

Abstract
Over the past two decades orthogonal acceleration time-of-flight has been the de facto analyzer of choice for solution and membrane soluble protein native mass spectrometry (MS) studies; this however is gradually changing. Here we compare three MS instruments, the Q-ToF, the Orbitrap and the FT-ICR to analyze, under native instrument and buffer conditions, the 7-transmembrane helical protein bacteriorhodopsin-octylglucoside micelle complex and the empty nanodisc (MSP1D1-Nd) using both MS and tandem-MS modes of operation. Bacteriorhodopsin can be released from the octylglucoside-micelle efficiently on all three instruments (MS-mode of operation) producing a narrow charge state distribution (z = 8+ to 10+) by either increasing the source lens or collision cell (or HCD) voltages. A lower center-of-mass collision energy (0.20–0.41 eV) is required for optimal bacteriorhodopsin liberation on the FT-ICR, in comparison to the Q-ToF and Orbitrap instruments (0.29–2.47 eV). The empty MSP1D1-Nd can be measured with relative ease on a three instruments, resulting in a highly complex spectrum of overlapping, polydisperse charge state; a consequence of varying levels of phospholipid incorporation. There is a measurable difference in MSP1D1-Nd charge state distribution (z = 15+ to 26+), average molecular weight (141.7 to 169.6 kDa) and phospholipid incorporation number (143 to 184) under low activation conditions. Utilizing tandem-MS, bacteriorhodopsin can be effectively liberated from the octylglucoside-micelle by collisional (Q-ToF and FT-ICR) or continuous IRMPD activation (FT-ICR). MSP1D1-Nd spectral complexity can also be significantly reduced by tandem-MS (Q-ToF and FT-ICR) followed by mild collisional or continuous IRMPD activation, resulting in a spectrum in which the charge state and phospholipid incorporation levels can easily be determined.
Graphical abstract
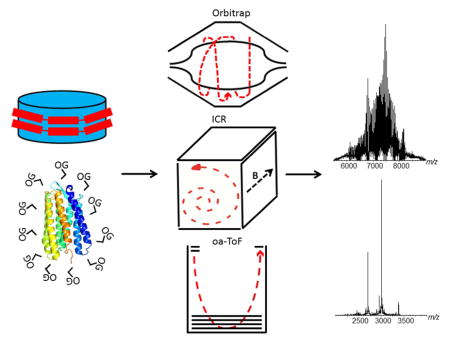
Introduction
Membrane proteins such as G-protein coupled receptors (GPCRs or seven–transmembrane domain receptors) 1 and ion channels 2 constitute approximately 50% of current therapeutic targets 3,4, making them an incredibly important class of molecule for structural characterization. Native mass spectrometry (MS) 5 can contribute to any structural biology effort since molecular weight (Mw), stoichiometry and purity information can be obtained rapidly with low sample consumption (typically sub-μg of material consumed). Over the past seven years tremendous progress has been made analyzing and characterizing membrane proteins and associated complexes within a detergent micelle 6 under native-MS conditions, since the original surfactant experiments performed by Loo 7 in 1994. Typically the membrane protein is encapsulated in a detergent micelle (above the critical micelle concentration; cmc 8) followed by protein liberation from the micelle by varying degrees of gas-phase activation 6a, b, 8b. To a lesser extent, amphipols 6c, 9 have also been applied as an effective medium to solubilize membrane proteins prior to native MS-analysis.
Nanodisc (Nd) technology is a versatile detergent-free medium to solubilize, handle and manipulate a membrane protein or complex, 10. Depending on the size of the membrane protein being studied, the Nd can be easily adapted by changing the size of the membrane scaffold protein (MSP) 10b. Engineering a biotin tag onto the MSP, the Nd (typically consisting of two MSPs and a membrane protein inserted into a phospholipid bilayer) enables immobilization on to a streptavidin coated surface plasmon resonance (SPR) chip and dose dependence screening experiments can be performed 11. From a pharmaceutical discovery point-of-view, this enables levels of non-MS based screening to be performed, which the micelle method does not currently offer. However, from a native-MS perspective, the Nd, especially the full Nd (membrane protein loaded) represents a significant challenge due to their inherent high levels of heterogeneity (vide infra).
The concept of native protein MS-measurement is over two decades old 12,13 and significant progress has been made on both the spectral quality and instrument ease of use. Since the initial reported native-MS experiments 13, the quadrupole time-of-flight (Q-ToF) 14, typically heavily modified 15, has naturally evolved into the de facto instrument of choice. However, over the past five years the Fourier Transform Ion Cyclotron Resonance (FT-ICR) instrument 16 and the Orbitrap 17 in the form of the extended mass range (EMR) 18,19 instrument, have proven to be more than capable of producing high quality data on soluble proteins and complexes, under native buffer and MS-conditions. The majority of native-MS experiments, regardless of the instrument platform, have been performed in the MS-only mode of operation; rarely has tandem-MS (MS/MS) been performed. Quadrupole ion selection and subsequent ion activation is a highly efficient orthogonal analytical MS technique enabling significant spectral simplification 20 and stoichiometric determination 21,19 of aqueous soluble protein complexes.
Herein, we have intentionally chosen three MS-instruments with very different ion optic geometries and detectors; the axially and temporally focusing Q-ToF; axially harmonic Orbitrap and the radially harmonic FT-ICR and assessed their abilities to analyze the 7-transmembrane helical membrane protein bacteriorhodopsin solubilized in the non-ionic detergent octylglucoside (OG) 22 and the empty MSP1D1-Nd particle. To date the Orbitrap and FT-ICR instruments have not been extensively used for native membrane protein analysis, although their usage is steadily increasing 23,24,25,26. The intention of this manuscript is to compare and contrast each instrument platform and highlight the benefits of each for native membrane protein and Nd analysis, by specifically focusing on the spectral quality produced in both MS and tandem-MS modes of operation.
Experimental
Materials
Purple membrane bacteriorhodopsin was purchased from Sigma-Aldrich (St. Louis, MO, USA; B-0184). Ammonium acetate purchased from Sigma-Aldrich (St. Louis, MO; A2706). Sinapinic acid was purchased from (St. Louis, MO; 85429). The detergent n-octyl-β-D-glucopyranoside was purchased from Affymetrics (Santa Clara, CA, USA; O311). Gold coated nanoESI needles (M956232AD1, Long Thin Wall; Waters MS-Technologies Centre, Manchester UK). Sulphur hexafluoride (SF6) was purchased from Airgas (Palmdale, CA). BioRad P6 spin columns were purchased from BioRad (732-6221, Hercule, CA).
Sample Purification
A modified version of the purple membrane bacteriorhodopsin purification procedure 22 was followed and is described in detail in the Supporting Information (Figures S1, S2, S3 & S4). The bacteriorhodopsin mutant M60A was provided by Nicholas Woodall (UCLA) and it’s preparation for native-MS is described in the Supporting Information. The empty MSP1D1-Nd preparation and purification have been described elsewhere 10b, 11, 27 and will not be discussed in detail here.
Mass Spectrometry Instrumentation
The Synapt G2 quadrupole-ion mobility (Waters MS-Technologies, Manchester, UK), Orbitrap Exactive-Plus EMR (ThermoScientific, Bremen, Germany) and a Solarix 15T Fourier Transform Ion Cyclotron Resonance (Bruker, Bremen, Germany) MS instruments were used to generate the native-MS and tandem-MS data reported herein. The instruments will be referred to throughout the proceeding text as Q-ToF, Orbitrap and FT-ICR. For general instrumental parameters and optimization please refer to the Supporting Information. Specific acquisition voltages and parameters are explicitly stated in either the discussion section or the figure legends. The bacteriorhodopsin and MSP1D1-Nd solutions, at concentrations of 5–10 uM, in 200 mM ammonium acetate (containing 1.1% w/v OG equating to 2 x cmc, for bacteriorhodopsin only) were loaded into gold-coated borosilicate capillaries and analyzed in positive nESI mode (10–40 nL/min; 0.9–1.0 kV).
Results and Discussion
Native-MS analysis of bacteriorhodospin and MSP1D1-Nd
Figure 1 shows the native MS data acquired for bacteriorhodopsin from the Q-ToF (Figure 1a); the Orbitrap (Figure 1b) and the FT-ICR (Figure 1c) instruments, with activation energies used to generate optimal spectral quality. There are high degrees of spectral consistency for bacteriorhodopsin liberation on all instruments. Bacteriorhodopsin (B-0184) is efficiently released from the OG-micelle in all cases, resulting in two major charge states (z = 10+, m/z 2706 & z = 9+, m/z 3006) corresponding to a monomeric Mw of 27 kDa which retains the Schiff-base bound retinal (+284 Da). Additional low intensity charge states (z = 11+, m/z 2460; z = 8+, m/z 3382; z = 7+, m/z 3866) are instrument specific, and a function of the activation energies used, are also observed. For example, the bacteriorhodopsin charge state distributions are similar on both the Orbitrap (Figure 1b) and the FT-ICR (Figure 1c). Under the native-MS conditions used for optimal bacteriorhodopsin liberation, there is a subtle but clear shift to higher m/z in charge state distribution from the Orbitrap and FT-ICR to the Q-ToF. In the spectra displayed in Figures 1a, b and c, the average charge for bacteriorhodopsin on the Orbitrap and FT-ICR instrument are z = 9.77+ and 9.70+ respectively, compared to the z = 9.44+ for the Q-ToF. The bacteriorhodopsin charge state distribution has shifted to a higher m/z value on the Q-ToF instrument due to the increased relative intensity of the z = 9+ and 8+ charge states and the appearance of the z = 7+ charge state (m/z 3866) suggesting that under these applied voltages (required to obtain equivalent spectral quality) charge stripping is occurring as a function of increased ion-neutral collisions within the TWIG trap cell. It can be assumed that charge stripping does not occur in the MS-source region, since the voltage potential (sample cone) was set intentionally low (25 V). Collision activation of the bacteriorhodopsin-OG micelle complex was performed in different regions of the instruments using different collision gases (SF6, N2 and Ar). It is therefore prudent to use Ecom (center-of-mass collision energy in eV 28) rather than Elab (laboratory frame collision energy), since Ecom takes into account the mass of the neutral (Mgas; SF6 146 Da, N2, 28 Da; Ar, 44 Da).
Figure 1.

Representative native-MS spectra acquired under optimized instrument conditions for bacteriorhodopsin (B-0184) on the: a) Q-ToF (source block temperature 30°C, sample cone 25 V, TWIG trap 100 V); b) Orbitrap (capillary temperature 200°C, source induced dissociation 20 V, HCD 100 V); c) FT-ICR (capillary temp 100°C, skimmer 25 V, collision cell 40 V). The effect of bacteriorhodopsin (B-0184) charge-state liberation (z = 9+) by source and collisional activation, as a function of source temperature on the: d) Q-ToF; e) Orbitrap; f) FT-ICR. The respective collision (or HCD) cell gases are: Q-ToF, SF6; Orbitrap, N2 and FT-ICR, Ar. SID refers to source induced dissociation; CID refers to collision induced dissociation. n=1.
| (1) |
Calculating Ecom, a normalized collision energy can be obtained for all instruments, under the assumption that the ion-neutral collisions are single events as a function of their mean-free-path (approximately 1.55e−6 m at 6 mbar, source; 0.5e−3 m at 2e−2 mbar, collision cell, based on typical Q-ToF operating pressures). Mprojectile ion is the Mw of the ion (bacteriorhodopsin in an average OG-micelle; 53 kDa).
On all three instruments, collisional activation was performed in either the ion source or the collision (or HCD) cell. Since all instruments use N2 as either the nESI gas, skimmer/cone gas or capillary transfer gas, it can be assumed that all ion collisions within the instrument sources are predominantly with N2. The collision gases used within the TWIG trap, HCD or hexapole collision cell, of the Q-ToF, Orbitrap and FT-ICR instruments are SF6, N2 and Ar, respectively. Bacteriorhodopsin release from the OG-micelle was investigated as a function of source temperature on all instruments (30–125°C, Q-ToF; 62–200°C Orbitrap; 30–125°C FT-ICR) as it was hypothesized that less activation energy is required to release the membrane protein from the detergent micelle at higher nESI source temperatures. For all instrument platforms, a source operated at elevated temperature results in dramatically increased levels of bacteriorhodopsin liberation as a function of source sample cone (Q-ToF; Figure 1d) source induced dissociation (SID; Orbitrap; Figure 1e) and source skimmer (FT-ICR; Figure 1f) activation. These observations can be easily rationalized; the increased source temperature imparts thermal and therefore internal energy into the membrane protein-micelle complex, therefore less activation energy (cone, skimmer or SID) is required for efficient detergent micelle decomposition and subsequent protein liberation. Optimal bacteriorhodopsin liberation is achieved at Ecom values of 0.95 eV (Q-ToF, 200 V, N2), 0.59 eV (Orbitrap, 125 V, N2) and 0.95 eV (FT-ICR, 200 V, N2).
This trend is also observed for bacteriorhodopsin liberation as a function of source temperature and HCD or collision cell activation on the Orbitrap and FT-ICR platforms (Figures 1e & 1f, respectively). At the source transfer temperatures of 200°C and 125°C for the Orbitrap (HCD, approximately 5e−2 mbar, N2) and FT-ICR (collision cell 4e−6 mbar, Ar) instruments respectively, optimal bacteriorhodopsin liberation is achieved at Ecom values ranging from 0.29 to 0.71 eV (60 to 150 V, N2; Figure 1e) and 0.20 to 0.41 eV (30 to 60 V, Ar; Figure 1f). At high activation voltages (Orbitrap, SID, 200 V; HCD 150 V) retinal release (−284 Da) was observed (data not shown). The Q-ToF however does not follow this trend (Figure 1d). More efficient liberation is observed at lower source temperatures (30°C and 60°C) for any given TWIG trap (2e−2 mbar, SF6) collision energy, than at elevated source temperatures (125°C). Optimal bacteriorhodopsin liberation on the Q-ToF is achieved at Ecom values ranging from 0.49 to 2.47 eV (20 to 100 V, SF6; Figure 1d).
There is a fundamental difference between the Q-ToF source geometry to that of the Orbitrap and FT-ICR platform. First, the Q-ToF source samples the nESI generated ions in an orthogonal manner (Z-Spray) as opposed to a more axial and direct sampling geometry implemented on the Orbitrap and FT-ICR. Secondly, the Q-ToF does not use a heated ion transfer capillary, whereas the Orbitrap (4.25 cm steel) and the FT-ICR (20 cm glass) instruments do. It is hypothesized herein that directing nESI-generated ions into heated transfer capillary and their subsequent transfer into the partial vacuum region of the nESI source is beneficial for membrane protein liberation, as opposed to spraying (orthogonally) into a partial vacuum region (through a source cone) held at 6 mbar 15, 29 at elevated temperatures. It should also be noted that the recorded source pressures on the Orbitrap and FT-ICR (both approximately 2 mbar) instruments are lower than that of the Q-ToF (6 mbar). Landreh 30 recently demonstrated the effects of source backing pressure (Q-ToF only) on membrane protein liberation from a detergent micelle. Lippens et al 31 have also demonstrated this temperature effect for nucleic acids. It therefore appears that heating the source block within the Q-ToF source may be detrimental to protein liberation from the bacteriorhodopsin-OG micelle. Investigating the effect of source temperature on the gas-phase conformation of bacteriorhodopsin was not performed on the Q-ToF, since Lippens et al 31 have also recently demonstrated that a heated 20 cm transfer capillary modification to a Synapt G2 was not detrimental to the gas-phase structure of a duplex nucleic acid molecule.
The empty MSP1D1-Nd can be analyzed with relative ease on all three instruments (Figure 2a, b & c) when acquired under native buffer conditions (200 mM ammonium acetate) resulting in a highly complex and polydisperse distribution of charge states, representing different numbers of DMPC phospholipid incorporations into the MSP1D1-Nd particle 23b, 32. Under the specific instrument temperature, pressure and voltage conditions used to acquire the spectra presented in Figures 2a, b & c, the level of spectral fidelity and complexity are comparable. The multiply charged distribution of the MSP1D1-Nd ions all appear within a broadly similar m/z range (6,000–9,000; Figures 2a, b & c and Figure S5, Supporting Information). One striking difference is the poor signal-to-noise (S/N) of the data acquired on the Q-ToF instrument (Figure 2a). Both spectra acquired on the Orbitrap and FT-ICR instruments are close to, or fully baseline resolved, suggesting superior levels of ion desolvation. The application of a modest polynomial background subtraction (polynomial order 25, 2% below curve) can dramatically reduce the raised baseline present in the Q-ToF spectrum (Figure S6, Supporting Information) resulting in a spectrum remarkably comparable to that acquired on the Orbitrap and FT-ICR instruments (Figures 2b & c).
Figure 2.

Representative native-MS spectra acquired under optimized instrument conditions for the empty MSP1D1-Nd acquired on the on the: a) Q-ToF (source temperature 30°C, sample cone 150V, TWIG trap 10V); b) Orbitrap (capillary temperature 200°C, source induced dissociation 80V, HCD 20V); c) FT-ICR (capillary temperature 100°C, skimmer 100V, collision cell 4V). The insets are heat-maps displaying the zero-charge deconvoluted molecular weight along the x-axis and the observable charge states which are present in the unprocessed data, on the y-axis. All spectral data are unprocessed apart from spectrum a, where a minimal smooth was applied (10×2 Savitsky-Golay, MassLynx 4.1). The empty MSP1D1-Nd average molecular weight (Mw in kDa) and average phospholipid (DMPC) count are noted.
The Q-ToF was operated with SF6 as the TWIG collision gas, and the Orbitrap and FT-ICR used N2 and Ar in the HCD and collision cell, respectively. It has been demonstrated extensively that larger, more massive collision gases (mono and polyatomic) result in superior ion desolvation and collisional cooling/focusing effects for large multi-protein complexes 18, 21a, c, 33, so the effect observed here could be interpreted as more efficient ion desolvation within the source regions of the Orbitrap and FT-ICR instruments, rather than in the instrument collision (or HCD) cells. Additionally, both the Orbitrap and FT-ICR instruments use on-axis nESI infusion, whereas the Q-ToF utilizes dual orthogonal nESI sampling (Z-Spray; vide supra). Again, the Orbitrap and FT-ICR possess heated transfer capillaries which transport the ions from atmosphere to partial vacuum. A heated transfer capillary modification to a Q-ToF platform 31 may prove beneficial for native Nd analysis. Conversely, the operating pressures and therefore the mean-free-path at which the MSP1D1-Nd (and bacteriorhodopsin) activation and subsequent desolvation (and protein liberation, in the case of bacteriorhodopsin) occurs are also very different and must be considered. The CID-activation on the Q-ToF, Orbitrap and FT-ICR occur at approximately 2e−2 mbar, SF6, 5e−2 mbar N2 and 4e−6 mbar Ar, respectively and the approximate mean-free-paths for the empty MSP1D1-Nd are 0.5e−5 m, 2e−6 m and 2e−2 m, in the Q-ToF, Orbitrap and FT-ICR collision cells, respectively. Depending on the Nd preparation, additional unresolved species are observed in the m/z range 8000–12,000 (Figures S9, S11 & S12, Supporting Information) which we attribute to MSP1D1-Nd dimers, consistent with the measurements made by atomic force microscopy (Figure S7, Supporting Information). Multimeric forms of a protein complex under native-MS conditions have previously been characterized 34.
For MSP1D1-Nd charge state assignment, Mw and phospholipid count determination, algorithmic deconvolution is essential. Efficient deconvolution of the empty MSP1D1-Nd spectral data can be achieved using the Bayesian-based deconvolution algorithm Maximum Entropy 35 or UniDec 23b, 32 (Supporting Information S8). The MSP1D1-Nd data acquired on the FT-ICR (Figure 2c) reflects the lowest average Mw species at 132.4 kDa and a mean phospholipid value of 129 DMPC molecules. The Orbitrap (Figure 2b) and Q-ToF (Figure 2a) show average Mw and mean phospholipid values of 141.8 kDa, 143 DMPC and 168.3 kDa and 182 DMPC, respectively. The phospholipid number and therefore Mw are highly source activation voltage specific (Figures S9, S11 & S12, Supporting Information). Previously reported mean DMPC numbers of this MSP1D1-Nd scaffold varies from 155 23a to 165 23b. The values calculated in Figures 2a–c are both higher and lower than previous reported values, but depending on instrument acquisition parameters, the mean phopholipid numbers can be highly consistent. For example, when the Q-ToF is operated with a sample cone of 200 V and a TWIG trap of 40 V and the Orbitrap is operated with a source induced dissociation value of 50 V and a HCD of 20 V, a mean lipid count of 164 and 153 respectively, are accurately derived (Figures S10 & S11, Supporting Information). These observed differences are real, reproducible and most likely reflect differences in instrument geometries, ion optics, vacuum regimes, collision gases and related ion activation gradients within the three instruments. The MSP1D1-Nd sample and batch were identical in all cases, therefore sample variability is minimized.
The charge state distributions of the MSP1D1-Nd sample (insets Figures 2a, b & c and Supporting Information Figure S5) also accurately reflect the differences in the measured Mw and mean lipid numbers due to charge stripping (DMPC loss). The FT-ICR data displays the lowest charge state values, z = 16+ to 22+ (highly skimmer voltage dependent; Figure 2c & Figures S5 & S12 Supporting Information) as opposed to the higher values observed on the Q-ToF instrument, z = 22+ to 26+ (Figure 2a, Figure S5 & S9). On all instruments, the MSP1D1-Nd charge state distribution can be forced to higher m/z. This has previously been reported for the FT-ICR instrument 23b, however, not for the Q-ToF or Orbitrap instruments. For each voltage-step increase on the trap TWIG (or HCD) cell, a significant amount of charge stripping occurs, with the concomitant reduction in overall MSP1D1-Nd Mw (Supporting Information Figure S9, S10 & S11). The mean phospholipid count (DMPC) also reduces, as does the observed charge states. Applying a TWIG trap activation voltage of 75 to 100 V (Figure S10, Supporting Information) the extent of charge stripping is such that discrete charged states (z = 9+, 10+, 11+, 12+ & 13+) can also be manually determined by the mass difference between adjacent DMPC adducts. Therefore, as a function of activation energy, it appears that the reduction in Mw and charge states are predominantly driven by DMPC ejection.
Tandem-MS and mild collision induced activation of bacteriorhodopsin-OG micelle and MSP1D1-Nd
The Q-ToF and FT-ICR instruments both possess a quadrupole ion guide, therefore precursor ion selection and subsequent ion activation can easily be achieved. This has previously been demonstrated whereby the complexity of an aqueous soluble polydisperse protein, αβ-crystallin complex, was significantly reduced 20. Protein complex stoichiometry have also been determined 36, as have protein stability experiments 37 and general gas-phase protein unfolding experiments 38 been performed. Figure 3a shows Q-ToF tandem-MS of m/z 3,500 (50 Da transmission window) of the bacteriorhodopsin-OG micelle (M60A mutant) and subsequent CID activation (sample cone 25 V, TWIG trap 70 V, Ecom 1.73 eV). Protein liberation is easily achieved, producing a spectrum displaying excellent signal-to-noise (S/N, 200/1). The S/N in a representative spectrum produced by TWIG trap activation with no tandem-MS selection (Figure 1a; bacteriorhodopsin, B-0184) is in the order of 50 to 60/1. As expected, the low m/z range (500–2000) is far less congested with detergent related ions in the tandem-MS generated spectrum (Figure S13, Supporting Information). Tandem-MS and subsequent CID activation was also performed at m/z values of 4000, 4500, 5000 and 5500 and bacteriorhodopsin (B-0184) liberation was still achieved (Q-ToF, Figure S14; FT-ICR Figure S15, Supporting Information). Interestingly, when higher m/z regions (4500–5500) are quadrupole selected and activated on the Q-ToF, bacteriorhodopsin dimer (54.1 kDa) is observed (Figure S14, Supporting Information). This would suggest that the bacteriorhodopsin purification protocol described by Nollert 22 does not fully monomerize bacteriorhodopsin, as described, or the dimerization observed is purely a nESI phenomenon. Interestingly, a dimeric species is not observed on the FT-ICR instrument (Figure S14, Supporting Information).
Figure 3.

Q-ToF tandem-MS (m/z 3500) of a) bacteriorhodopsin M60A-OG micelle followed by collision cell activation (sample cone 25 V, TWIG trap SF6 70 V); b) FT-ICR quadrupole tandem-MS selection (m/z 3500; skimmer 25 V) of bacteriorhodopsin (B-0184) followed by continuous IRMPD activation for 1 s at 70% power; c) continuous IRMPD activation of the entire m/z range (m/z 1000–6000; skimmer 25 V) for 0.5 s at 75% power; d) Q-ToF quadrupole tandem-MS selection (m/z 6500; sample cone 50 V) and collision cell activation (TWIG trap SF6 40 V) and e) FT-ICR quadrupole tandem-MS selection (m/z 6000; skimmer 50 V) and continuous IRMPD activation (0.2 s, 50% power) of the empty MSP1D1-Nd. Individual charge states are annotated on all spectra. Insets (d & e) display the “zoomed-in” regions of the MSP1D1-Nd z = 18+ charge state (Q-ToF) and z = 23+ charge state (FT-ICR) with annotated fine structure corresponding to the intact molecular weight (kDa) and associated phospholipid (DMPC) number.
Reducing the evident spectral complexity of the Q-ToF acquired MSP1D1-Nd data was performed by tandem-MS of region m/z 6500, followed by mild CID activation (Figure 3d, TWIG trap 40 V; inset shows the fine structure of the z = 18+). At first glance the spectrum is less complex and distinct charge states are present, however, automated devonvolution using UniDec 32 or MaxEnt 35 were not successful; surprisingly a useable zero-charge spectrum cannot be obtained algorithmically. Manual peak-selection can be performed to produce a number of candidate zero-charge molecular weight values (Figure 3d), all of which are in close agreement to the determined Mw and mean phospholipid number presented in Figure 2a and Figures S9 & S10. Six distinct species can be identified manually (Figure 3d inset; 145.6 kDa, 146.2 kDa, 146.9 kDa, 147.6 kDa, 148.3 kDa and 148.9 kDa, with charge states ranging from z = 17+ to 21+ and phospholipid counts 149, 159, 151, 152, 153 and 154) all differing in Mw by approximately one DMPC molecule (678 Da). The charge states of the major species within Figure 3d are determined by measuring the mass difference between each adjacent DMPC adduct (Figure 3d inset). Figure S16 (Supporting Information) shows the empty MSP1D1-Nd spectra produced upon Q-ToF tandem-MS and activation of m/z 7500. The assigned Mws and phospholipid counts are even higher at 156.0 kDa, 157.6 kDa and 157.4 kDa; 164, 165 and 167, respectively, and observed charge states are expectedly lower. These observations would suggest that quadrupole isolation can select and transmit a very narrow ensemble of Nd-structures; high charge states (lower Mw values; Figure S3d) are present in the lower m/z spectral regions, and conversely lower charge states (higher Mw values; Figure S16 Supporting Information) are present in the higher m/z spectral regions. Tandem-MS selection experiments were also performed on the FT-ICR instrument (Figures S17a & b, Supporting Information) and interestingly, less activation (CID) voltage (0–10 V) is required to generate a charge state resolved MSP1D1-Nd spectrum. The manually derived charge states (z = 17+ to 21+), average Mw (133.7 kDa) and phospholipid incorporation number (131) are highly consistent with those described in Figures 2c, S5 & S12 (Supporting Information).
Laser liberation of bacteriorhodopsin from the OG-micelle
Another mechanism for ion activation is infrared multi photon dissociation (IRMPD) 39. There is a growing body of research where IRMPD (continuous irradiation) has been used in combination with electron capture dissociation (ECD) to aid top-down characterization of integral membrane proteins 40. However, very little research has been performed using IRMPD activation of native proteins 41 let alone membrane proteins encapsulated within detergent micelles 41,42. Figure 3b shows the FT-ICR tandem-MS of m/z 3500 (50 Da transmission window) and subsequent continuous IRMPD irradiation (1.0 s, 70% power; Synrad 30-W CO2 laser). Efficient bacteriorhodopsin (B-0184) ejection from the OG-micelle is achieved. Figure 3c shows continuous IRMPD irradiation (0.5 s, 75% power; Synrad 30-W CO2 laser) of the entire m/z range (no quadrupole selection) and again, efficient bacteriorhodopsin ejection is observed. There is a factor of eight improvement in S/N for full spectral irradiation (S/N 124) compared to that of quadrupole selection (S/N 16). However, in both cases the charge state distribution is highly consistent with those produced by TWIG trap, HCD or hexapole activation (Figures 1a, 1b & 1c) and also quadrupole selection followed by collisional activation (Figure 3a and Supporting Information S14 & S15). If one considers continuous IRMPD irradiation of trapped ions as a slow (0.5 s) heating process 42 of the detergent encapsulated membrane protein, which produces a charge state distribution highly comparable to that of CID activation (another ion heating process but over a much shorter time frame 28b, ms; Figures 1a, b & c), it can be inferred that both processes efficiently remove the OG-detergent, thus liberating bacteriorhodopsin, whose gas-phase conformation is comparable. This would obviously require confirmation by an orthogonal technique such as ion mobility. Additionally, upon laser activation, there is no evidence of retinal loss (−284 Da) from bacteriorhodopsin.
Tandem-MS and laser activation of the empty MSP1D1-Nd
Continuous IRMPD irradiation of the entire m/z range of an empty MSP1D1-Nd has previously been demonstrated 23b and was shown to produce significant levels of charge reduction. Herein we have demonstrated tandem-MS prior to IRMPD activation results in a vastly simplified empty MSP1D1-Nd spectrum (Figure 3e and Figure S17c, d & e, Supporting Information), comparable to that produced by Q-ToF quadrupole selection and subsequent CID activation (Figure 3d and Figure S16, Supporting Information). The FT-ICR upper quadrupole selection limit is lower than that of the Q-ToF, therefore only m/z 6000 could be selected, transmitted and subsequently irradiated. The observed charge states, upon laser irradiation are higher (z = 20+ to 24+; Figure 3e) than those observed by Q-ToF CID activation (z = 17+ to 21+; Figure 3d), which is clearly a result of a lower FT-ICR quadrupole selection range, but highly consistent with the algorithmically derived charge states displayed in Figure S12 (equivalent skimmer values, 50 V). One striking difference between the CID and IRMPD laser activated spectra, are the levels of DMPC polydispersity. Based on the spectra presented herein, the resolved charge states produced by mild CID activation possess up to six major species (Figure 3d inset) as opposed to two major and two minor species produced by IRMPD activation (Figure 3e inset). It must be noted that in both IRMPD laser activation examples (bacteriorhodopsin and MSP1D1-Nd) very low source skimmer voltages are implemented, 25 V and 50 V respectively, suggesting that bacteriorhodopsin liberation and MSP1D1-Nd spectral simplification are purely a result of laser activation and not source induced activation. Based on the MSP1D1-Nd data (Figure 3e inset) it could also be inferred that IRMPD activation has the ability to gently remove higher levels of non-specific DMPC associations when compared to CID activation (Figure 3d inset). These results are in alignment with those recently reported by Mikhailov 41–42, suggesting that IRMPD activation is a very efficient mechanism of liberating bacteriorhodopsin and equally importantly, spectral simplification of the empty MSP1D1-Nd data.
Conclusion
Utilizing three very different mass spectrometer platforms, we have compared and contrasted their performances and the spectral fidelity acquired for bacteriorhodopsin ejected from an OG-micelle and the empty MSP1D1-Nd particle, in both MS and tandem-MS modes of operation. The observed charge state distribution of (z = 9+ to 11+) for bacteriorhodopsin is highly consistent on all three instruments. Lower activation energies (based on Ecom) are required to eject bacteriorhodopsin from the OG-micelle on both the Orbitrap (0.29–0.71 eV) and FT-ICR (0.20–0.41 eV) instruments, compared to that of the Q-ToF (0.49–2.47 eV). Consequently, charge stripping and therefore an additional charge state is observed (z = 7+) on the Q-ToF. Based on the data presented herein, it can be concluded that bacteriorhodopsin is most easily liberated from the OG-micelle on the FT-ICR instrument. This has significant implications, since it further emphasizes that the FT-ICR instrument as a bona fide MS platform for native-MS gas-phase structural biology experiments. The empty MSP1D1-Nd displays a measurable range of phospholipid incorporation on the three instruments, ranging from 143 to 184 DMPC molecules, under low activation conditions, indicating significant differences in the levels of instrument activation and resultant desolvation and Mw values. Some care and judgment therefore must be applied when the Mw and the phospholipid counts of the Nd species are determined. Both the Q-ToF and FT-ICR instruments possess a quadrupole filter, allowing for ion selection and subsequent activation. It was successfully demonstrated that bacteriorhodopsin could be liberated from the OG-micelle, by first quadruple selection of a 50 Da mass window, followed by ion activation, by either CID or continuous IRMPD irradiation. Continuous IRMPD activation (FT-ICR) was also performed over the entire m/z range and successful bacteriorhodopsin liberation was achieved where the S/N was demonstrated to be comparable to that of quadrupole ion selection followed by continuous IRMPD activation. In all cases, retinal remained Schiff-base bound to bacteriorhodopsin. Quadrupole selection followed by ion activation (CID and IRMPD) also resulted in significantly reduced spectral complexity of the MSP1D1-Nd sample, allowing manual charge state, Mw and phospholipid assignment to be achieved; this is not the case for the MS-only MSP1D1-Nd spectra where algorithmic deconvolution is required. It is clearly apparent that tandem-MS is a very effective data acquisition mode affording dramatic levels of spectral simplification.
From the presented data, it is clear that multiple instrument platforms lend themselves particularly well to native membrane protein and Nd analysis. Traditionally, the Q-ToF platform was the de-facto instrument of choice for native protein MS analysis and detection, due to its ease of use; ability to readily modify instrument pressures 15, 43 and the extended m/z range of the oa-ToF analyzer 15. Recently however, published data on the Nd and membrane proteins would suggest that the FT-ICR 23–25 and Orbitrap 25b, 26 instruments have clear potential, and arguably, distinct advantages, in terms of levels of native protein desolvation (for both aqueous and membrane soluble); reduced background noise and increased S/N levels. It is intriguing to speculate as to whether the heated transfer capillary in both the Orbitrap and FT-ICR sources aid in the ion desolvation process, or whether the higher analyzer vacuum levels (10e−9 to 10e−10 mbar values in the FT-ICR and Orbitrap respectively) and ion residence times (0.2 s and above in the ICR depending on the transient time) contribute to the apparent spectral quality of the highly polydisperse MSP1D1-Nd sample. This can certainly be tested on the FT-ICR platform, since it allows for exquisite levels of ion control and manipulation. We predict that the FT-ICR platform will become a more routine instrument for both aqueous and membrane soluble protein native-MS analyses.
Supplementary Material
Acknowledgments
The authors are incredibly grateful to Idlir Liko and Carol Robinson (Oxford University, UK) for their open and transparent discussions regarding native MS analysis of membrane proteins within Amgen Therapeutic Discovery. Michael Marty and Justin Benesch (Oxford University, UK) are thanked for their discussions regarding operation, of the UniDec algorithm. Samer Chmait, John Hui and Hannah Catterall (Amgen) are thanked for their discussions during the bacteriorhodopsin purification, MALDI-ToF and data analysis. Nicholas Woodall (UCLA) is thanked for the generous gift of the mutant (M60A) bacteriorhodopsin. Philip Tagari (Amgen) is acknowledged for his support of the nanodisc project within Therapeutic Discovery. Support from the American Society for Mass Spectrometry Postdoctoral Research Award to HL is gratefully acknowledged. Support from the US National Institutes of Health (R01GM103479 and S10RR028893) and the US Department of Energy (UCLA/DOE Institute for Genomics and Proteomics; DE-FC03-02ER63421) to JAL is acknowledged.
References
- 1.Katritch V, Cherezov V, Stevens RC. Annual review of pharmacology and toxicology. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, Swain NA. J Med Chem. 2013;56:593–624. doi: 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- 3.(a) Drews J. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]; (b) Terstappen GC, Reggiani A. Trends in pharmacological sciences. 2001;22:23–26. doi: 10.1016/s0165-6147(00)01584-4. [DOI] [PubMed] [Google Scholar]; (c) Arinaminpathy Y, Khurana E, Engelman DM, Gerstein MB. Drug Discov Today. 2009;14:1130–1135. doi: 10.1016/j.drudis.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopkins AL, Groom CR. Nature reviews Drug discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 5.(a) van den Heuvel RH, Heck AJ. Curr Opin Chem Biol. 2004;8:519–526. doi: 10.1016/j.cbpa.2004.08.006. [DOI] [PubMed] [Google Scholar]; (b) Benesch JL, Ruotolo BT, Simmons DA, Robinson CV. Chem Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]; (c) Sharon M, Robinson CV. Annu Rev Biochem. 2007;76:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]; (d) Heck AJ. Nature methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]; (e) Benesch JL, Ruotolo BT. Curr Opin Struct Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, Seeger MA, Miguel RN, Korkhov VM, van Veen HW, Venter H, Walmsley AR, Tate CG, Robinson CV. Nature methods. 2009;6:585–587. doi: 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Laganowsky A, Reading E, Hopper JT, Robinson CV. Nat Protoc. 2013;8:639–651. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hopper JT, Yu YT, Li D, Raymond A, Bostock M, Liko I, Mikhailov V, Laganowsky A, Benesch JL, Caffrey M, Nietlispach D, Robinson CV. Nature methods. 2013;10:1206–1208. doi: 10.1038/nmeth.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hopper JT, Robinson CV. Angew Chem Int Ed Engl. 2014;53:14002–14015. doi: 10.1002/anie.201403741. [DOI] [PubMed] [Google Scholar]; (f) Reading E, Liko I, Allison TM, Benesch JL, Laganowsky A, Robinson CV. Angew Chem Int Ed Engl. 2015;54:4577–4581. doi: 10.1002/anie.201411622. [DOI] [PubMed] [Google Scholar]
- 7.Loo RR, Dales N, Andrews PC. Protein Sci. 1994;3:1975–1983. doi: 10.1002/pro.5560031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Sharon M, Ilag LL, Robinson CV. J Am Chem Soc. 2007;129:8740–8746. doi: 10.1021/ja067820h. [DOI] [PubMed] [Google Scholar]; (b) Barrera NP, Di Bartolo N, Booth PJ, Robinson CV. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- 9.Leney AC, McMorran LM, Radford SE, Ashcroft AE. Anal Chem. 2012 doi: 10.1021/ac302223s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Bayburt TH, Grinkova YV, Sligar SG. Nano Letters. 2002;2:853–856. [Google Scholar]; (b) Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 11.Xu H, Hill JJ, Michelsen K, Yamane H, Kurzeja RJ, Tam T, Isaacs RJ, Shen F, Tagari P. Biochimica et biophysica acta. 2015;1848:1974–1980. doi: 10.1016/j.bbamem.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Loo JA. Journal of Mass Spectrometry. 1995;30:180–183. [Google Scholar]
- 13.Loo JA. Mass Spectrometry Reviews. 1998;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 14.Morris HR, Paxton T, Dell A, Langhorne J, Berg M, Bordoli RS, Hoyes J, Bateman RH. Rapid Commun Mass Spectrom. 1996;10:889–896. doi: 10.1002/(SICI)1097-0231(19960610)10:8<889::AID-RCM615>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 15.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 16.(a) Mao Y, Valeja SG, Rouse JC, Hendrickson CL, Marshall AG. Anal Chem. 2013;85:4239–4246. doi: 10.1021/ac303525n. [DOI] [PubMed] [Google Scholar]; (b) Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang H, Cui W, Gross ML. Int J Mass Spectrom. 2013:354–355. doi: 10.1016/j.ijms.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li H, Wolff JJ, Van Orden SL, Loo JA. Anal Chem. 2014;86:317–320. doi: 10.1021/ac4033214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makarov A. Anal Chem. 2000;72:1156–1162. doi: 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- 18.Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJ. Nature methods. 2012;9:1084–1086. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- 19.Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL. Anal Chem. 2013;85:11163–11173. doi: 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- 20.(a) Benesch JL, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Chem Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]; (b) Benesch JL. J Am Soc Mass Spectrom. 2009;20:341–348. doi: 10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 21.(a) Lorenzen K, Versluis C, Van Duijn E, Van den Heuvel R, Heck AJR. Internation journal of mass spectrometry. 2007;268:198–206. [Google Scholar]; (b) Lorenzen K, Olia AS, Uetrecht C, Cingolani G, Heck AJ. J Mol Biol. 2008;379:385–396. doi: 10.1016/j.jmb.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Campuzano I, Giles K. Nanospray Ion Mobility Mass Spectrometry of Selected High Mass Species. Nanoproteomics: Methods and Protocols. In: Toms SA, Weil R, editors. Methods in Molecular Biology. Vol. 790. Humana Press, a part of Springer Science+Business Media, LLC; New York: 2011. pp. 57–70. [DOI] [PubMed] [Google Scholar]
- 22.Nollert P. Methods. 2004;34:348–353. doi: 10.1016/j.ymeth.2004.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Marty MT, Zhang H, Cui W, Blankenship RE, Gross ML, Sligar SG. Anal Chem. 2012;84:8957–8960. doi: 10.1021/ac302663f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marty MT, Zhang H, Cui W, Gross ML, Sligar SG. J Am Soc Mass Spectrom. 2014;25:269–277. doi: 10.1007/s13361-013-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campuzano I, Li H, Loo JA, Svitel G, Schnier P. Proc. 62nd ASMS Conf. Mass spectrometry and Applied Topics; Baltimore. 2014; 2014. p. MP184. [Google Scholar]
- 25.(a) Campuzano I, Li H, Bagal D, Schnier P, Loo JA. Proc. 63rd ASMS Conf. Mass spectrometry and Applied Topics; St Louis. 2015; 2015. TOD. [Google Scholar]; (b) Gault J, Mize T, Damoc E, Belov ME, Makarov A, Robinson CV. Proc. 63rd ASMS Conf. Mass spectrometry and Applied Topics; St Louis. 2015; 2015. TOD. [Google Scholar]
- 26.(a) Marty MT, Hoi KK, Gault J, Robinson CV. Angew Chem Int Ed Engl. 2016;55:550–554. doi: 10.1002/anie.201508289. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gault J, Donlan JA, Liko I, Hopper JT, Gupta K, Housden NG, Struwe WB, Marty MT, Mize T, Bechara C, Zhu Y, Wu B, Kleanthous C, Belov M, Damoc E, Makarov A, Robinson CV. Nature methods. 2016;13:333–336. doi: 10.1038/nmeth.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grinkova YV, Denisov E, Sligar SG. Protein Engeering Design and Selection. 2010;23:843–848. doi: 10.1093/protein/gzq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Jorgensen TJ, Delforge D, Remacle J, Bojesen G, Roepstorff P. Internation journal of mass spectrometry. 1999;188:63–85. [Google Scholar]; (b) Sleno L, Volmer DA. J Mass Spectrom. 2004;39:1091–1112. doi: 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- 29.Tahallah N, Pinkse M, Maier CS, Heck AJ. Rapid Commun Mass Spectrom. 2001;15:596–601. doi: 10.1002/rcm.275. [DOI] [PubMed] [Google Scholar]
- 30.Landreh M, Liko I, Uzdavinys P, Coincon M, Hopper JT, Drew D, Robinson CV. Chem Commun (Camb) 2015;51:15582–15584. doi: 10.1039/c5cc07045g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lippens JL, Mangrum JB, McIntyre W, Redick B, Fabris D. Rapid Commun Mass Spectrom. 2016;30:773–783. doi: 10.1002/rcm.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, Robinson CV. Anal Chem. 2015;87:4370–4376. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Tito MA, Robinson CV. 2000-GB2625 WO. 2000; (b) Sobott F, Robinson CV. Internation journal of mass spectrometry. 2004;236:25–32. [Google Scholar]; (c) Campuzano I, Giles K, Hughes C. 2008102163 WO. 2008
- 34.(a) van Berkel WJ, van den Heuvel RH, Versluis C, Heck AJ. Protein Sci. 2000;9:435–439. doi: 10.1110/ps.9.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hernandez H, Robinson CV. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 35.Ferrige AG, Seddon MJ, Green BN, Jarvis SA, Skilling J. Rapid Commun Mass Spectrom. 1992;6:707–711. [Google Scholar]
- 36.Zhou M, Sandercock AM, Fraser CS, Ridlova G, Stephens E, Schenauer MR, Yokoi-Fong T, Barsky D, Leary JA, Hershey JW, Doudna JA, Robinson CV. Proc Natl Acad Sci U S A. 2008;105:18139–18144. doi: 10.1073/pnas.0801313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hopper JT, Oldham NJ. J Am Soc Mass Spectrom. 2009;20:1851–1858. doi: 10.1016/j.jasms.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 38.(a) Ruotolo BT, Hyung SJ, Robinson PM, Giles K, Bateman RH, Robinson CV. Angew Chem Int Ed Engl. 2007;46:8001–8004. doi: 10.1002/anie.200702161. [DOI] [PubMed] [Google Scholar]; (b) Bornschein RE, Niu S, Eschweiler J, Ruotolo BT. J Am Soc Mass Spectrom. 2015 doi: 10.1007/s13361-015-1250-7. [DOI] [PubMed] [Google Scholar]; (c) Bornschein RE, Ruotolo BT. Analyst. 2015 doi: 10.1039/c5an01242b. [DOI] [PubMed] [Google Scholar]
- 39.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 40.(a) Novak P, Haskins WE, Ayson MJ, Jacobsen RB, Schoeniger JS, Leavell MD, Young MM, Kruppa GH. Anal Chem. 2005;77:5101–5106. doi: 10.1021/ac040194r. [DOI] [PubMed] [Google Scholar]; (b) Jacobsen RB, Sale KL, Ayson MJ, Novak P, Hong J, Lane P, Wood NL, Kruppa GH, Young MM, Schoeniger JS. Protein Sci. 2006;15:1303–1317. doi: 10.1110/ps.052040406. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ryan CM, Souda P, Bassilian S, Ujwal R, Zhang J, Abramson J, Ping P, Durazo A, Bowie JU, Hasan SS, Baniulis D, Cramer WA, Faull KF, Whitelegge JP. Mol Cell Proteomics. 2010;9:791–803. doi: 10.1074/mcp.M900516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Thangaraj B, Ryan CM, Souda P, Krause K, Faull KF, Weber AP, Fromme P, Whitelegge JP. Proteomics. 2010;10:3644–3656. doi: 10.1002/pmic.201000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mikhailov VA, Mize T, Bush MF, Robinson CV. Proceedings of the 61st ASMS Conference on Mass Spectrometry and Applied Topics; Minneapolis. 2013; 2013. [Google Scholar]
- 42.Mikhailov VA, Liko I, Mize TH, Bush MF, Benesch JL, Robinson CV. Anal Chem. 2016;88:7060–7067. doi: 10.1021/acs.analchem.6b00645. [DOI] [PubMed] [Google Scholar]
- 43.Chernushevich IV, Thomson BA. Anal Chem. 2004;76:1754–1760. doi: 10.1021/ac035406j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.