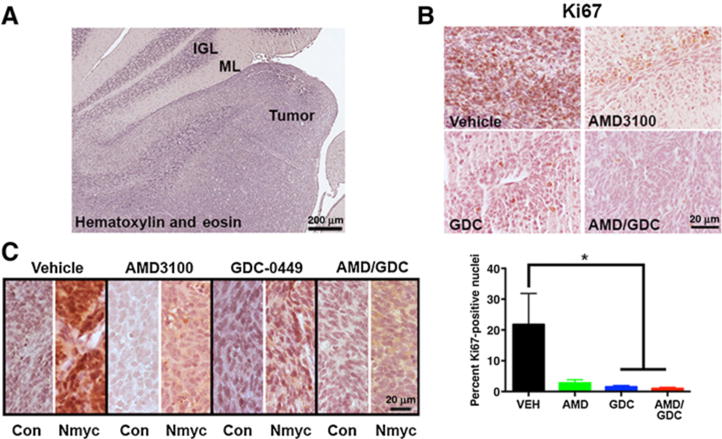
Combined AMD3100 and GDC-0449 is effective in orthotopic xenografts. A, Representative image of a hematoxylin and eosin–stained section through the cerebellum of a tumor-bearing mouse. The adjacent internal granule cell layer (IGL) and molecular layer (ML) can be seen alongside the tumor. Scale bar, 200 μm. B, Representative images of Ki67 staining for proliferating cells (brown) in each of the treatment groups along with quantification of the fraction of proliferating cells. *, P < 0.05 as compared with vehicle (VEH) using unpaired t test; n = 3 to 5 independent tumors from each treatment group. C, Representative images of Nmyc immunostaining from each of the treatment groups. Con, control. B and C, Scale bar, 20 μm.
We were next interested in the potential mechanisms of resistance to GDC-0449. First, we looked directly at GDC-0449 effects on Gli2 expression in tumor tissue. Not only is Gli2 a target of SHH signaling, amplification of GLI2 is a known mechanism of resistance to GDC-0449 in basal cell carcinoma (11) and medulloblastoma (34). We found that GDC-0449 potently inhibited the expression of Gli2, consistent with direct inhibition of Smo by GDC-0449 and the absence of Gli2 amplification as a mechanism of resistance (Supplementary Fig. S2A). GDC-0449 resistance in both laboratory-based and clinical settings can also result from emergence of tumor subclones harboring mutations in SMO or the downstream mediator SUFU (34). Resistance-promoting SMO mutations are typically in its ligand-binding pocket (human residues D473G, H231R, W281C, Q477E). These mutations reduce the affinity of drug:target interactions (9). Activating mutations in SUFU result in persistent SHH pathway signaling independent of Smo activity (12). To determine whether tumor regrowth in the GDC-0449 treatment group was the result of acquired mutations in murine Smo or Sufu, we sequenced the DNA encompassing the ligand-binding pocket region of the murine Smo gene and the entirety of murine Sufu following GDC-0449 treatment and tumor regrowth. We found no evidence of additional Smo or Sufu mutations in 10 recurrent tumors after GDC-0449 treatment alone or in combination with AMD3100 (Supplementary Fig. S2B).
Resistance to GDC-0449 has also been ascribed to modulatory pathways like the PI3K pathway (35, 36). To determine whether resistance to GDC-0449 might relate to increased PI3K pathway activity, we performed Western blot analysis of tumor tissue lysates for two different phosphorylation sites on Akt. Recurrence after GDC-0449 treatment was not associated with any change in pT308 (Supplementary Fig. S2C). There was a trend toward increased phosphorylation at S473 in GDC-0449 treated tumors, but this did not reach statistical significance and was abrogated by cotreatment with AMD3100. Thus, additional mechanisms might drive regrowth after GDC-0449 treatment.
Tumor regrowth is driven by TPC function. As TPC function was correlated with high levels of surface CXCR4 expression, and as tumor regrowth was blocked by cotreatment with AMD3100, we sought to determine whether treatment altered surface localization of CXCR4. We measured the proportion of CXCR4hi and CXCR4lo cells recovered from in vivo–treated tumors. Vehicle- and AMD3100-treated tumors exhibited the same biphasic staining pattern (Fig. 5A), with the majority of cells exhibiting high levels of surface CXCR4 expression. In contrast, GDC-0449 treatment either alone or in combination with AMD-3100 significantly shifted the cell population from CXCR4hi to CXCR4lo, with a significant decrease in the magnitude of total CXCR4 surface signal (Fig. 5B) and total CXCR4 protein (Fig. 5C). The effect of GDC-0449 on CXCR4 internalization is similar to what has been previously reported for cyclopamine (21) and again suggested that the SmoA1 mutation does not completely abrogate sensitivity to GDC-0449. The pattern of surface CXCR4 staining was similar between GDC-0449–treated and dual-treated tumor cells, indicating that the added antitumor effect of combining AMD3100 with GDC-0449 was not a result of a further loss of surface CXCR4. Together, these data suggest that inhibition of Smo by GDC-0449 promotes the downregulation of CXCR4 from the cell surface, but only in a subset of tumor cells. The persistence of high levels of surface CXCR4 in the remaining cells suggested the possibility that these cells are resistant to the effects of GDC-0449 and may be the source of TPC activity that drives regrowth.
Figure 5.
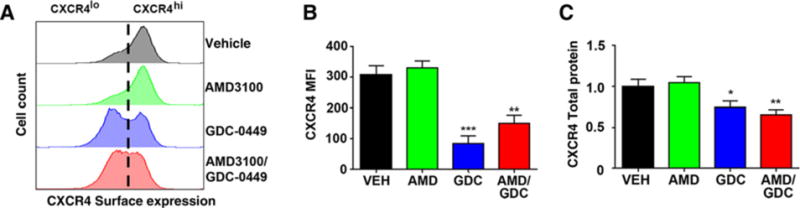
GDC-0449 promotes downregulation of surface CXCR4 in a subset of tumor cells. In vivo–treated tumors were processed into single-cell suspensions and analyzed by flow cytometry or Western blotting. A, Surface expression of CXCR4 was determined using flow cytometry. Representative histograms are shown. To the left of the vertical dotted line is the CXCR4lo and to the right of the dotted line the CXCR4hi subpopulations. B, Total CXCR4 fluorescence intensity (MFI) was quantified from flow cytometry data (n = 3–5 independent tumors from each treatment group). **, P < 0.005; ***, P < 0.001, as compared with vehicle (VEH) using unpaired t test. C, Total CXCR4 was measured using Western blotting. Total protein was normalized to actin and to vehicle-treated tumor samples. *, P < 0.05; **, P < 0.01 as compared with vehicle using unpaired t test; n = 3 to 5 independent tumors from each treatment group.
To determine whether combined SHH and CXCR4 inhibition abrogated TPC function, we subtransplanted cells derived from tumors in each of the in vivo treatment groups into naïve animals to directly measure their in vitro clonogenic capacity and in vivo tumorigenicity. Consistent with published studies (37), clonogenic cell frequency was approximately 0.2% in tumor-derived heterogeneous cell suspensions from vehicle-treated tumors (Fig. 6A). Monotherapy with AMD3100 or GDC-0449 alone had no effect on the clonogenic cell frequency. In contrast, cotreatment with AMD3100 and GDC-0449 significantly diminished the clonogenic cell frequency to less than 0.05%, suggesting that this therapy uniquely targets the medulloblastoma stem-like cell pool. Consistent with these ELDA results, vehicle- and single agent–treated tumors retained their tumorigenic potential and reinitiated tumors in more than 50% of recipient animals following transplantation of only 5,000 cells (Fig. 6B). The histology of subtransplanted tumors closely resembled those of the original tumor (Fig. 6C). Strikingly, tumors formed in only 20% of animals that received cells from tumors treated with both AMD3100 and GDC-0449. In all cases, cells isolated from these subtransplanted tumors (in cases when they grew) exhibited uniformly high levels of surface CXCR4, highlighting the functional relevance of surface CXCR4 levels to recurrence (Fig. 6D).
Figure 6.
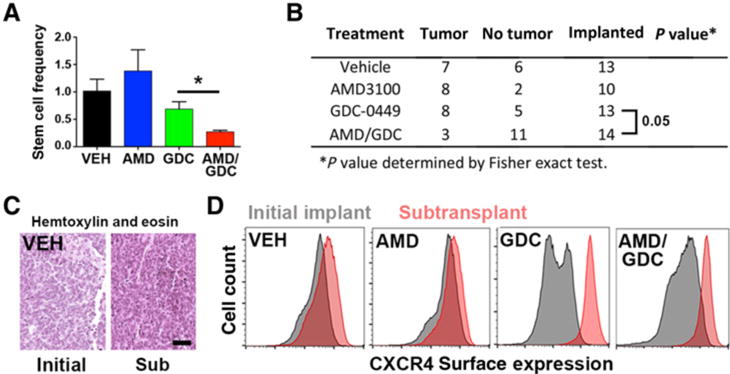
Tumor-propagating activity can be abrogated by combined SHH and CXCR4 antagonism. A, Clonogenic cell frequency was calculated from ELDA assays performed with unsorted single-cell suspensions isolated from in vivo–treated tumors (n = 3–5 independent tumors from each treatment group). *, P < 0.05 as determined by unpaired t test for the comparison between GDC alone and dual-treated tumors. VEH, vehicle. B, In vivo–treated tumors were processed into a single-cell suspension and subtransplanted into naïve immunocompetent C57Bl/6J animals (5,000 cells/implant). Tumor formation was gauged as growth of a tumor at least 5 mm in any dimension within 6 months of implantation date. Dual-treated tumors were significantly impaired for growth in naïve animals, P = 0.05 as determined by Fisher exact test for the comparison between dual treatment and GDC-treated tumors. C, Tumor tissue from in vivo–treated tumors was compared with tumor tissue in subtransplanted (Sub) animals by hematoxylin and eosin staining. Scale bar, 20 μm. D, Tumor tissue from in vivo–treated tumors (gray histograms) or from tumor tissue in subtransplanted animals (red histograms) was processed into a single-cell suspension prior to staining for surface CXCR4 and flow cytometry. All tumors that formed after subtransplantation expressed high levels of CXCR4 on the cell surface.
Recurrence is driven by the activity of a stem-like subpopulation of tumor cells. This stem-like state is maintained through epigenetic mechanisms, including the balance between activating and repressive histone marks in the promoters of stem cell genes (38). To determine whether the effect of combined AMD3100 and GDC-0449 on in vitro clonogenicity and in vivo tumorigenicity was correlated with differences in the epigenetic regulation of stem cell gene expression, we measured the abundance of activating H3K4 and repressive H3K27 trimethylation marks in the promoters of stem cell genes from CXCR4hi specimens isolated from in vivo–treated tumors. Similar to what we observed when comparing the CXCR4hi and CXCR4lo populations, there was no correlation between the effects of treatment on clonogenic cell frequency or in vivo tumorigenicity and levels of H3K4 trimethylation, which were similar regardless of treatment (Fig. 7A). However, consistent with the hypothesis that combined GDC-0449 and AMD3100 uniquely suppress the TPC phenotype, increased H3K27 trimethylation repressive marks were observed in the promoters of each of the stem cell genes in the combined treatment condition (Fig. 7B). Consistent with this effect, combined AMD3100 and GDC-0449 treatment decreased the expression of both Bmi1 and Sox2 in intracerebellar xenografts compared with all other treatment groups (Fig. 7C).
Figure 7.
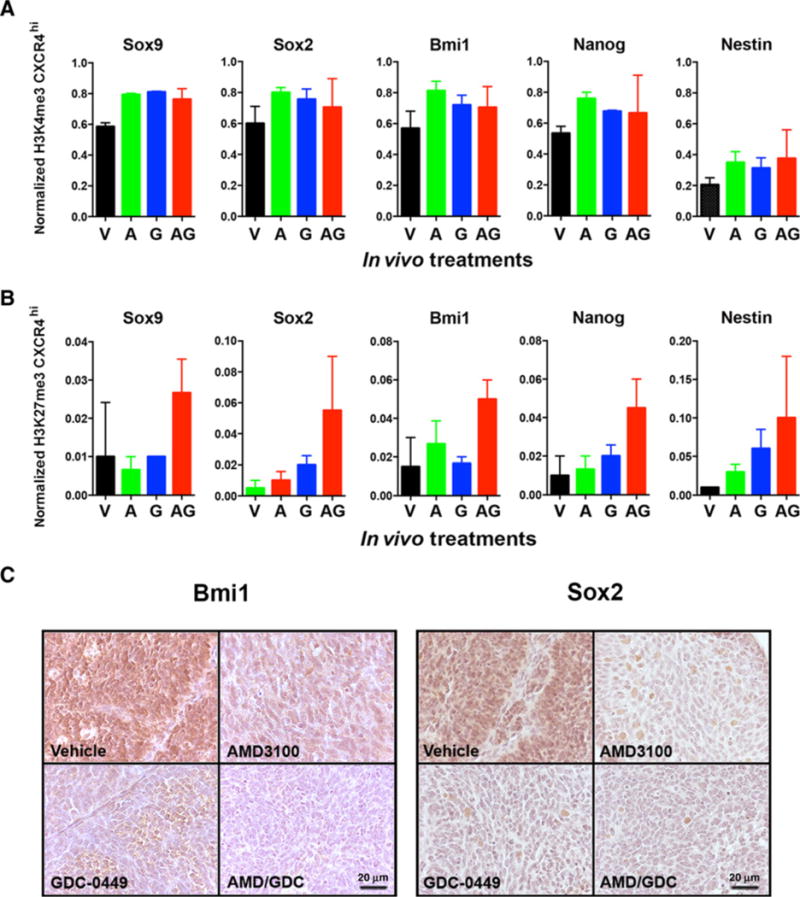
Combined SHH and CXCR4 antagonism reprograms histone marks in stem cell genes in CXCR4hi TPCs. CXCR4hi single-cell suspensions derived from in vivo–treated tumors [vehicle (V), AMD3100 (A), GDC-0449 (G), and AMD3100/GDC-0449 (AG)] were prepared for chromatin immunoprecipitation for histone H3K4 trimethylation (A) or histone H3K27 trimethylation (B). Treatment significantly increased H3K27 trimethylation (P value for treatment effect = 0.000617). C, Representative images of Bmi1 and Sox2 IHC in tumors from each treatment group. Scale bar, 20 μm.
Discussion
In this study, using two different treatment paradigms in two different mouse strains, we show that combined CXCR4 and SHH pathway antagonism displays synergistic antitumor activity in both flank and intracerebellar orthotopic xenograft models of medulloblastoma. The enhanced therapeutic effect was associated with decreased proliferation and a unique reprogramming of TPC potential of a subpopulation of cells that exhibited high levels of surface CXCR4 expression. As both AMD3100 and GDC-0449 are already available for human use, testing this treatment regimen should move rapidly into clinical trials.
The data presented here suggest a model of activity with at least three components that could be addressed in future studies to better define the mechanisms by which SHH and CXCR4 converge on the maintenance of a TPC phenotype and to further advance their effective cotargeting. The first is the apparent sensitization of SmoA1 cells to intermittent AMD3100 by GDC-0449 treatment. Intermittent AMD3100 had no activity as a single agent. This is in contrast to continuous AMD3100 treatment in the PTC heterozygous mouse model and continuous AMD3100 or AMD3465 in the Daoy model (16, 17, 21). Treatment with GDC-0449 resulted in a significant loss of surface CXCR4 expression in most, but not all, cells. The importance of residual surface CXCR4 to tumor regrowth following GDC-0449 treatment was clearly shown by the synergistic effect that combined GDC-0449 and AMD3100 had on tumor regrowth, in vitro clonogenicity, and in the in vivo subtransplanted tumorigenic potential of treated cells.
The second component of the model is the clear relationship observed between surface CXCR4 and TPC phenotype and function. This was evident in the correlation between surface CXCR4 and expression of CD15, stem cell marker genes, clonogenicity, and in vivo tumorigenicity. Although GDC alone had no effect on TPC function, it did promote a split in the tumor cell population to those in which there was maintenance of both high levels of surface CXCR4 expression and TPC activity and those with lower levels of surface CXCR4 expression and TPC activity. The relationship between surface CXCR4 and TPC activity is likely to relate to the abnormal mode of CXCR4 signaling previously described in multiple types of brain tumor cells, in which there is prolonged suppression of cAMP levels through sustained heterotrimeric G protein signaling (17, 39, 40). In several cases, this appears to be the result of decreased GRK expression and activity. Here, the absence of CXCR7 expression, a negative regulator of CXCR4 surface localization and CXCR4-induced heterotrimeric G protein signaling, suggests that this may be an additional mechanism by which very high levels of surface CXCR4 are achieved. High levels of CXCR4 expression in the absence of CXCR7 were also seen in human medulloblastoma, emphasizing the relevance of these findings to human disease.
The third component of the model relates to whether the combined effects of AMD3100 and GDC-0449 on histone modification, stem cell gene expression, and TPC function are mediated directly on the persistent CXCR4hi population or on the CXCR4lo population to block a TPC-promoting effect they have on the CXCR4hi cells. If the effect is direct, GDC-0449 might sensitize CXCR4hi cells to the effects of AMD3100 by altering CXCR4 coupling to downstream pathways that converge on epigenetics and gene expression. Again, this could involve SHH pathway effects on CXCR4 modulators that determine the bias between heterotrimeric-dependent and -independent events downstream of receptor activation (41). If the effect is indirect, the effects of combined AMD3100 and GDC-0449 might be mediated through the CXCR4lo subpopulation. Although these cells do no possess significant TPC activity themselves, they emerge upon GDC-0449 treatment and thus may be a unique target of AMD3100. If this is the case, they may provide paracrine support for the TPC activities in the CXCR4hi population.
The mechanistic details of these models will be defined in future studies. Although this work is ongoing, the results presented here support the combined administration of GDC-0449 and AMD3100 for patients with SHH subtype medulloblastoma and other cancers where these pathways are coexpressed.
Supplementary Material
Acknowledgments
We would like to thank Rajarshi Sengupta, Jasmin Sponagel, Cameron Hill, Tao Sun, Erica Maria Lantelme, and Dorjan Brinja for technical assistance. We thank Albert Kim, Milan Chheda, and Kristen Kroll for critical reading of the manuscript. We thank the Alvin J. Siteman Cancer Center and the Department of Pathology and Immunology at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO, for the use of the Siteman Flow Cytometry core.
Grant Support
This work was supported by the NIH RO1CA118389 (J.B. Rubin), F32 CA162558 (S.A. Ward), and the Hyundai Hope on Wheels Scholar Program (J.B. Rubin). Use of the flow cytometry core at The Siteman Cancer Center was supported in part by NCI Cancer Center support grant #P30 CA091842.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: S.A. Ward, J.B. Rubin
Development of methodology: S.A. Ward, J.B. Rubin
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S.A. Ward, N.M. Warrington, S. Taylor, N. Kfoury
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S.A. Ward, N.M. Warrington, S. Taylor, N. Kfoury, J. Luo, J.B. Rubin
Writing, review, and/or revision of the manuscript: S.A. Ward, N.M. Warrington, S. Taylor, N. Kfoury, J. Luo, J.B. Rubin
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): N.M. Warrington, S. Taylor, N. Kfoury, J.B. Rubin
Study supervision: J.B. Rubin
References
- 1.Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, et al. Alex’s Lemonade Stand Foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2015;16(Suppl 10):x1–x36. doi: 10.1093/neuonc/nou327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ning MS, Perkins SM, Dewees T, Shinohara ET. Evidence of high mortality in long term survivors of childhood medulloblastoma. J Neurooncol. 2015;122:321–7. doi: 10.1007/s11060-014-1712-y. [DOI] [PubMed] [Google Scholar]
- 3.Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–84. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2010;29:1408–14. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19:6305–12. doi: 10.1158/1078-0432.CCR-13-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33:2646–54. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Juhasz ML, Marmur ES. Systematic review of Vismodegib toxicity profile in the treatment of advanced basal cell carcinomas compared to other systemic therapies in dermatology. J Drugs Dermatol. 2014;13:729–33. [PubMed] [Google Scholar]
- 9.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–53. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27:327–41. doi: 10.1016/j.ccell.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao X, Ponomaryov T, Ornell KJ, Zhou P, Dabral SK, Pak E, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623–35. doi: 10.1158/0008-5472.CAN-14-2999-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gururangan S, Robinson G, Ellison DW, Wu G, He X, Lu QR, et al. Gorlin syndrome and desmoplastic medulloblastoma: report of 3 cases with unfavorable clinical course and novel mutations. Pediatr Blood Cancer. 2015;62:1855–8. doi: 10.1002/pbc.25560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaillant C, Monard D. SHH pathway and cerebellar development. Cerebellum. 2009;8:291–301. doi: 10.1007/s12311-009-0094-8. [DOI] [PubMed] [Google Scholar]
- 15.Klein RS, Rubin JB, Gibson HD, DeHaan EN, Alvarez-Hernandez X, Segal RA, et al. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development. 2001;128:1971–81. doi: 10.1242/dev.128.11.1971. [DOI] [PubMed] [Google Scholar]
- 16.Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003;100:13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang L, Jackson E, Woerner BM, Perry A, Piwnica-Worms D, Rubin JB. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007;67:651–8. doi: 10.1158/0008-5472.CAN-06-2762. [DOI] [PubMed] [Google Scholar]
- 18.Micallef IN, Stiff PJ, Stadtmauer EA, Bolwell BJ, Nademanee AP, Maziarz RT, et al. Safety and efficacy of upfront plerixafor + G-CSF versus placebo + G-CSF for mobilization of CD34(+) hematopoietic progenitor cells in patients >/=60 and <60 years of age with non-Hodgkin’s lymphoma or multiple myeloma. Am J Hematol. 2013;88:1017–23. doi: 10.1002/ajh.23561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004;37:1253–62. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- 20.Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, et al. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR- 4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44:1667–73. doi: 10.1128/aac.44.6.1667-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengupta R, Dubuc A, Ward S, Yang L, Northcott P, Woerner BM, et al. CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer Res. 2012;72:122–32. doi: 10.1158/0008-5472.CAN-11-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun T, Warrington NM, Luo J, Brooks MD, Dahiya S, Snyder SC, et al. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J Clin Invest. 2014;124:4123–33. doi: 10.1172/JCI71048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Woerner BM, Warrington NM, Kung AL, Perry A, Rubin JB. Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res. 2005;65:11392–9. doi: 10.1158/0008-5472.CAN-05-0847. [DOI] [PubMed] [Google Scholar]
- 25.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 26.Chen D, Xia Y, Zuo K, Wang Y, Zhang S, Kuang D, et al. Crosstalk between SDF-1/CXCR4 and SDF-1/CXCR7 in cardiac stem cell migration. Sci Rep. 2015;5:16813. doi: 10.1038/srep16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M, Chang CJ, Lathia JD, Wang L, Pacenta HL, Cotleur A, et al. Chemokine receptor CXCR4 signaling modulates the growth factor-induced cell cycle of self-renewing and multipotent neural progenitor cells. Glia. 2011;59:108–18. doi: 10.1002/glia.21080. [DOI] [PubMed] [Google Scholar]
- 28.Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–47. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward RJ, Lee L, Graham K, Satkunendran T, Yoshikawa K, Ling E, et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 2009;69:4682–90. doi: 10.1158/0008-5472.CAN-09-0342. [DOI] [PubMed] [Google Scholar]
- 30.Nicolis SK. Cancer stem cells and “stemness” genes in neuro-oncology. Neurobiol Dis. 2007;25:217–29. doi: 10.1016/j.nbd.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Davidow L, Arvanites AC, Blanchard J, Lam K, Xu K, et al. Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chem Biol. 2012;19:972–82. doi: 10.1016/j.chembiol.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma M, Wu W, Li Q, Li J, Sheng Z, Shi J, et al. N-myc is a key switch regulating the proliferation cycle of postnatal cerebellar granule cell progenitors. Sci Rep. 2015;5:12740. doi: 10.1038/srep12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metcalfe C, Alicke B, Crow A, Lamoureux M, Dijkgraaf GJ, Peale F, et al. PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer Res. 2013;73:7034–42. doi: 10.1158/0008-5472.CAN-13-1222. [DOI] [PubMed] [Google Scholar]
- 37.Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. 2014;26:33–47. doi: 10.1016/j.ccr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mack SC, Hubert CG, Miller TE, Taylor MD, Rich JN. An epigenetic gateway to brain tumor cell identity. Nat Neurosci. 2016;19:10–9. doi: 10.1038/nn.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warrington NM, Woerner BM, Daginakatte GC, Dasgupta B, Perry A, Gutmann DH, et al. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res. 2007;67:8588–95. doi: 10.1158/0008-5472.CAN-06-2220. [DOI] [PubMed] [Google Scholar]
- 40.Woerner BM, Luo J, Brown KR, Jackson E, Dahiya SM, Mischel P, et al. Suppression of G-protein-coupled receptor kinase 3 expression is a feature of classical GBM that is required for maximal growth. Mol Cancer Res. 2012;10:156–66. doi: 10.1158/1541-7786.MCR-11-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubin JB. Chemokine signaling in cancer: one hump or two? Semin Cancer Biol. 2009;19:116–22. doi: 10.1016/j.semcancer.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.