

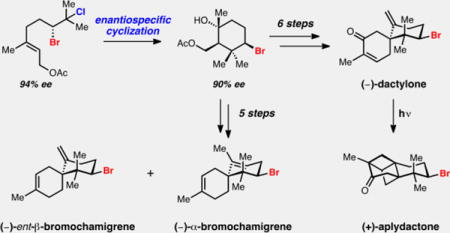
Abstract
The brominated chamigrene sesquiterpenes constitute a large subclass of bromocyclohexane containing natural products, yet no general enantioselective strategy for the synthesis of these small molecules exists. Herein we report a general strategy for accessing this family of secondary metabolites including the enantioselective synthesis of (−)-α- and (−)-ent-β-bromochamigrene, (−)-dactylone, and (+)-aplydactone. Access to these molecules is enabled by a stereospecific bromopolyene cyclization initiated by the solvolysis of an enantioenriched vicinal bromochloride.
Keywords: asymmetric synthesis, halogenation, natural products, solvolysis, terpenes
Dihalides Light the Way
A highly general approach to the brominated chamigrene sesquiterpenes has been realized via a stereospecific bromopolyene cyclization of an enantioenriched bromochloride. The total synthesis of (+)-aplydactone has been accomplished for the first time via an intramolecular [2+2] cycloaddition.
Of the roughly 300 natural products that have been isolated and structurally characterized containing a bromocyclohexane motif (1, Figure 1a),[1a–c] more than 50 are represented by the brominated chamigrene sesquiterpenes (2, Figure 1a). Most members of this family differ in their level of saturation, halogenation, and oxygenation (3–8, Figure 1b). The structural variety within the halogenated chamigrenes is complemented by diverse biological activities, with noteworthy examples including antibacterial,[2] antifungal,[3] antiviral,[4] anthelmintic,[5] and anticancer[6] effects. To date, the total synthesis of (+)-elatol 6 by Stoltz and Grubbs[7a] constitutes the only enantioselective synthesis of a member of the halogenated chamigrene sesquiterpenes. A general enantioselective approach for the synthesis of this class of molecules has yet to be reported.[7b–e] Along these lines, we set out to develop a strategy that would enable rapid construction of the spirocyclic core and thus facilitate elaboration to structurally disparate members of this family of small molecules for further chemical and biological investigations.
Figure 1.
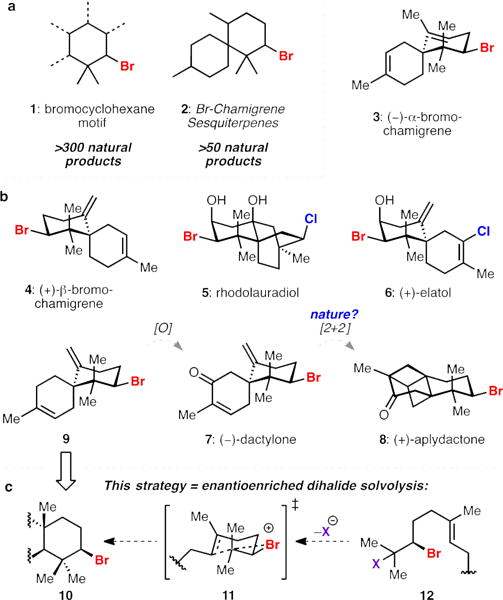
The halogenated chamigrene sesquiterpenes. a) Ubiquitous bromocyclohexane motif and bromochamigrene skeleton; b) representative members of the natural product class; c) this approach.
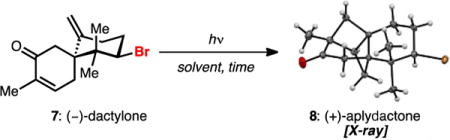
We were particularly intrigued by the cancer preventive agent dactylone (7)[8] and a related congener, aplydactone (8).[9] Dactylone has been shown to suppress phenotype expression at non-cytotoxic doses in human lung, colon, and skin tumor cell lines and therefore holds promise as a molecular tool for anticancer studies.[8c] Dactylone and aplydactone were originally isolated from the sea hare Aplysia dactylomela in 1987, however the structure of aplydactone was not disclosed until 2001 with the assistance of X-ray crystallography.[9] In that report, it was suggested that 8 may arise from an intramolecular [2 + 2] cycloaddition of 7. Accordingly, we selected dactylone 7 as a synthetic target with the objective of also interrogating its conversion to aplydactone 8. We hoped this investigation would shed light on the feasibility of this transformation both in a biosynthetic and laboratory setting. We were confident that 7 could be obtained directly from its corresponding deoxygenated precursor 9. This diene, while not yet having been isolated, is representative of the simplest members of the brominated chamigrenes, specifically its isomeric natural product counterparts, α– and β–bromochamigrene[3,10,11] (3 and 4, Figure 1b). We thus devised a strategy that was capable of providing facile access to numerous brominated spirodienes in enantioenriched form.
We targeted isoprenoid-derived bromocycle 10 that could be rapidly elaborated to the spirocyclic chamigrene core (10 to 9, Figure 1c), providing a general way of accessing this class of natural products. While a bromonium-induced carbocationic cyclization would provide a direct means to access candidate carbocycles, no enantioselective bromopolyene cyclization protocols have been reported on natural product-relevant scaffolds.[12] Cognizant of this methodological gap, we saw an opportunity to use enantioenriched vicinal dihalides as progenitors of bromonium ions. Enantiopure bromonium ions have been previously generated from enantioenriched bromohydrin derivatives and have been captured in an intra- and intermolecular fashion.[12c,d,13b] However, to our knowledge the generation and subsequent trapping of an enantioenriched bromonium ion from a vicinal dihalide has not been reported. Recently our laboratory disclosed an enantioselective Schiff base catalyzed di- and interhalogenation reaction of allylic alcohols, providing access to highly enantioenriched bromochlorides,[14a] dibromides, and dichlorides.[14b–d] We hypothesized that subjecting these species to ionizing conditions could facilitate the generation of an enantioenriched halonium ion that could be captured intramolecularly for productive cyclization (12 to 10 via 11, Figure 1c).
This plan called for the enantioselective dihalogenation of allylic alcohol 13 (Scheme 1), which can be prepared in one step from commercially available geranyl acetate.[15] Using conditions previously disclosed by our laboratory, (R,S)-Schiff base 14-catalyzed bromochlorination proceeded in good yield (66%) and with excellent enantioselectivity (94% ee) on 4.4 g scale to deliver bromochloride 15. Two-step deoxygenation[14d] followed by reacylation provided >1 g of bromochloro-geranyl acetate 16.
Scheme 1.
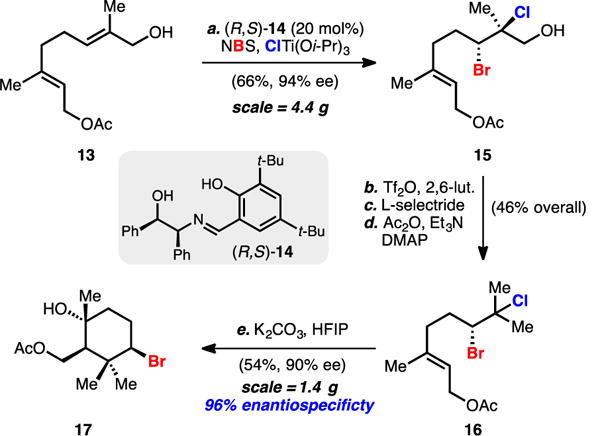
Enantioselective dihalogenation and key solvolytic bromopolyene cyclization. Reagents and conditions: a) NBS (1.05 equiv), ClTi(Oi-Pr)3 (1.1 equiv), 20 mol % (R,S)-14, hexanes, –20 ˚C, 66%; b) triflic anhydride (1.2 equiv), 2,6-lutidine (2.0 equiv), CH2Cl2, –78 ˚C, 85%; c) L-selectride (5.5 equiv), THF, –78 ˚C to r.t., 58%; d) acetic anhydride (1.0 equiv), 4-dimethylaminopyridine (0.05 equiv), triethylamine (1.2 equiv), CH2Cl2, r.t., 94%; e) potassium carbonate (1.5 equiv), hexafluoroisopropanol (0.05 M), r.t., 54%.
With the requisite precursor in hand, we subjected 16 to ionizing conditions in basic hexafluoroisopropanol (HFIP)[13] optimistic that an enantiopure non-racemizing bromonium ion could be generated and captured by the pendant allylic acetate. To our delight, the cyclization proceeded smoothly and afforded bromocarbocycle 17 in 54% yield as a single isolated diastereomer with near-perfect enantiospecificity (90% ee 17; 96% enantiospecificity). Analogous dibromoalcohols were observed to racemize under the ionizing conditions and were unstable to the deoxygenation conditions. To our knowledge this is the first example of the solvolysis and intramolecular capture of an enantioenriched dihalide.
We next sought to elaborate enantioenriched carbocycle 17 to the spirocyclic framework relevant to the brominated chamigrene sesquiterpenes (Scheme 2). We envisioned that an isoprene Diels–Alder transformation with a corresponding exocyclic enone could provide the desired scaffold. Bromocycle 17 was cleanly dehydrated to the exocyclic olefin[16] with subsequent oxidative cleavage[17] affording ketone acetate 18 in good yield (62% over two steps). Facile elimination of the acetate to the exocyclic enone 19 occurred in the presence of DBU with mild heating, but attempts at its isolation led to rapid formation of unusual hetero-Diels–Alder dimer 20 upon concentration and standing. Circumventing isolation of this unstable intermediate, enone 19 was generated in situ and treated with dimethylaluminum chloride and excess isoprene,[18] and the desired Diels–Alder adduct was easily obtained as a 4.3:1 (52% 21 + 12% 22) mixture of separable epimers. Access to these diastereomeric spiroketones served as a convenient diversification point in our synthesis providing access to the doubly unsaturated, isomeric α- and ent-β-bromochamigrenes (4 and 3, respectively).
Scheme 2.
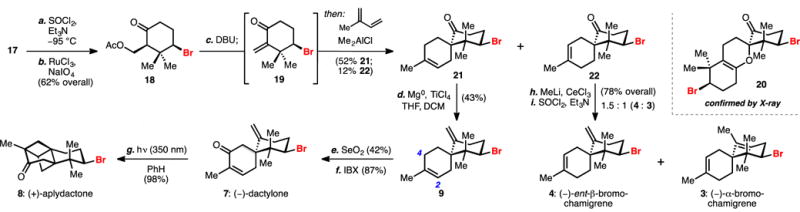
Synthesis of (–)-dactylone, (+)-aplydactone, and (–)-α- and (–)-ent-β-bromochamigrene. Reagents and conditions: a) thionyl chloride (1.5 equiv), triethylamine (5 equiv), CH2Cl2, –196 ˚C to –95 ˚C; b) ruthenium(III) chloride (0.02 equiv), sodium periodate (1.5 equiv), MeCN/CCl4/H2O, r.t., 62% from 17; c) 1,8-diazabicycloundec-7-ene (1.1 equiv), isoprene (20 equiv), Me2AlCl (3.5 equiv), PhMe, –78 ˚C to –10 ˚C 52% 21 + 12% 22; d) magnesium (8.0 equiv), titanium tetrachloride (2.0 equiv), THF/CH2Cl2, 0 ˚C to r.t., 43%; e) selenium dioxide (1.5 equiv), dioxane, 80 ˚C, 42%; f) 2-iodoxybenzoic acid (2.0 equiv), DMSO, r.t., 87%; g) hv (350 nm), PhH, r.t., 98%; h) methyllithium (4.5 equiv), cerium(III) chloride (5.0 equiv), THF, –78 ˚C to 0 ˚C; i) thionyl chloride (2.1 equiv), triethylamine (5.4 equiv), CH2Cl2, –78 ˚C to 0 ˚C, 78% combined.
Transformation of spiroketones 21 and 22 to their corresponding exocyclic olefins was next required for the synthesis of ent-β-bromochamigrene 4 and dactylone 7. An initial survey of standard methylenation conditions[19] failed to convert any of the spiroketone (21) to the desired olefin 9. We reasoned that failed olefinations were due to the combination of the extremely hindered neopentyl ketone and the bulkiness of reagents. We thus turned our attention to conditions successful at methylenating highly hindered substrates[20] utilizing dichloromethane as a methylene equivalent in combination with TiCl4 and Mg0. These conditions were sufficient to convert 21 into the doubly unsaturated spirocycle 9 in 43% yield. An exhaustive investigation of known olefination conditions did not provide superior results. Surprisingly, attempts to convert epimeric spiroketone 22 to (–)-ent-β-bromochamigrene (4) using the conditions listed above resulted in diminished yields compared to its corresponding isomer. However, a two-step protocol via the addition of methylcerium dichloride[7] followed by dehydration in the presence of thionyl chloride afforded (−)-α- and (−)-ent-β-bromochamigrene as a 1.5:1 mixture in 78% yield. Conversion of spirodiene 9 to dactylone 7 called for a selective allylic oxidation of the methylene at C4 to the ketone oxidation state. Unfortunately, conditions to directly affect this transformation[21] failed to selectively oxidize the allylic methylene at C4 over C2. Selenium dioxide[22] efficiently produced the C4 allylic alcohol, which was readily oxidized to the corresponding enone with IBX, completing the first synthesis of (–)-dactylone 7. Cognizant of the reported failed attempts of the isolation team to convert dactylone 7 to aplydactone 8 under long-term UV irradiation,[9] we nevertheless subjected dactylone 7 to known enone [2+2] conditions under irradiation with 350 nm light. To our delight, dactylone 7 underwent clean conversion to aplydactone 8 (see Table 1 for X-ray structure) over a period of 36 hours, which we were able to isolate in near-quantitative yield (98%) after silica gel chromatography.[23]
Table 1.
[a] Yield based on 1H NMR using 1,4-dinitrobenzene as an internal standard. [b] Isolated yield.
| ||||
|---|---|---|---|---|
| entry | solvent | hv | time | % conv.7 (% yield 8) |
| 1 | THF | 254 nm | 15 m | 100 (7a) |
| 2 | PhH | 350 nm | 20 h | 85 (ca. 85) |
| 3 | PhH | 350 nm | 36 h | 100(98b) |
| 4 | CDCI3 | California sunlight | 8 days | 15 (ca. 15) |
Irradiation of dactylone 7 with 254 nm light led to formation of small amounts of 8 as a complex mixture (Table 1, entry 1), indicating that judicious choice of light source is crucial to the success of the intramolecular [2+2] cycloaddition. Standard enone-olefin 350 nm irradiation delivered 8 with the necessity for prolonged reaction times (entries 2 and 3). Exposure of dactylone 7 to ambient sunlight over a period of 8 days (entry 4) resulted in the formation of aplydactone 8, albeit in small quantities (15% conversion).[24] This observation raises an interesting question about the role of sunlight in the biosynthesis of aplydactone, in particular whether or not enzymatic machinery is necessary in its biogenesis. It is worth noting that photochemical transformations, specifically those utilizing sunlight, have been directly implicated in the biosynthesis of marine natural products.[25]
The enantioselective total synthesis of (–)-α- and (–)-ent-β-bromochamigrene, (–)-dactylone, and (+)-aplydactone was enabled by the gram-scale enantiospecific solvolytic cyclization of an enantioenriched bromochloride. Access to this interhalogenated motif was enabled by the highly chemo-, diastereo-, and enantioselective Schiff base catalyzed bromochlorination of allylic alcohol 13. This work highlights a highly general approach to the halogenated chamigrene sesquiterpenes and we anticipate that it will find use in the synthesis of additional members of this class. Studies along those lines as well as a comprehensive investigation into the solvolytic cyclization of enantioenriched dihalides are in progress and will be reported in due course.
Supplementary Material
Acknowledgments
We are grateful to Dr. A. Oliver (University of Notre Dame) for X-ray crystallographic analysis and Dr. S. Lynch (Stanford University) for assistance with NMR spectroscopy. This work was supported by Stanford University, the National Institutes of Health (R01 GM114061), and the National Science Foundation (DGE-114747 GRF to AJB).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Gribble GW. Naturally Occurring Organohalogen Compounds-A Comprehensive Survey. Springer-Verlag: Wien; 1996. [PubMed] [Google Scholar]; b Gribble GW. Naturally Occurring Organohalogen Compounds–A Comprehensive Update. Springer-Verlag; Wien: 2010. [Google Scholar]; c Wang BG, Gloer JB, Ji NY, Zhao JC. Chem Rev. 2013;113:3632–3685. doi: 10.1021/cr9002215. [DOI] [PubMed] [Google Scholar]
- 2.a) Vairappan CS, Daitoh M, Suzuki M, Abe T, Masuda M. Phytochemistry. 2001;58:291–297. doi: 10.1016/s0031-9422(01)00243-6. [DOI] [PubMed] [Google Scholar]; b) ibid. :517–523. [Google Scholar]
- 3.König GM, Wright AD. J Nat Prod. 1997;60:967–970. doi: 10.1021/np970181r. [DOI] [PubMed] [Google Scholar]
- 4.Kimura J, Kamada N, Tsujimoto Y. Bull Chem Soc Jpn. 1999;72:289–292. [Google Scholar]
- 5.Davyt D, Fernandez R, Suescun L, Mombrú AW, Saldaña J, Domínguez L, Coll J, Fujii MT, Manta E. J Nat Prod. 2001;64:1552–1555. doi: 10.1021/np0102307. [DOI] [PubMed] [Google Scholar]
- 6.a) Juagdan EG, Kalidindi R, Scheuer P. Tetrahedron. 1996;53:521–528. [Google Scholar]; b) Rashid MA, Gustafson KR, Cardellina JH, Boyde MR. Nat Prod Lett. 1995;6:255–259. doi: 10.1080/10575630108041253. [DOI] [PubMed] [Google Scholar]
- 7.a) White DE, Stewart IC, Grubbs RH, Stoltz BM. J Am Chem Soc. 2008;130:810–811. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]; For racemic approaches to brominated chamigrenes, see:; b) Wolinsky LE, Faulkner DJ. J Org Chem. 1976;41:597–600. [Google Scholar]; c) Ichinose I, Kato T. Chem Lett. 1979:61–62. [Google Scholar]; a racemic synthesis of α-bromochamigrene has been claimed, however the reported NMR spectra disagree with our and previous isolation data:; d) Cui Q, Kang L, Yang HS, Xu XH. Chinese Chem Lett. 2009;20:554–556. [Google Scholar]; for an approach involving chiral resolution, see:; e) Martin JD, Pérez C, Ravelo JL. J Am Chem Soc. 1986;108:7801–7811. doi: 10.1021/ja00284a052. [DOI] [PubMed] [Google Scholar]; for an insightful review on sterecontrolled halogenation in natural product synthesis, see:; f) Chung W, Vanderwal CD. Angew Chem Int Ed. 2016;55:4396–4434. doi: 10.1002/anie.201506388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Federov SN, Reshetnyak AP, Shchedrin AP, Il’in SG, Struchkov YT, Stonik VA, Elyakov GB. Dokl Akad Nauk SSSR. 1989;305:877–879. [Google Scholar]; b) Lyakhova EG, Federov SN, Shubina L, Radchenko OS, Kalinovsky AI, Dvitrenok PS, Stonik VA. Russ Chem Bull Int Ed. 2003;52:970–974. [Google Scholar]; c) Federov SN, Shubina LK, Bode AM, Stonik VA, Dong Z. Cancer Res. 2007;67:5194–5920. doi: 10.1158/0008-5472.CAN-06-3723. [DOI] [PubMed] [Google Scholar]
- 9.Federov SN, Radchenko OS, Shubina LK, Kalinovsky AI, Gerasimenko AV, Popov DY, Stonik VA. J Am Chem Soc. 2001;123:504–505. doi: 10.1021/ja003254t. [DOI] [PubMed] [Google Scholar]
- 10.10-bromo-α-chamigrene herein referred to as α-bromochamigrene; for isolation reports see:; a) Howard BM, Fenical W. Tetrahedron Lett. 1976;17:2519–2520. [Google Scholar]; b) Suzuki M, Furusaki A, Kurosawa E. Tetrahedron. 1979;35:823–831. [Google Scholar]; c) König GM, Wright AD. J Nat Prod. 1994;57:477–485. [Google Scholar]; for isolation of the (+)-enantiomer, see:; d) Guella G, Öztunç A, Mancini I, Pietra F. Tetrahedron Lett. 1997;38:8261–8264. [Google Scholar]
- 11.10-bromo-β-chamigrene herein referred to as β-bromochamigrene; for isolation reports see ref. 10d and:; a) König GM, Wright AD. Phytochem Anal. 1997;8:167–172. [Google Scholar]; b) Ji NY, Li XM, Li K, Ding LP, Gloer JB, Wang BG. J Nat Prod. 2007;70:1901–1905. doi: 10.1021/np070378b. [DOI] [PubMed] [Google Scholar]; c) Li XD, Ding W, Miao FP, Ji NY. Magn Reson Chem. 2012;50:174–177. doi: 10.1002/mrc.2870. [DOI] [PubMed] [Google Scholar]
- 12.a) Snyder SA, Treitler DS. Angew Chem Int Ed. 2009;48:7899–7903. doi: 10.1002/anie.200903834. [DOI] [PubMed] [Google Scholar]; Up to 36% ee has been achieved in an enantioselective bromopolyene cyclization with a stoichiometric chiral promoter:; b) Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]; For formal enantioselective bromopolyene cyclizations see:; c) Braddock DC, Marklew JS, Thomas AJF. Chem Comm. 2011;47:9051–9053. doi: 10.1039/c1cc13619d. [DOI] [PubMed] [Google Scholar]; d) Braddock DC, Marklew JS, Foote KM, White AJP. Chirality. 2013;25:692–700. doi: 10.1002/chir.22194. [DOI] [PubMed] [Google Scholar]; e) Snyder SA, Treitler DS, Schall A. Tetrahedron. 2010;66:4796–4804. [Google Scholar]
- 13.For a classic solvolysis study using HFIP see:; a) Schadt FL, Bentley TW, Schleyer PvR. J Am Chem Soc. 1976;98:7667–7674. [Google Scholar]; For a recent study on the solvolysis of dihalide derivatives using HFIP see:; b) Denmark SE, Burk MT, Hoover AJ. J Am Chem Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]
- 14.a) Hu DX, Seidl FJ, Bucher CB, Burns NZ. J Am Chem Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]; b) Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]; c) Landry ML, Hu DX, McKenna GM, Burns NZ. J Am Chem Soc. 2016;138:5150–5158. doi: 10.1021/jacs.6b01643. [DOI] [PMC free article] [PubMed] [Google Scholar]; For use of this methodology in total synthesis see:; d) Bucher CB, Deans RM, Burns NZ. J Am Chem Soc. 2015;137:12784–12787. doi: 10.1021/jacs.5b08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umbreit MA, Sharpless KB. J Am Chem Soc. 1977;99:5526–5528. [Google Scholar]
- 16.Snyder SA, Treitler DS, Brucks AP. J Am Chem Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]
- 17.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936–3938. [Google Scholar]
- 18.Antonsen S, Skattebøl L, Stenstrøm Y. Molecules. 2014;19:29664–20670. [Google Scholar]
- 19.a) Tebbe FN, Parshall GW, Reddy GS. J Am Chem Soc. 1978;100:3611–3613. [Google Scholar]; b) Clawson L, Buchwald SL, Grubbs RH. Tetrahedron Lett. 1984;25:5733–5736. [Google Scholar]; c) Cannizzo LF, Grubbs RH. J Org Chem. 1985;50:2386–2387. [Google Scholar]; d) Petasis NA, Bzowej EI. J Am Chem Soc. 1990;112:6392–6394. [Google Scholar]; e) Wittig G, Schöllkopf U. Chem Ber. 1954;87:1318–1330. [Google Scholar]
- 20.Yan T, Tsai C, Chien C, Cho C, Huang P. Org Lett. 2004;6:4961–4963. doi: 10.1021/ol0478887. [DOI] [PubMed] [Google Scholar]
- 21.a) Nicolaou KC, Claiborne CF, Nantermet PG, Couladouros EA, Sorensen EJ. J Am Chem Soc. 1994;116:1591–1592. [Google Scholar]; b) Wilde NC, Isomura M, Mendoza A, Baran PS. J Am Chem Soc. 2014;136:4909–4912. doi: 10.1021/ja501782r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Iwata C, Fusaka T, Maezaki N, Nakamura S, Shinoo Y, Yamada M, Tanaka T. Chem Pharm Bull. 1988;36:1638–1645. [Google Scholar]; d) Salmond WG, Barta ME, Havens JL. J Org Chem. 1978;43:2057–2059. [Google Scholar]; e) Kawamura S, Chu H, Baran PS. Nature. 2016;532:90–93. doi: 10.1038/nature17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen L, McKerrall SJ, Kuttruff CA, Ungeheuer F, Felding J, Baran PS. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- 23.See Supporting Information.
- 24.For classic reports of sunlight mediated intramolecular [2+2] cycloadditons see:; a) Ciamician G, Silber P. Ber. 1908;41:1928–1935. [Google Scholar]; b) Büchi G, Goldman IM. J Am Chem Soc. 1957;79:4741–4748. [Google Scholar]
- 25.a) Ireland C, Faulkner J. Tetrahedron. 1981;37:233–240. [Google Scholar]; b) Look SA, Fenical W. J Am Chem Soc. 1984;106:5026–5027. [Google Scholar]; c) Shin J, Fenical W. J Org Chem. 1991;56:1227–1233. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.