

Abstract
Unlocking the therapeutic potential of the glucocorticoid receptor (GR) has motivated a search for small molecules that selectively modulate GR’s ability to activate or repress gene transcription. Recently, breakthrough studies in the field of genomics have reinvigorated debate over long-standing transcriptional models explaining how GR controls tissue-specific gene expression. We highlight these genomic studies with the dual goals of advancing understanding of nuclear receptor-mediated transcription and stimulating thought on the development of anti-inflammatory and immunosuppressive ligands for GR that have reduced harmful effects on metabolism.
Keywords: Glucocorticoid receptor, nuclear receptor, transcription, functional genomics, cistromics
GR: a nuclear receptor with widespread physiological impact
GR, or NR3C1 (see Glossary), is a transcription factor (TF) that regulates gene expression in nearly every cell of the body. A member of the nuclear receptor (NR) superfamily, its ligand-binding domain confers transcriptional regulation by endogenous and synthetic lipophilic molecules [1]. Glucocorticoid (GC) activates GR [2]. A class of adrenal cortex steroid hormones named for its glucose regulating properties, site of production and compound structure, i.e. glucose + cortex + steroid, it was first linked to metabolism upon determining that removal of the adrenal gland in diabetic animals lowered glucose levels in the blood [3].
Whereas GR is expressed ubiquitously, cortisol and corticosterone, the major GC hormones in human and mouse, respectively, elicit tissue-specific effects (Figure 1). Consistent with its naming, GC stimulates glucose production in liver. It also affects energy homeostasis by inhibiting insulin-dependent glucose uptake in muscle and adipose, promoting the release of amino acids and free fatty acids from muscle and adipose tissue breakdown, and inhibiting insulin release from pancreatic β cells [4]. Systemic metabolic changes from GCs ultimately increase blood glucose levels. GCs also reduce inflammatory responses of immune cells, affect cardiovascular function in coronary arteries and alter mental and emotional states through action in the central nervous system (CNS) [5]. These actions are especially important during periods of acute stress, when elevated release of GC into the bloodstream helps to mobilize stored fuel for the ‘fight or flight’ response, maintain a ready supply of energy from glucose for the brain, and maintain vascular stability to prevent potentially life-threatening hypotension. It is noteworthy that some of the actions of GCs may result from cross talk between tissues. For example, GCs promote lipid catabolism in adipose tissue, yet whether they drive lipolysis by directly or solely acting on adipocytes remains unclear [6–8]. It should also be mentioned that GCs exert rapid effects occurring in minutes that are insensitive to inhibitors of DNA transcription and protein synthesis [9]. These non-genomic effects are thought to regulate the early stages of the stress response, though more study is needed to clarify this and their potential on impact physiology.
Figure 1. Tissue-specific functions of GC signaling to GR.
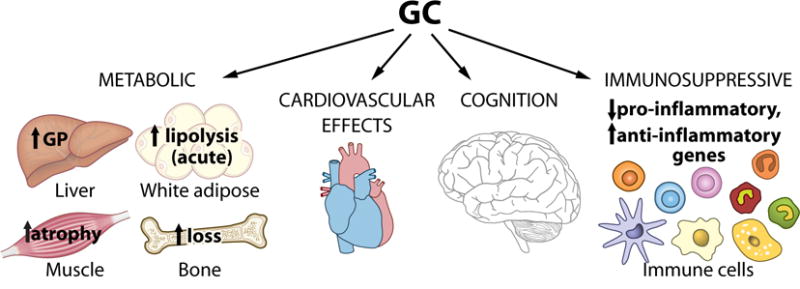
GCs mediate distinct biological effects in different tissues and cell types to systemically influence metabolism, cardiovascular function, cognition and inflammation. They impact energy homeostasis by increasing glucose production (GP) in the liver and promoting catabolic processes in muscle, adipose and bone. Their immunosuppressive effects are conferred by the repression of pro-inflammatory genes and activation of anti-inflammatory genes in white blood cells.
Genomic studies of GR have provided new insights into the tissue-specific effects of GCs. GR targets genes for transcriptional regulation by recognizing and binding to a particular DNA-sequence motif. Although widely expressed, GR occupies only a subset of its genomic motifs in any given cell type because most are buried in repressive chromatin structure that renders them inaccessible [10]. As chromatin structure is organized differently for each cell-type [11], the genomic occupancy and transcriptional output of GR are cell-type specific. How GR interacts with the native genome to regulate gene expression is the main focus of this review.
Health issues arising from defects in GC signaling to GR
Serious health issues result from abnormal GC signaling that can lead to death if untreated, underscoring the importance of GR in human physiology. Primary adrenal insufficiency or Addison’s disease arises from adrenal gland problems that cause production of cortisol and possibly aldosterone, the other major adrenal cortex steroid hormone and important regulator of blood pressure, to become too low to meet the body’s needs. Adrenal damage from the body’s immune system and infection in the developing world are often to blame. More common is secondary adrenal insufficiency. Characterized by disrupted signaling through the hypothalamic-pituitary-adrenal (HPA) axis, the pituitary gland fails to produce enough adrenocorticotropin hormone (ACTH) to stimulate cortisol synthesis and secretion. Replacing the absent hormones treats weakness, hypoglycemia, hypotension and other potential symptoms that are affiliated with adrenal insufficiencies [12]. Opposite of too little cortisol, Cushing’s disease commonly arises when a tumor causes too much cortisol to be produced. It can reside in the adrenal gland itself or a secondary ectopic site usually in the brain that leads to overproduction of ACTH or corticotropin-releasing hormone (CRH) by the hypothalamus. In addition to hyperglycemia, osteoporosis and muscle atrophy, a hallmark of Cushing’s disease is obesity from a redistribution of adipose tissue that drives central adiposity at the expense of peripheral fat [13].
Rare mutations can impair GR’s molecular function and alter tissue sensitivity to GCs in humans, resulting in primary generalized GC resistance (PGGR) and hypersensitivity (PGGH) [14]. Familial and sporadic PGGR, or Chrousos Syndrome, is characterized by general and partial insensitivity of tissues to GC and compensatory hyperactivation of the HPA axis. PGGH represents the opposite, with GC hypersensitivity and consequent hypoactivation of the HPA axis. As the cost of whole genome sequencing declines, it is becoming feasible to examine the relationship between mutations and polymorphisms in the GR gene and the considerable variation between patients in their response to GC treatment.
GR ligands are commonly prescribed drugs
GCs and their synthetic analogs are among the most widely prescribed drugs in the world. Their anti-inflammatory and immunosuppressive properties are important for the treatment of rheumatioid arthritis, cerebral edema, asthma and other allergic reactions, and the prevention of organ transplant rejection and graft-versus-host disease [15,16]. GCs are more potent than nonsteroidal anti-inflammatory drugs (NSAIDS, e.g. aspirin), yet up to a third of patients with severe asthma may be unresponsive to them [17]. GC resistance is likely a result of complex interactions between an individual’s genetic makeup and environment. In addition, GCs cause undesirable metabolic effects that can prematurely end treatment. Features of metabolic syndrome including obesity, dyslipidemia, insulin resistance and type 2 diabetes mellitus are prevalent, as well as osteoporosis and muscle atrophy [4].
Metabolic dysfunction arising from the chronic use of GC medication, termed Cushing’s syndrome, is related to Cushing’s disease, but more common in western societies that frequently prescribe GC drugs. Though many of the problems stemming from chronic GC excess can be explained by the acute effects of GC to induce insulin resistance, raise plasma glucose and increase protein catabolism, the underlying molecular mechanism(s) explaining how prolonged activation of GR causes weight gain is poorly understood. Indeed, GCs acutely induce lipolysis in human adipose tissue [13], and consistent with their catabolic effects, cause rats to lose weight over time [18,19]. A possible explanation for this paradox may involve depot-specific responses to excess GC that simultaneously trigger the breakdown and growth of limb and abdominal adipose tissue, respectively [13]. GCs facilitate adipocyte differentiation [20,21], though whether this contributes to the obesity of Cushing’s syndrome is unknown. While more work is needed to resolve the paradox, it serves as a reminder of the limitations of rodent models to provide insight into human physiology.
Tissue-specific function for GR
Intense effort has been dedicated to understanding how ubiquitously expressed GR elicits complex tissue-specific effects by controlling distinct gene programs in different cell types. Not surprisingly, mechanisms involving both GC availability and GR function provide answers. Cell-type-specific GC availability is modulated by a pair of hydroxysteroid 11-beta dehydrogenase (HSD11B) enzymes that control intracellular cortisol/cortisone levels. HSD11B1 converts the inert GC cortisone to cortisol, while HSD11B2 catalyzes the reverse reaction. Through distinct tissue-specific expression profiles, they amplify or mute responses to the circulating cortisol level set by the HPA axis [22]. HSD11B2 is found in kidney, lung, colon, salivary glands and HSD2 neurons, all of which are responsive to aldosterone, an activating ligand for the mineralocorticoid nuclear receptor (MR). Because cortisol also activates MR, HSD11B2 prevents its illicit activation by decreasing intracellular cortisol concentrations. HSD11B1 is expressed in key metabolic tissues such as adipose to amplify GC signaling. For example, HSD11B1 overexpression in mouse BAT decreases BAT thermogenic activity, while pharmacological HSD11B1 inhibition or knockdown enhances its function [23], consistent with corticosterone inhibition of BAT in rodents [24], and demonstrating that local GC levels matter. Of note, local levels of GCs may also be affected by extra-adrenal GC synthesis. For example, local GC production by the intestinal epithelium may regulate intestinal immune responses [25].
Although encoded by a single gene, GR displays considerable heterogeneity through the combined effects of alternative mRNA splicing, alternative translation initiation, and complex post-translational modification. Differential expression of the various GR isoforms may contribute to tissue-specific functions [26,27]. GRβ, the best characterized splice variant, uses of an alternate exon 9 that disrupts the structure of helices 11 and 12 in the ligand-binding domain, a region required for co-regulator recruitment [28]. Constitutively localized in the nucleus, GRβ cannot activate gene transcription in response to GC, suggesting that it acts as an endogenous dominant negative of GRα, the classic GR referred to in most studies. Though generally expressed at lower levels than GRα, cellular signals affecting the expression ratio of GRα to GRα show clinical associations with GC sensitivity, autoimmune disease, and lipid metabolic profiles [26]. Amino-terminal truncations of GR occur through the use of seven alternative translation initiation sites. While none directly prohibit DNA or ligand binding, they can alter GR conformation in a manner that affects its subcellular localization and transcriptional activity [29]. Furthermore, extensive phosphorylation of the N-terminal domain of GR adds to the complexity. Up to six serine residues in human GR are phosphorylated in vitro by either mitogen-activated protein kinases (MAPKs), cyclin-dependent kinases (CDKs) or glycogen synthase kinase 3 (GSK3) [26]. Phosphorylation of Ser-211 by p38 is associated with co-activator recruitment and transcriptional activation [30,31], whereas phosphorylation of Ser-226 by c-Jun N-terminal kinases (JNKs) [32] or CDK5 [33] impairs transcriptional activity [30,34]. Intriguingly, ChIP with phospho-specific GR antibodies suggests differential recruitment [34], however the affects of phosphorylation and other post-translational modifications on genome-wide binding for GR remain to be tested. Indeed, a future challenge is the development of molecular tools that can distinguish between the different isoforms to determine their genomic functions.
Ultimately, a more complete understanding of GR and its complex tissue-specific functions requires insight into how chromatin structure influences its ability to access genomic sites for gene regulation. A detailed discussion describing how GR interacts with the native genome follows, with a goal of dispelling two widespread models for negative regulation by GR.
GR occupies open chromatin through sequence-specific binding
Upon binding ligand in the cytoplasm, GR moves to the nucleus to target genes for transcriptional regulation (Figure 2A). It binds as a homodimer, and possibly as a homotetramer [35], to a palindromic DNA sequence roughly approximated as g/aGnACAnnnTGTnCt/c. GR binding in vivo has been dramatically informed by the advent of genome-wide approaches [36,37]. Early chromatin immunoprecipitation with deep sequencing (ChIP-seq) studies set the stage for new and surprising determinants for GR binding in diverse cell types and tissues obtained from mice and humans [10,38–44]. For a field focused on proximal gene promoters, the discovery that most GR-binding sites (GBSs) reside outside of these regulatory regions was eye opening. Also unforeseen was that GBSs vastly outnumber GC-regulated genes. The identification of thousands to tens-of-thousands of binding sites in any particular cell type surpasses the number of gene targets by an order of magnitude or more, with the caveat that the number of targets may increase if GR-regulated non-coding RNAs are discovered in large numbers. This difference is partly explained by findings showing that GC-regulated genes are enriched for multiple GBSs. However, an unexpectedly large number of sites do not co-localize with the histone modifications, chromatin remodelers and transcriptional cofactors associated with active enhancers [45]. It is tempting to consider these as experimental noise or artifact, but they are enriched for the GR motif, suggesting sequence-specific binding. While the size and complexity of the GR cistrome is greater than seemingly needed, the existence of sites without transcriptional characteristics may be understood by principles of constructive neutral evolution [46].
Figure 2. Genomic occupancy of GR monomers and dimers regulates gene transcription.
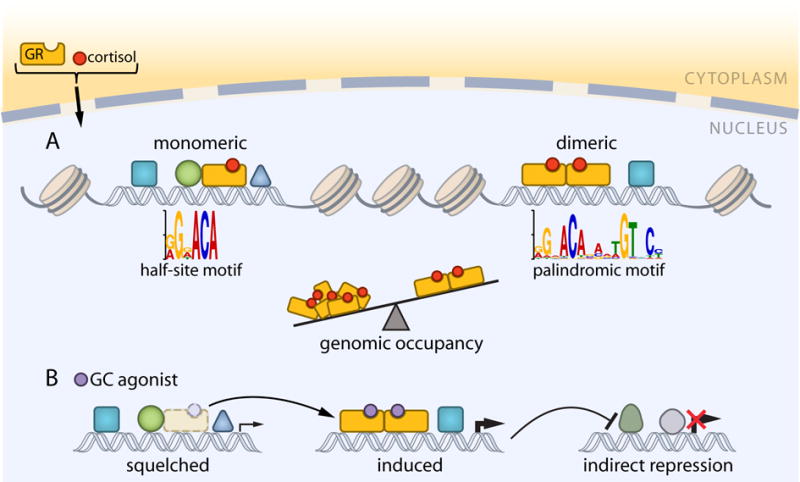
(A) Under normal physiological settings, cortisol enables GR to activate transcription through monomeric and dimeric interaction with half-site and palindromic motifs scattered throughout the genome. Neither monomers nor dimers can efficiently access motifs buried in repressive chromatin. Thus, most of their binding sites reside in open chromatin established by lineage determining TFs. Genomic occupancy is tilted toward monomers given that monomeric sites outnumber dimeric by 5:1 in liver. (B) In response to GC drugs, induced gene expression is associated with increased GR occupancy at dimeric sites, whereas down-regulated and unchanged genes correlate with a concomitant loss of GR at monomeric sites. While the genome-wide balance remains tilted toward monomers, gain of occupancy at one set of sites at the expense of another suggests a squelching mechanism for GC-mediated repression of gene expression. Indirect repression can also result from the primary induction by dimeric GR of genes whose products repress transcription. Genomic data do not support a mechanism for direct GC-mediated repression.
Genomic studies have afforded new insight into the influence of chromatin structure on GR binding. An important clue came from genome-wide mapping of chromatin structure using DNase I hypersensitive site sequencing (DNase-seq), and comparing the locations of open chromatin to GBSs [10]. DNase-seq in the absence and presence of ligand revealed that up to 95% of GR occupancy is directed to preexisting regions of accessible chromatin. Similar results were gathered in vivo with an adrenalectomized mouse model [39], suggesting that GR on its own cannot efficiently counteract histone-mediated repression to access DNA in nucleosomes. As this is a distinguishing feature of pioneer TFs [47], the ability of GR to serve a pioneering role on its own is limited [48]. Yet, GR binding further remodels chromatin to increase accessibility for other TFs through a mechanism termed assisted loading, originally advanced to describe how TFs targeting the same DNA motifs can stimulate, rather than inhibit, each other’s occupancy via dynamic TF-DNA interaction [49]. The model depends on the ability of TFs to trigger chromatin remodeling [50], and provides insight into the collaborative binding widely observed at neighboring cis-regulatory sequences [51–55], and conversely, helps to explain why enhancers typically contain several closely-spaced TF-binding sites.
Along with open chromatin, it is important to emphasize that the palindromic GR motif is a critical determinant of genomic occupancy. Prior to the emergence of genomics, characterization of GR’s recognition sequence had been primarily determined by in vitro approaches. Thus, it was reassuring that motif analyses of GR ChIP-seq data revealed robust enrichment of the palindromic sequence in vivo. However, due to the limited resolution of ChIP-seq, strong enrichment of additional co-localized motifs was also observed, confounding a definitive interpretation of GR’s genomic substrate. This was resolved by ChIP with lambda exonuclease digestion and sequencing (ChIP-exo), a technique designed to unequivocally identify bound motifs through unbiased, high resolution mapping of TFs to the genome [56]. With ChIP-exo, several groups demonstrated GR binding at the expected palindromic sequence in native chromatin [57–59].
GR monomers occupy the genome sequence-specifically and through tethering
Binding sites lacking the expected motif are present as a minor fraction in most TF ChIP-seq datasets, but can still account for thousands of sites in some cistromes. While experimental artifact such as antibody cross reactivity with chromatin-bound proteins other than GR may partially explain these, it is possible that TF binding in the absence of a motif may be biologically relevant. Of interest to GR, bound sites lacking the motif may represent protein-protein interaction between GR and other DNA-bound TFs to form so called tethered sites [60,61]. Indirect DNA binding is thought to explain GR’s effects on the expression of reporter genes lacking GR motifs, but unambiguous determination of the interacting protein(s) is challenging because presumed tethered sites often reside at composite enhancers bound by multiple TFs. With its superior resolution, ChIP-exo has identified tethering partners for GR monomers [58,59]. GR monomers were also found to interact sequence-specifically with the GR half-site motif, consistent with earlier in vitro studies [62–64]. It is intriguing that tethered sites strongly co-localize with monomeric, genomic sites, suggesting that transient contacts between monomers and nearby TFs are promoted and/or stabilized by GR interaction with half-site motifs. This idea, termed half-site-facilitated tethering, designates DNA as the primary recruiter of monomeric GR (Figure 2A), and stably bound TF neighbors as targets for subsequent protein-protein interaction through GR tracking on chromatin [62,65]. As the evidence for TF tethering mounts in vivo [66], better understanding of its prevalence and role(s) is needed.
GR is a transcriptional activator
How GR activates some genes while repressing others remains unsettled after decades of study. While significant controversy remains regarding GR-mediated repression, a consensus model for transcriptional activation has emerged: GR activates transcription through sequence-specific binding to the genome at palindromic motifs. This is strongly supported by genomic studies. Dimeric sites preferentially associate with ligand-activated gene expression on a genome scale [57,58,63], and become occupied by transcriptional cofactors, activating histone modifications and RNAPII in response to GR binding during cell differentiation [67,68]. While these data are correlative, self-transcribing active regulatory region sequencing (STARR-seq) has been used to directly examine the transcriptional regulatory properties of GBSs. The assay interrogates enhancer function in a direct, quantitative, and high-throughput manner by placing DNA fragments from any source downstream of a minimal promoter and introducing the reporter library into cells [69]. Analysis of GR ChIP DNA by STARR-seq revealed that 95% of the fragments conferring GC regulation increased reporter gene expression in response to exogenous GC [70]. Moreover, sequence analysis identified the GR palindromic motif as the sole predictor of GC regulation, and ChIP-exo revealed a characteristic dimeric profile at the GC-regulated regions, demonstrating that GC-induced enhancers encode dimeric sites for GR.
Like ligand, DNA functions as an allosteric regulator of GR by modulating its activity downstream of genomic occupancy to induce a select fraction of the GR transcriptome [64,71–73]. More generally, GR activates gene expression by integrating signals to nucleate the assembly of cofactors and the general transcription machinery. Recruitment of the Mediator Complex [43,68], a cofactor that facilitates long-range chromatin interactions between TFs and the transcription initiation machinery [74,75], implicates GR in the formation of DNA loops that potentially connect GBSs with promoters to activate distant genes. Similarly, DNA looping was invoked to explain the observation that dimeric sites are frequently surrounded by additional GBSs that lack both the palindromic motif and the ability to confer transcriptional regulation in response to GC [70]. The model posits that many ChIP-seq peaks represent chromatin-bound GR tethered to remotely bound TFs via looping, but this requires testing by chromatin confirmation capture techniques. It is also possible that monomeric binding by GR helps to explain the clustered arrangement of GBSs. Monomeric sites are enriched near dimeric sites in liver, yet they outnumber dimeric sites by 5:1 [58]. In comparison with GR dimers, monomers are sub-optimized for DNA binding [62,64] and transcriptional activation [71,76,77]. This may suggest that they are intermediates in the evolution of dimeric sites. However, with the discovery that genomic recognition sequences are sub-optimized for TF affinity to favor tissue specificity at the expense of activity [78], it is also possible that monomeric sites are important for the tissue-specific functions of GR. Consistent with this, chromatin-bound monomers colocalize more frequently with lineage dependent TFs than dimers [58,59], which may be a general property of steroid nuclear receptors [60,79].
Glucocorticoid-mediated repression and GR
About half of the genes affected by GC treatment are down regulated independent of the experimental system under study. This is important given that the immunosuppressive properties of GCs are mediated, at least in part, by transcriptional inhibition of pro-inflammatory genes in immune cells [80]. Unlike transactivation, a clear mechanism for GR transrepression has not emerged from genomic data. What appears clear, however, is that the two prominent models for repression, namely 1) tethering of GR monomers and 2) binding of GR to repressive DNA motifs termed negative glucocorticoid response elements, or nGREs [81,82], are not supported by an unbiased examination of GBSs. ChIP-seq in primary macrophages [83] and liver tissue [39] revealed similar enrichment of GR near both ligand-activated and ligand-repressed genes. Despite expectations, sequence analyses failed to find motifs distinguishing the putatively activating and repressing GBSs, and thus failed to implicate potential TFs mediating GR-dependent repression through tethering. Furthermore, the nGRE was not enriched in these studies nor any other published GR ChIP-seq dataset, challenging the idea that GR binds directly to this motif under physiological conditions. It is striking that STARR-seq failed to identify GBSs that repress reporter gene activity in response to exogenous GC [70]. A technical deficiency is unlikely given that STARR-seq found sequences conferring negative regulation by steroid hormone in flies [84]. In this case, the repression occurred independently of receptor binding to the regulatory sequence, suggesting an indirect cause. A potential concern of STARR-seq is that current iterations measure reporter activity from plasmids, and substantial differences exist between enhancer activity encoded on episomes versus chromosomes [85]. While it is formally possible that STARR-seq is unable to detect GR-mediated repression requiring a chromosomal context, this is unlikely for tethering, which was originally described using transiently transfected reporter plasmids.
It is increasingly argued that the immunosuppressive effects of GCs are conferred indirectly by GR through the activation of genes encoding proteins that inhibit expression of pro-inflammatory genes [86–88]. Genomic studies offer another mechanism compatible with the idea that GC-mediated repression is a secondary effect of GR function (Figure 2B). Treatment of mice with a GC drug caused liver gene expression changes that associated with a redistribution of GR and RNAPII from monomeric to dimeric GR-binding sites, with lost and gained sites enriched near repressed and induced genes, respectively [58]. This can be explained by expanding the classic squelching model to include the redistribution of TFs in addition to co-activators in response to external stimuli. Consistent with observations showing ligand-stimulated degradation of GR [89], the model proposes that both monomers and dimers are available in limiting amounts so that gain of occupancy at one set of sites leads to loss at another. Furthermore, by attributing GC-repressed transcription to the loss of activating GR monomers, the model maintains the logic of sequence-specific binding for GR genomic function. As squelching has been suggested to explain the transcriptional repression resulting from other NR ligands [90,91], its role in NR function needs further examination.
Concluding remarks and future perspectives
The development of ligands that provide immunosuppressive benefits without unwanted metabolic effects remains as an unmet goal of GR research. Traditionally, this has meant searching for ligands that repress pro-inflammatory genes in immune cells without activating catabolic genes in liver, fat, muscle and bone. While the genomic data do not support a path to such ligands, they raise questions that could increase GR’s therapeutic value if resolved (Outstanding Questions Box). For example, is GC-mediated repression of gene transcription dependent on squelching or an as yet to be determined molecular mechanism? Of particular interest to inflammation, does squelching occur in immune cells to affect expression of pro-inflammatory genes? Are stress-induced releases of cortisol into the bloodstream sufficient to redistribute monomers and dimers on the genome in a similar manner to what has been observed for GC drugs, or relatedly, do ultradian and circadian GC fluctuations alter GR interaction with the genome over the course of a day? Given that GC-mediated repression may result secondarily from GR action, can DNA loops map direct gene targets? Ultimately, a productive therapeutic approach may involve cell-selective delivery of GC drugs to minimize their complex tissue-specific effects.
OUTSTANDING QUESTIONS BOX.
Is GC-mediated repression of gene transcription dependent on squelching or through an undetermined molecular mechanism?
How do the ultradian, circadian and stress-induced releases of cortisol into the bloodstream modulate GR occupancy of the genome?
Does GR drive long-range chromatin interactions, and can DNA loops be used to distinguish primary versus secondary gene targets?
Does cortisol signaling involve interplay between GR and MR?
An overlooked area of inquiry from a genomics viewpoint is the potential interplay between GR and the mineralocorticoid receptor (MR). MR is unique among nuclear receptors because it can bind to multiple steroid hormones including aldosterone, cortisol and progesterone [92]. This is likely to have physiological consequences [93]. MR and GR bind with high affinities to cortisol, which circulates at concentrations that are 10–1000-fold higher than aldosterone, and HSD11B2 is absent in some tissues with high MR expression such as myocardium and hippocampus. MR can form heterodimers with GR [94,95], and with DNA-binding domains that share 94% similarity, both receptors target the same DNA motif. Unfortunately, genomic data for MR are limited by the lack of good ChIP-grade antibodies, yet how and where it binds the genome may provide new mechanisms of GC signaling that could be leveraged for therapeutic gain.
TRENDS BOX.
Glucocorticoids (GCs) are among the most widely prescribed drugs in the world. They target the glucocorticoid receptor (GR) to counteract harmful inflammation associated with autoimmune and allergic reactions and organ transplant rejection, yet their negative effects can halt treatment.
GR, a transcription factor present in nearly every human cell, drives programs of tissue-specific gene expression with widespread physiological impact including effects on energy homeostasis.
Emerging genomic data reveal that GR activates transcription through monomeric and dimeric interaction with specific DNA motifs. Transcriptional inhibition by GCs may be mediated by secondary mechanisms.
GR’s genomic function suggests that developing immunosuppressive-selective ligands without unwanted metabolic effects may require targeting delivery of GC drugs to specific cell types to minimize their complex tissue-specific effects.
Acknowledgments
We thank Hee-Woong Lim, Paul Titchenell, Matt Emmett and Mitch Lazar for insightful comments, and Mary Leonard for figure artwork. We apologize to researchers whose relevant studies were not discussed because of space limitations. Research on GR in the laboratory of D.J.S. is supported by NIH grant R01 DK098542.
GLOSSARY
- Addison’s disease
Also called adrenal insufficiency, it is a disorder that occurs when the body produces insufficient amounts of the adrenal gland hormone cortisol and possibly aldosterone. It is a rare condition, only 1 in 100,000 people has it. Affected individuals can lead a normal life with hormone replacement therapy. President John F. Kennedy had the disease
- ChIP-exo
An experimental technique that combines ChIP with lambda exonuclease digestion and sequencing. Exonuclease digestion of ChIP DNA before sequencing generates nuclease-protected regions, or footprints, for chromatin-bound proteins. ChIP-exo improves the spatial resolution of ChIP-seq approximately 5–10-fold, facilitating the determination of bound sequences and the identification DNA motifs
- ChIP-seq
An experimental technique that couples chromatin immunoprecipitation (ChIP) with deep sequencing (seq) to map the location of histone and nonhistone proteins across the genome in an unbiased manner. It can also be used to interrogate DNA modifications such as methylation. With a resolution of approximately 200 base pairs, it is best suited to determine where, rather than what, a transcription factor binds genome-wide
- Cistrome
The set of genomic loci occupied by a particular protein, e.g. GR, or associated with an epigenomic modification, e.g. histone 3 lysine 27 acetylation. It is further specified by cell type, tissue, species, physiological state, etc
- Cushing’s disease and syndrome
Both occur when the body is exposed to excessively high levels of glucocorticoid over a prolonged time period, leading to metabolic dysfunction including obesity, diabetes, osteoporosis and muscle wasting. They are sometimes distinguished by the source of glucocorticoid. Cushing’s disease results from the overproduction of endogenous cortisol most often caused by a tumor residing in the adrenal gland or a secondary ectopic site, usually in the brain, that drives excessive secretion of the hormones that stimulate cortisol production and release. Cushing’s syndrome arises from the chronic use of glucocorticoid medication, and is more common than the disease in western societies
- GR (NR3C1)
The molecular target of glucocorticoids and a type I nuclear receptor, GR is released from a cytoplasmic chaperone complex upon binding ligand and trans-located to the nucleus, where it regulates gene transcription through sequence-specific interaction with the genome. It modulates numerous gene programs in a cell-type-specific manner to widely impact development and physiology
- Nuclear receptor superfamily
With 48 human members, it is class of transcription factors that bind and respond to hormones (steroid and thyroid hormones), vitamins (A and D), metabolic intermediates (e.g. fatty acids, bile acids, sterols) and xenobiotics. Nuclear receptors have a modular design that is typified by an N-terminal AF-1 domain (weak transcriptional activation domain), DNA-binding domain, hinge region, ligand-binding domain (ligand-dependent transcriptional activation), and a variable C-terminal domain. Through sequence-specific DNA binding as momomers, homodimers and/or heterodimers, they regulate gene expression by recruiting additional transcriptional regulators such as co-activator and co-repressor complexes
- STARR-seq
An acronym for “self-transcribing active regulatory region sequencing,” it is a high-throughput experimental method for evaluating the ability of DNA fragments from any source, e.g. ChIP DNA, to enhance transcription
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long CN, Lukens FD. The effects of adrenalectomy and hypophysectomy upon experimental diabetes in the cat. J Exp Med. 1936;63:465–490. doi: 10.1084/jem.63.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fardet L, Fève B. Systemic glucocorticoid therapy: a review of its metabolic and cardiovascular adverse events. Drugs. 2014;74:1731–1745. doi: 10.1007/s40265-014-0282-9. [DOI] [PubMed] [Google Scholar]
- 5.Sapolsky RM, et al. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 6.Mueller KM, et al. Adipocyte Glucocorticoid Receptor Deficiency Attenuates Aging- and HFD-Induced Obesity and Impairs the Feeding-Fasting Transition. Diabetes. 2017;66:272–286. doi: 10.2337/db16-0381. [DOI] [PubMed] [Google Scholar]
- 7.Oberlin D, Buettner C. How does leptin restore euglycemia in insulin-deficient diabetes? J Clin Invest. 2017;127:450–453. doi: 10.1172/JCI91880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perry RJ, et al. Mechanism for leptin’s acute insulin-independent effect to reverse diabetic ketoacidosis. J Clin Invest. 2017;127:657–669. doi: 10.1172/JCI88477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang CL, et al. The novel strategy of glucocorticoid drug development via targeting nongenomic mechanisms. Steroids. 2015;102:27–31. doi: 10.1016/j.steroids.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 10.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yue F, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Napier C, Pearce SHS. Current and emerging therapies for Addison’s disease. Curr Opin Endocrinol Diabetes Obes. 2014;21:147–153. doi: 10.1097/MED.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 13.Lee MJ, et al. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. 2014;1842:473–481. doi: 10.1016/j.bbadis.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charmandari E, et al. Primary generalized familial and sporadic glucocorticoid resistance (Chrousos syndrome) and hypersensitivity. Endocr Dev. 2013;24:67–85. doi: 10.1159/000342505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito K, et al. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;117:522–543. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Kirwan J, Power L. Glucocorticoids: action and new therapeutic insights in rheumatoid arthritis. Curr Opin Rheumatol. 2007;19:233–237. doi: 10.1097/BOR.0b013e3280d6471a. [DOI] [PubMed] [Google Scholar]
- 17.Leung DYM, Bloom JW. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2003;111:3–22. doi: 10.1067/mai.2003.97. quiz 23. [DOI] [PubMed] [Google Scholar]
- 18.Rafacho A, et al. Functional alterations in endocrine pancreas of rats with different degrees of dexamethasone-induced insulin resistance. Pancreas. 2008;36:284–293. doi: 10.1097/MPA.0b013e31815ba826. [DOI] [PubMed] [Google Scholar]
- 19.Shpilberg Y, et al. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis Model Mech. 2012;5:671–680. doi: 10.1242/dmm.008912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park YK, Ge K. Glucocorticoid Receptor Accelerates, but Is Dispensable for, Adipogenesis. Mol Cell Biol. 2017;37 doi: 10.1128/MCB.00260-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong JC, et al. A glucocorticoid- and diet-responsive pathway toggles adipocyte precursor cell activity in vivo. Sci Signal. 2016;9:ra103. doi: 10.1126/scisignal.aag0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomlinson JW, et al. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, et al. Essential roles of 11β-HSD1 in regulating brown adipocyte function. J Mol Endocrinol. 2013;50:103–113. doi: 10.1530/JME-12-0099. [DOI] [PubMed] [Google Scholar]
- 24.Strack AM, et al. Corticosterone decreases nonshivering thermogenesis and increases lipid storage in brown adipose tissue. Am J Physiol. 1995;268:R183–191. doi: 10.1152/ajpregu.1995.268.1.R183. [DOI] [PubMed] [Google Scholar]
- 25.Noti M, et al. Extra-adrenal glucocorticoid synthesis in the intestinal epithelium: more than a drop in the ocean? Semin Immunopathol. 2009;31:237–248. doi: 10.1007/s00281-009-0159-2. [DOI] [PubMed] [Google Scholar]
- 26.Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem. 2011;286:3177–3184. doi: 10.1074/jbc.R110.179325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandevyver S, et al. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev. 2014;35:671–693. doi: 10.1210/er.2014-1010. [DOI] [PubMed] [Google Scholar]
- 28.Nagy L, Schwabe JWR. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–342. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 30.Chen W, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol Baltim Md. 2008;22:1754–1766. doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garza AMS, et al. Site-specific phosphorylation induces functionally active conformation in the intrinsically disordered N-terminal activation function (AF1) domain of the glucocorticoid receptor. Mol Cell Biol. 2010;30:220–230. doi: 10.1128/MCB.00552-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoh M, et al. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal phosphorylation. Mol Endocrinol Baltim Md. 2002;16:2382–2392. doi: 10.1210/me.2002-0144. [DOI] [PubMed] [Google Scholar]
- 33.Kino T, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol Baltim Md. 2007;21:1552–1568. doi: 10.1210/me.2006-0345. [DOI] [PubMed] [Google Scholar]
- 34.Blind RD, Garabedian MJ. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol. 2008;109:150–157. doi: 10.1016/j.jsbmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Presman DM, Hager GL. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor? Transcription. 2017;8:32–39. doi: 10.1080/21541264.2016.1249045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greulich F, et al. There goes the neighborhood: Assembly of transcriptional complexes during the regulation of metabolism and inflammation by the glucocorticoid receptor. Steroids. 2016;114:7–15. doi: 10.1016/j.steroids.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacta MA, et al. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu Rev Physiol. 2016;78:155–180. doi: 10.1146/annurev-physiol-021115-105323. [DOI] [PubMed] [Google Scholar]
- 38.Everett LJ, et al. Integrative genomic analysis of CREB defines a critical role for transcription factor networks in mediating the fed/fasted switch in liver. BMC Genomics. 2013;14:337. doi: 10.1186/1471-2164-14-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grøntved L, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013;32:1568–1583. doi: 10.1038/emboj.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy TE, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19:2163–2171. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siersbæk R, et al. Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J. 2011;30:1459–1472. doi: 10.1038/emboj.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.So AYL, et al. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007;3:e94. doi: 10.1371/journal.pgen.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steger DJ, et al. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010;24:1035–1044. doi: 10.1101/gad.1907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu CY, et al. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PloS One. 2010;5:e15188. doi: 10.1371/journal.pone.0015188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shlyueva D, et al. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 46.Sorrells TR, Johnson AD. Making sense of transcription networks. Cell. 2015;161:714–723. doi: 10.1016/j.cell.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaret KS, et al. Chromatin Scanning by Dynamic Binding of Pioneer Factors. Mol Cell. 2016;62:665–667. doi: 10.1016/j.molcel.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swinstead EE, et al. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell. 2016;165:593–605. doi: 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voss TC, et al. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell. 2011;146:544–554. doi: 10.1016/j.cell.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Workman JL, Kingston RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 51.Goldstein I, et al. Transcription factor assisted loading and enhancer dynamics dictate the hepatic fasting response. Genome Res. 2016 doi: 10.1101/gr.212175.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heinz S, et al. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madsen MS, et al. Peroxisome proliferator-activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading. Mol Cell Biol. 2014;34:939–954. doi: 10.1128/MCB.01344-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soccio RE, et al. Genetic Variation Determines PPARγ Function and Anti-diabetic Drug Response In Vivo. Cell. 2015;162:33–44. doi: 10.1016/j.cell.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu B, et al. Coactivator-Dependent Oscillation of Chromatin Accessibility Dictates Circadian Gene Amplitude via REV-ERB Loading. Mol Cell. 2015;60:769–783. doi: 10.1016/j.molcel.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Z, et al. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat Commun. 2015;6:8323. doi: 10.1038/ncomms9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim HW, et al. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. 2015;25:836–844. doi: 10.1101/gr.188581.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Starick SR, et al. ChIP-exo signal associated with DNA-binding motifs provides insight into the genomic binding of the glucocorticoid receptor and cooperating transcription factors. Genome Res. 2015;25:825–835. doi: 10.1101/gr.185157.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gertz J, et al. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell. 2013;52:25–36. doi: 10.1016/j.molcel.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langlais D, et al. The Stat3/GR Interaction Code: Predictive Value of Direct/Indirect DNA Recruitment for Transcription Outcome. Mol Cell. 2012;47:38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 62.Gebhardt JCM, et al. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods. 2013;10:421–426. doi: 10.1038/nmeth.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schiller BJ, et al. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. 2014;15:418. doi: 10.1186/s13059-014-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watson LC, et al. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. 2013;20:876–883. doi: 10.1038/nsmb.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen J, et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. 2014;156:1274–1285. doi: 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, et al. Discrete functions of nuclear receptor Rev-erbα couple metabolism to the clock. Science. 2015;348:1488–1492. doi: 10.1126/science.aab3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cohen DM, et al. ATF4 licenses C/EBPβ activity in human mesenchymal stem cells primed for adipogenesis. eLife. 2015;4:e06821. doi: 10.7554/eLife.06821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siersbæk R, et al. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014;7:1443–1455. doi: 10.1016/j.celrep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 69.Arnold CD, et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–1077. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 70.Vockley CM, et al. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell. 2016;166:1269–1281.e19. doi: 10.1016/j.cell.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schöne S, et al. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat Commun. 2016;7:12621. doi: 10.1038/ncomms12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomas-Chollier M, et al. A naturally occurring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proc Natl Acad Sci U S A. 2013;110:17826–17831. doi: 10.1073/pnas.1316235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams M, et al. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol Baltim Md. 2003;17:2583–2592. doi: 10.1210/me.2002-0305. [DOI] [PubMed] [Google Scholar]
- 77.Jewell CM, et al. Complex human glucocorticoid receptor dim mutations define glucocorticoid induced apoptotic resistance in bone cells. Mol Endocrinol Baltim Md. 2012;26:244–256. doi: 10.1210/me.2011-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farley EK, et al. Suboptimization of developmental enhancers. Science. 2015;350:325–328. doi: 10.1126/science.aac6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joseph R, et al. Integrative model of genomic factors for determining binding site selection by estrogen receptor-α. Mol Syst Biol. 2010;6:456. doi: 10.1038/msb.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab TEM. 2013;24:109–119. doi: 10.1016/j.tem.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hudson WH, et al. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. 2013;20:53–58. doi: 10.1038/nsmb.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Surjit M, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 83.Uhlenhaut NH, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49:158–171. doi: 10.1016/j.molcel.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shlyueva D, et al. Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol Cell. 2014;54:180–192. doi: 10.1016/j.molcel.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 85.Inoue F, et al. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017;27:38–52. doi: 10.1101/gr.212092.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ayroldi E, et al. Targeting glucocorticoid side effects: selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? A perspective FASEB J Off Publ Fed Am Soc Exp Biol. 2014;28:5055–5070. doi: 10.1096/fj.14-254755. [DOI] [PubMed] [Google Scholar]
- 87.Hübner S, et al. The glucocorticoid receptor in inflammatory processes: transrepression is not enough. Biol Chem. 2015;396:1223–1231. doi: 10.1515/hsz-2015-0106. [DOI] [PubMed] [Google Scholar]
- 88.Vandevyver S, et al. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013;154:993–1007. doi: 10.1210/en.2012-2045. [DOI] [PubMed] [Google Scholar]
- 89.Wallace AD, Cidlowski JA. Proteasome-mediated Glucocorticoid Receptor Degradation Restricts Transcriptional Signaling by Glucocorticoids. J Biol Chem. 2001;276:42714–42721. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- 90.Guertin MJ, et al. Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol Endocrinol Baltim Md. 2014;28:1522–1533. doi: 10.1210/me.2014-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Step SE, et al. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARγ-driven enhancers. Genes Dev. 2014;28:1018–1028. doi: 10.1101/gad.237628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gomez-Sanchez E, Gomez-Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol. 2014;4:965–994. doi: 10.1002/cphy.c130044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meijer OC, de Kloet ER. A Refill for the Brain Mineralocorticoid Receptor: The Benefit of Cortisol Add-On to Dexamethasone Therapy. Endocrinology. 2017;158:448–454. doi: 10.1210/en.2016-1495. [DOI] [PubMed] [Google Scholar]
- 94.Mifsud KR, Reul JMHM. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc Natl Acad Sci U S A. 2016;113:11336–11341. doi: 10.1073/pnas.1605246113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trapp T, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptors increases the functional diversity of corticosteroid action. Trends Pharmacol Sci. 1996;17:145–149. doi: 10.1016/0165-6147(96)81590-2. [DOI] [PubMed] [Google Scholar]