

1. Introduction
The neuronal ceroid lipofuscinoses (NCLs) are a group of mostly recessively-inherited neurodegenerative diseases that primarily affect children (Mole et al., 2011). The pathologic hallmark of these diseases is an accumulation of fluorescent material in the lysosomes of affected individuals and onset is typically marked by seizures and/or visual problems, which become progressively more severe, eventually accompanied by dementia and loss of locomotor function. Progression is relentless, and these diseases generally result in death. To date, defects in 14 different genes have been definitively associated with patients diagnosed with NCL disease (Table 1).
Table 1. Description and reported incidence of NCL diseases.
| Gene name and disease | Gene product | NCL type and alternate presentation | Incidence | Population | Defined | Reference |
|---|---|---|---|---|---|---|
| PPT1 | palmitoyl protein thioesterase 1 | Infantile NCL | 5 per 100,000 | Finland | clinical | (Uvebrant and Hagberg, 1997) |
| 0.16 per 100,000 | Italy | Extrapolated from (Santorelli et al., 2013) | ||||
| 0.05 per 100,000 | Czech Republic | clinical | (Elleder et al., 1997) | |||
| 0.6-0.7 per million | Sweden, Norway, Finland | clinical | (Uvebrant and Hagberg, 1997) | |||
| 0.46 per 100,000 | West Germany | clinical | (Claussen et al., 1992) | |||
| 0.36 per 100,000 | Italy | clinical | (Cardona and Rosati, 1995) | |||
| TPP1 | tripeptidyl peptidase 1 | Late-infantile, classical NCL and spinocerebellar ataxia, autosomal recessive 7 | 0.15 per 100,000 | Portugal | genetic | (Teixeira et al., 2003) |
| 0.5 per 100,000 | Netherlands | clinical | (Taschner et al., 1999) | |||
| 0.62 per 100,000 | Czech Republic | clinical | Extrapolated from (Elleder et al., 1997) | |||
| 0.28 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | ||||
| 9 per 100,000 | Newfoundland | genetic | (Moore et al., 2008) | |||
| CLN3 | CLN3 protein | Juvenile NCL | 0.5 per 100,000 | Portugal | genetic | (Teixeira et al., 2003) |
| 4.8 per 100,000 | Finland | clinical | (Mitchison et al., 1995) | |||
| 0.02 per 100,000 | Czech Republic | clinical | extrapolated from (Elleder et al., 1997) | |||
| 1.6 per 100,000 | Denmark | clinical | (Ostergaard and Hertz, 1998) | |||
| 0.5 per 100,000 | Newfoundland | genetic | (Moore et al., 2008) | |||
| 0.15 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | ||||
| 1.45 per 100,000 | Netherlands | clinical | (Taschner et al., 1999) | |||
| DNAJC5 | DnaJ (Hsp40) homolog, subfamily C, member 5 | Autosomal dominant adult NCL | ||||
| CLN5 | CLN5 protein | Finnish variant late infantile, NCL | 0.07 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | |
| CLN6 | CLN6 protein | Variant late infantile NCL | 0.20 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | |
| 0.62 per 100,000 | Czech Republic | clinical | extrapolated from (Elleder et al., 1997) | |||
| MFSD8 | Major Facilitator Superfamily Domain Containing 8 | Variant late infantile NCL | 2.6 per 100,000 | Newfoundland | genetic | (Moore et al., 2008) |
| 0.14 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | ||||
| CLN8 | CLN8 protein | Variant late infantile NCL and Northern epilepsy | 0.07 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | |
| CTSD | cathepsin D | Congenital NCL | 0.01 per 100,000 | Italy | extrapolated from (Santorelli et al., 2013) | |
| GRN | progranulin | Adult-onset NCL and aphasia, primary progressive, frontotemporal lobar degeneration with ubiquitin-positive inclusions | ||||
| ATP13A2 | ATPase type 13A2 | Juvenile-onset NCL | ||||
| CTSF | cathepsin F | Autosomal recessive adult onset NCL | ||||
| KCTD7 | potassium channel tetramerization domain containing 7 | Infantile-onset NCL | ||||
| SGSH | N-sulfoglucosamine sulfohydrolase | Adult-onset NCL and MPS IIIA | ||||
| total NCL | 13.6 per 100,000 | Newfoundland | genetic | (Moore et al., 2008) | ||
| total NCL | 13 per 100,000 | Finland | (Santavuori et al., 1974) | |||
| 0.56 per 100,000 | Italy | clinical | (Cardona and Rosati, 1995) | |||
| 1.28 per 100,000 | West Germany | clinical | (Claussen et al., 1992) | |||
| 1.61 per 100,000 | Western Scotland | clinical | (Crow et al., 1997) | |||
| 1.95 per 100,000 | Netherlands | clinical | (Taschner et al., 1999) | |||
| 2.8 per 100,000 | Norway | (Augestad and Flanders, 2006) | ||||
| 1.3 per 100,000 | Czech Republic | clinical | (Elleder et al., 1997) | |||
| 1.2 per 100,000 | Italy | (Santorelli et al., 2013) | ||||
There are a number of estimates for the incidence of NCLs as a collective group in different European populations, and these range from ∼0.6 (Italy) to ∼14 (Newfoundland) per 100,000 live births (Table 1) (Claussen et al., 1992; Cardona and Rosati, 1995; Mitchison et al., 1995; Crow et al., 1997; Elleder et al., 1997; Uvebrant and Hagberg, 1997; Ostergaard and Hertz, 1998; Rider and Rider, 1999; Taschner et al., 1999; Teixeira et al., 2003; Augestad and Flanders, 2006; Moore et al., 2008; Santorelli et al., 2013). For individual NCLs, studies have examined PPT1 (previously denoted as CLN1), TPP1 (previously denoted as CLN2) and CLN3 and results are also population dependent (Table 1). However, interpretation of epidemiological data for NCLs is complicated by the fact that many earlier studies were conducted before the identification of the respective disease genes, thus patients were defined by clinical criteria. While some NCLs can be accurately identified from the ultrastructure of the storage material, there is significant overlap in clinical presentation between forms with distinct genetic origins. In addition, to date epidemiological studies have been confined to European or European-derived populations and little is known about the NCL prevalence and distribution in non-European populations.
The Exome Aggregation Consortium (ExAC) database is a collated set of whole exome sequencing data from more than 60,000 individuals (ExomeAggregationConsortium et al., 2015). With a large number of sequenced exomes and a broad sampling of ethnicities, it is possible to use the ExAC database to estimate the incidence of diseases of interest (Appadurai et al., 2015; Calvete et al., 2015; Ropers and Wienker, 2015; Minikel et al., 2016). In this study, we conducted an analysis of the ExAC database to obtain population frequencies for mutations in different ethnic populations in 12 genes most commonly associated with NCL disease. Using this approach, NCL carrier frequencies estimated for the USA correlate well with estimates based on disease incidence. Importantly, the use of genomic data allows the identification of numerous NCL variants that are annotated in public databases as pathogenic but which appear to be neutral polymorphisms. This highlights a serious problem that has clinical implications.
2. Materials and methods
2.1. Variant extraction and annotation
Two databases were used to calculate the prevalence of NCL gene variants in the general population, ExAC v3.0 (ExomeAggregationConsortium et al., 2015) and the Human Gene Mutation Database (HGMD, ver. 2014.3) (Stenson et al., 2014), the latter being an annotated collection of published human gene mutations. Variants that were common to both databases were extracted based on their chromosome, position, reference allele and alternative allele. If multiple alternative alleles at the same chromosome and position are reported in HGMD, they were considered separately in the analysis. The ExAC database excludes individuals with severe pediatric disease when possible (ExomeAggregationConsortium et al., 2015). Given that homozygosity for pathogenic alleles in most NCL genes leads to a clear pediatric disorder, allele frequency reported in ExAC should provide a good estimate for carrier frequency. Consistent with this, only one allele ATP13A2 (p.Ser277Cys) that we classified to be pathogenic is present as a homozygote among the more than 60,000 samples in ExAC. The pathogenic call is retained here as this gene is directly associated with a non-pediatric phenotype (Parkinson's disease).
HGMD phenotypes were used to help identify variants that are relevant to NCL disease. NCL genes are frequently annotated with multiple disease phenotypes in HGMD thus we have established criteria for respective acceptance or exclusion of these phenotypes in general or specific cases:
2.1.1. Homozygosity of null mutations in GRN results in NCL disease (Smith et al., 2012) while heterozygosity is a cause of frontotemporal dementia (FTD) (Baker et al., 2006; Cruts et al., 2006). We include variants associated with the latter in our dataset with the rationale that homozygosity would likely result in NCL disease – in fact, FTD might be regarded as an attenuated NCL resulting from haploinsuffficiency for GRN. We also included neurological phenotypes allied to FTD (e.g., Alzheimer's, Parkinson's) given clinical overlap between these respective disorders.
2.1.2. HGMD annotation can associate NCL genes with multiple diseases, and we have classified these as NCL, NCL-related (e.g., neurodegenerative disorders) and Other (Supplementary Table 1). NCL diseases frequently present with a wide range of clinical phenotypes, reflecting a spectrum of “severity” of mutations, ranging from null alleles to hypomorphs that retain varying degrees of biological function. Therefore, we have included both NCL and NCL-related phenotypes in our final dataset.
2.1.3. CTSD is annotated in HGMD with NCL as well as Alzheimer's disease and other phenotypes. While mutations in this gene are an established cause of NCL disease (Siintola et al., 2006), reported association of CTSD polymorphisms with other phenotypes including Alzheimer's (Papassotiropoulos et al., 2002) are controversial (Ntais et al., 2004) and thus these phenotypes are excluded from our study.
2.2. Variant function prediction and pathogenicity classification
The variant set common to both ExAC and HGMD, was annotated using ANNOVAR (Wang et al., 2010). Using the annotated variant set, we established a decision process (Fig. 1) to predict pathogenic alleles.
Figure 1. Decision flowchart for identifying pathogenic NCL variants.

First, we defined nonsense mutations, exonic insertion/deletions resulting in frameshift variants and mutations of conserved splice junctions as pathogenic. There is a small possibility that truncated proteins resulting from such variants may retain function and this may contribute to a false positive error, but in most cases, such variants are likely to have significant deleterious effects on the respective proteins.
Second, for nonsynonymous missense variants, we considered both ClinVar (Landrum et al., 2016) annotation and functional missense predictions to identify pathogenic alleles. ClinVar is a public archive that reports the clinical significance of human gene variants and is based in part on genotype analysis of patients. For functional predictions, we generated an aggregate score based on the results from eight functional prediction programs provided by ANNOVAR (Table 2). Alleles with aggregate scores > 0 were defined as pathogenic and < 0 as non-pathogenic. Alleles with scores = 0 without ClinVar annotation were examined further in detail and the rationale for our final assessment is listed in Supplementary Table 2. For most variants, ClinVar annotation agreed well with predicted functional consequences. In three cases, there was a disagreement between ClinVar and functional predictions. For these cases, a knowledge-based final decision was established by evaluating the supporting evidence underlying ClinVar annotation and on allele frequency relative to established pathogenic alleles.
Table 2. Aggregate scores based on functional prediction program results.
| Prediction Program | Numerical conversion of prediction program output for Aggregate Score | Category Definition | |||
|---|---|---|---|---|---|
| 1 | 0.5 | 0 | -1 | ||
| SIFT | D | no prediction | T | D: Deleterious (sift<=0.05); T: tolerated (sift>0.05) | |
| PolyPhen 2 HDIV | D | P | no prediction | B | D: Probably damaging (>=0.957); P: possibly damaging (0.453<=pp2_hdiv<=0.956); B: benign (pp2_hdiv<=0.452) |
| PolyPhen 2 Hvar | D | P | no prediction | B | D: Probably damaging (>=0.909); P: possibly damaging (0.447<=pp2_hdiv<=0.909); B: benign (pp2_hdiv<=0.446) |
| LRT | D | U | no prediction | N | D: Deleterious; N: Neutral; U: Unknown |
| MutationTaster | D or A | no prediction | N or P | A: disease_causing_automatic; D: disease_causing; N: polymorphism; P: polymorphism_automatic | |
| FATHMM | D | no prediction | T | D: Deleterious; T: Tolerated | |
| MetaSVM | D | no prediction | T | D: Deleterious; T: Tolerated | |
| MetaLR | D | no prediction | T | D: Deleterious; T: Tolerated | |
Third, for synonymous, intronic, and in-frame insertion/deletions variants, a knowledge-based final decision was established by evaluating the supporting evidence from the study reporting the variant and on allele frequency relative to established pathogenic alleles.
Overall, our strategy has some similarities with established criteria for interpretation of sequence variants (Richards et al., 2015). Clear null variants are defined as described above and considered very strong evidence for pathogenicity and are equivalent to Richards et al's PVS1 variants. Functional predictions based on ANNOVAR are equivalent to Richards et al's PP3 category for supporting evidence for pathogenicity. Other criteria outlined in Richards et al, e.g., segregation (PP1), experimental test for loss of function (PS3) and prevalence compared to controls (PS4) have not, for the most part, been explored with NCL variants and/or may not be possible given the rarity of the diseases and lack of functional understanding of gene product functions.
2.3. USA NCL carrier frequency calculation
For each variant in each ExAC ethnic category, carrier frequencies were calculated by dividing the number of heterozygous individuals by the total number of individuals genotyped at that position. Then the carrier frequencies of all variants within a NCL gene were summed to obtain the carrier frequency of the gene.
To calculate USA carrier frequencies, the number of living NCL patients currently registered with the Batten Disease Support and Research Association (BDSRA) was used. Note that we calculate minimum carrier frequencies based on incidence because not all NCL patients within the USA are registered with the BDSRA. To calculate carrier frequency based upon homozygous recessive inheritance, we determined incidence of NCL per number of live births based upon 4 million births per year in the USA:
where N is the number of living NCL patients in the USA.
3. Results
3.1 Polymorphisms annotated as pathogenic in public repositories
Key to the successful interpretation of genomic data in this study is the ability to differentiate between pathogenic alleles and polymorphisms or other alleles that have no functional consequence. We achieved this by first cross referencing variants extracted from ExAC with the HGMD database (Table 3). After identifying likely null mutants (nonsense mutations, frameshifting mutations, and splicing mutations), we used ANNOVAR to obtain ClinVar annotation and functional predictions for non-synonymous missense variants (Fig. 1, Methods). Analysis of our data reveals a number of variants which are reported to be pathogenic but have a predicted frequency from ExAC that is not consistent with observed frequency using collated data from patient genotyping studies (The NCL Mutation Database, http://www.ucl.ac.uk/ncl/mutation.shtml). Thus, these variants are likely to be neutral polymorphisms.
Table 3. Intersect of ExAC and HGMD variants.
| Number of variants in | ||||
|---|---|---|---|---|
| Gene Name | ExAC | HGMD | ExAC∩ HGMD | HGMD in ExAC |
| PPT1 | 363 | 67 | 24 | 35.8% |
| TPP1 | 829 | 104 | 27 | 26.0% |
| CLN3 | 1107 | 55 | 16 | 29.1% |
| DNAJC5 | 291 | 2 | 0 | 0.0% |
| CLN5 | 268 | 39 | 16 | 41.0% |
| CLN6 | 335 | 62 | 22 | 35.5% |
| MFSD8 | 449 | 32 | 9 | 28.1% |
| CLN8 | 243 | 28 | 15 | 53.6% |
| CTSD | 491 | 7 | 2 | 28.6% |
| GRN | 4583 | 139 | 30 | 21.6% |
| ATP13A2 | 1331 | 27 | 12 | 44.4% |
| CTSF | 487 | 5 | 2 | 40.0% |
| Total | 10777 | 567 | 175 | 30.9% |
A good example is rs374681194 (CLN6, Arg252His), which was annotated in HGMD to be associated with NCL. While there was no ClinVar annotation, predictive methods strongly suggested that this was a pathogenic allele (aggregation score 5.5). However, this is a relatively common allele with 21 heterozygotes in ExAC. Given that 52 heterozygotes were identified for other pathogenic alleles in CLN6 in total, Arg252His should represent ∼30% of pathogenic CLN6 alleles. However, when analyzing collated patient CLN6 mutation data, this allele was found to occur only twice in two compound heterozygotes out of 132 patients genotyped. Given that the single study in which Arg252His was reported (Kousi et al., 2012) did not functionally validate this missense variant, it is probably a polymorphism. Another example is the intronic change rs117284255 (PPT1 c.363-4G>A), which was also reported to be a pathogenic allele (Kousi et al., 2012). Analysis of ExAC indicates that this variant is ∼50-times more frequent than the most common documented pathogenic PPT1 allele (rs137852700, Arg151X) but it was only found in 3/220 patients (one homozygote and two compound heterozygotes) (Kousi et al., 2012). Again, the frequency of this variant is not consistent with patient surveys which suggests that this change is not pathogenic. There are other examples where allele frequency in ExAC is not consistent with patient genotyping studies and these are indicated in Supplementary Table 2.
3.2 Carrier frequencies and influence of ethnicity/race
After excluding the potential neutral, non-pathogenic variants, we calculated the carrier frequencies for each NCL gene. The ExAC dataset is derived from multiple populations: 8.6% African (AFR), 9.5% Latino (AMR), 7.1% East Asian (EAS), 5.4% Finnish (FIN), 55.0% Non-Finnish European (NFE), 13.6% South Asian (SAS) and 0.7% Other (OTH). NCL incidence is known to be related to ethnicity/race therefore we determined mutation incidence within each population (Table 4). For PPT1, the highest incidence was found in the Finnish population, with a carrier frequency of 1/75 which is in excellent agreement with the reported carrier frequency for PPT1 in Finland of 1/70 (Uvebrant and Hagberg, 1997). A single mutation (Arg122Trp) was found in the Finnish population. While this mutation was also observed in the NFE population, there were other mutations (e.g., Arg151X and Val181Met) that were also frequently identified. TPP1 mutations were most frequently encountered in the NFE population (1/392) although incidence was only slightly lower in the Latino population (1/458). For CLN3, the whole exome sequencing approach does not genotype the most frequently observed mutation in this disease, a ∼1kb deletion (Munroe et al., 1997) (Chr16: 28485965 – 28486930, GRCh38), which accounted for 79% of the pathogenic alleles (594 out of 753 alleles, NCL Mutation Database). We extrapolated incidence of the 1kb deletion based on other CLN3 mutations identified in ExAC, i.e., our estimated CLN3 carrier frequency including deletions = the frequency of CLN3 alleles in ExAC * 753/(753-594). Note that this extrapolation makes the assumption that the proportion of CLN3 mutations represented by the 1kb deletion in different ethnic populations parallels that observed in the NCL Mutation Database. While this is likely true for the NFE population, it may not be the case for other populations. CLN3 mutations were most prevalent in the NFE population (1/380) but were also found at appreciable levels in the AFR and FIN groups (Table 4). Other NCLs were typically found at lower incidence than PPT1, TPP1 and CLN3. Notable exceptions are CLN6 in EAS and SAS (1/308 and 1/634, respectively), CLN5 in FIN (1/547), GRN in EAS (1/288) and ATP13A2 in AMR and SAS (1/177 and 1/123, respectively). Note that for all non-European populations, there is no epidemiological data for NCLs. These results highlight different genes should be prioritized in clinical analysis of potential NCL cases according to patients' ancestry.
Table 4. Carrier frequency for NCL gene mutations in different ethnic populations from ExAC and in US.
| Population | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | African/African American (AFR) | Latino (AMR) | East Asian (EAS) | Finnish (FIN) | Non-Finnish European (NFE) | South Asian (SAS) | Other (OTH) | PREDICTED US |
| PPT1 | 1/573 | 1/2893 | 1/865 | 1/75 | 1/319 | 1/4127 | 1/108 | 1/435 |
| TPP1 | 1/865 | 1/458 | 1/1442 | 1/1101 | 1/392 | 1/1650 | nd | 1/459 |
| CLN3* | 1/4668 | 1/5538 | nd | 1/2644 | 1/1802 | 1/3904 | nd | 1/2401 |
| CLN3* | 1/985 | 1/1169 | nd | 1/558 | 1/380 | 1/824 | nd | 1/506 |
| CLN5 | 1/5182 | 1/1444 | 1/2163 | 1/547 | 1/1054 | 1/8253 | nd | 1/1307 |
| CLN6 | 1/1723 | 1/5770 | 1/308 | Nd | 1/1385 | 1/634 | nd | 1/1247 |
| MFSD8 | nd | 1/2886 | nd | Nd | 1/2562 | nd | nd | 1/3299 |
| CLN8 | 1/1718 | 1/5787 | 1/1433 | 1/826 | 1/2084 | 1/2058 | nd | 1/2164 |
| CTSD | nd | nd | nd | Nd | 1/33249 | 1/8256 | nd | 1/39990 |
| GRN | 1/5171 | 1/1446 | 1/288 | 1/1634 | 1/1445 | 1/917 | nd | 1/1231 |
| ATP13A2 | nd | 1/177 | nd | Nd | 1/2333 | 1/123 | nd | 1/599 |
| CTSF | nd | nd | nd | Nd | 1/16661 | nd | nd | 1/26786 |
Results for CLN3 extrapolated to include common 1kb deletion.
3.3 Comparison of genomic data with NCL incidence in the United States (US)
Using the carrier frequency estimates for AFR, AMR, EAS, SAS and NFE, we calculated a weighted carrier frequency that reflects the overall USA population (Table 4) based upon 2014 demographic information (Colby and Ortman, 2014): AFR, 12.4%; AMR, 17.4%; Asian (average EAS and SAS), 5.2%; NFE, 62.2%, OTH, 2.8%. In Figure 2, we compare this weighted estimated incidence for the USA with an observed incidence (Table 5) based upon the number of currently living NCL cases registered with a parent's advocate group, the Batten Disease Support and Research Association (BDSRA, http://bdsra.org/). Carrier frequencies for the different NCL types based on ExAC analysis correlate well between estimated frequency based on disease incidence (r2=0.67) (Figure 2, Panel A). As expected, PPT1, TPP1 and CLN3 are predicted to be the most frequently encountered NCLs, while CTSD is the least common (Table 4). Overall, we predict a carrier frequency for all NCLs combined in the USA of ∼1/85. Carrier frequencies calculated for all ExAC variants that intersect with HGMD (i.e., unfiltered for predicted pathogenicity) are highly inaccurate, providing overestimates for most NCL genes that are as much as >100-fold too high in the case of CLN8 (Figure 2, Panel B).
Figure 2. Correlation between observed and estimated incidence of NCLs in the US.
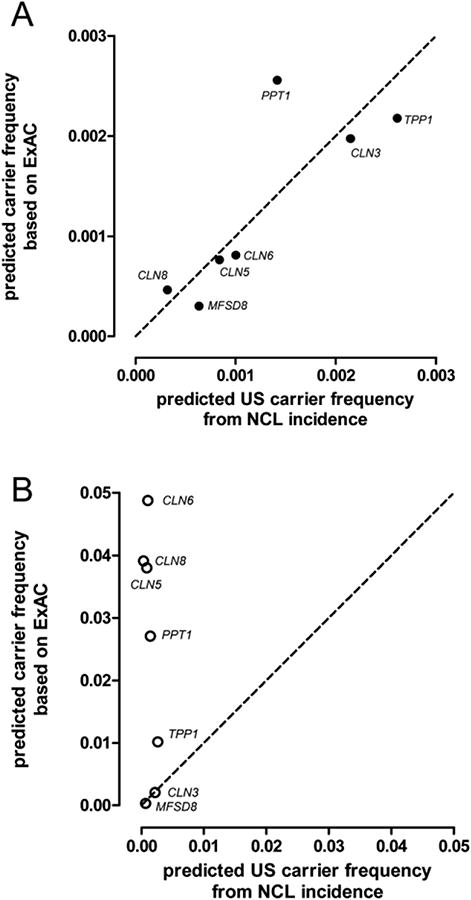
Panel A, ExAC variants found in HGMD after filtering for pathogenicity. Panel B, all ExAC variants found in HGMD. Dashed line, predicted carrier frequency based on incidence = predicted carrier frequency derived from ExAC variant analysis.
Table 5. Epidemiology of NCLs in the USA based on steady state occurrence (December 2015).
Number of living NCL patients currently registered with the Batten Disease Support and Research Association (BDSRA).
| Patients in the US | Average survival | Patients born per year | Minimum carrier frequency (1 per n individuals) | |
|---|---|---|---|---|
| PPT1 | 20 | 10 | 2 | 1/707 |
| TPP1 | 68 | 10 | 6.8 | 1/383 |
| CLN3 | 115 | 25 | 4.6 | 1/466 |
| CLN5 | 7 | 10 | 0.7 | 1/1195 |
| CLN6 | 10 | 10 | 1 | 1/1000 |
| MFSD8 | 4 | 10 | 0.4 | 1/1581 |
| CLN8 | 1 | 10 | 0.1 | 1/3162 |
4. Discussion
4.1 Carrier frequency for NCL diseases
By considering both the demographic composition of the USA and the carrier frequencies for different ethnic groups based upon ExAC analysis, we estimated overall carrier frequencies within the USA for the different NCL genes. These frequencies correlate well with estimates based on current numbers of living NCL patients within the USA obtained from an NCL advocate organization, the BDSRA, although the latter are likely to be an underestimate because not all NCL patients are registered with this organization.
4.2 Potential sources of underestimate
There are several potential sources of error associated with this approach and we conducted control experiments to ensure they will not strongly affect our estimates. First, it is possible that not all known NCL-associated mutations are present within the HGMD. To test this possibility, we examined the HGMD for each individual gene for known NCL mutations identified in patient sequencing studies collated in the NCL Mutation Database. When considering only nonsense and missense mutations, 239 of the 271 known NCL gene mutations (88.2%) are included in HGMD. These mutations collectively accounted for 93.6% of known NCL cases in the NCL Mutation Database (Supplementary Table 3). Thus, it appears the vast majority of known NCL-associated mutations are present within the HGMD. A converse limitation of using the HGMD to screen ExAC variants is that our analysis will not identify unknown pathogenic NCL alleles that have not been reported in patients. While this may contribute to underestimates of carrier frequency in populations, such mutations are likely uncommon compared to established mutations identified from analyses of patient cohorts.
Second, whole exome sequencing does not detect large deletions or intronic mutations outside of splice junctions (see discussion with respect to CLN3 in Results) and there may be errors in calling insertion-deletion (Indel) variants (Fang et al., 2014). In addition, for each NCL gene, there may be private mutations that are present at an incidence that is too low to be detected in the ExAC database. For example, approximately half (62/107) of pathogenic alleles identified in TPP1 are only identified once (Supplementary Table 2). To investigate these possibilities, we extrapolated a total expected allele incidence for TPP1 based on two common alleles (Sleat et al., 1999), Arg208X and a splice junction allele c.509-1G>C that together account for 51% of observed mutations in late infantile NCL patients (333 alleles/613 total, NCL Mutation Database). We observed a total of 64 Arg208X and c.509-1G>C alleles in ExAC and based on the observed ratio, we would expect to identify a total of 118 TPP1 alleles in ExAC (64 × 613/333). In good agreement with this prediction, we identified 110 total pathogenic alleles in TPP1, indicating that a failure to detect intronic changes, large deletions or rare mutations will not have a strong effect on the carrier frequency estimates, at least for TPP1.
4.3 Errors in reporting of variant pathogenicity
Importantly, we have identified a number of NCL gene alleles that are reported to be pathogenic, but have a frequency in the ExAC dataset that is inconsistent with patient sequencing studies (e.g. CLN6, Arg252His and PPT1 c.363-4G>A). In total, we annotated 38 alleles of the 173 obtained from HGMD as non-pathogenic and similar observations have been made in other disease genes (Dorschner et al., 2013). Typically, errors arise when variants are identified in patients and are assigned to be pathogenic without functional validation or detailed pedigree analysis. Many studies attempt to prevent erroneous assignment of pathogenicity by parallel genotype analysis of unaffected controls. For example, in our earlier study of the molecular pathology of classical late infantile NCL (LINCL) (Sleat et al., 1999), we sequenced TPP1 exons in 60 LINCL patients, and in the absence of next-generation sequencing resources, we used 27 controls to eliminate polymorphisms. This approach is effective if variants are common within the patient population but if they are rare, the numbers of such controls typically examined are unlikely to be adequate.
To determine the scope of potential error associated with NCL gene variants that are included in HGMD but appear to be non-pathogenic, we compared carrier frequencies in the USA after filtering for pathogenicity (Fig. 2, Panel A) with results when we used all HGMD NCL and NCL-related phenotypes without filtering (Fig. 2, Panel B). For some of the NCL genes (CLN3 and MFSD8), unfiltered ExAC and patient-based estimates agree. However, for the other NCL genes, a failure to filter the HGMD phenotypes results in overestimates of carrier frequencies that range from ∼4-fold (TPP1) to >100-fold (CLN8). Similar overestimates for other genes have been observed (ExomeAggregationConsortium et al., 2015; Minikel et al., 2016) and this highlight a general problem when variants of unclear significance are reported as pathogenic alleles without validation (Bell et al., 2011).
In conclusion, the results of this study provide a clear picture of the relative and absolute epidemiology of NCL diseases in the USA and several non-European ethnic groups. These results are valuable to both basic NCL research and clinical applications. It should also be possible to extrapolate the ExAC variant data to calculate NCL carrier frequencies for other countries with defined ethnic subpopulations. In addition, we have identified numerous variants that appear to be benign or tolerated but are annotated as pathogenic in the literature and eventually in databases such as HGMD. This propagates errors that may potentially have serious clinical consequences (Yong, 2016) and continues to be an area of concern (Richards et al., 2015).
Supplementary Material
Acknowledgments
We would like to thank Dr. Margie Frazier of the Batten Disease Support and Research Association for help in obtaining NCL patient statistics for the US. We also thank the two anonymous reviewers for their valuable comments. This study was funded by NIH grants NS088786 (DES) and NS037918 (PL).
References
- Appadurai V, DeBarber A, Chiang PW, Patel SB, Steiner RD, Tyler C, Bonnen PE. Apparent underdiagnosis of Cerebrotendinous Xanthomatosis revealed by analysis of ∼60,000 human exomes. Mol Genet Metab. 2015 doi: 10.1016/j.ymgme.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augestad LB, Flanders WD. Occurrence of and mortality from childhood neuronal ceroid lipofuscinoses in norway. J Child Neurol. 2006;21:917–22. doi: 10.1177/08830738060210110801. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernandez B, Fernandez V, Dominguez F, Salas C, Romero-Laorden N, Garcia-Donas J, Carrillo J, Perona R, Trivino JC, Andres R, Cano JM, Rivera B, Alonso-Pulpon L, Setien F, Esteller M, Rodriguez-Perales S, Bougeard G, Frebourg T, Urioste M, Blasco MA, Benitez J. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-Fraumeni-like families. Nat Commun. 2015;6:8383. doi: 10.1038/ncomms9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona F, Rosati E. Neuronal ceroid-lipofuscinoses in Italy: an epidemiological study. Am J Med Genet. 1995;57:142–3. doi: 10.1002/ajmg.1320570206. [DOI] [PubMed] [Google Scholar]
- Claussen M, Heim P, Knispel J, Goebel HH, Kohlschutter A. Incidence of neuronal ceroid-lipofuscinoses in West Germany: variation of a method for studying autosomal recessive disorders. Am J Med Genet. 1992;42:536–8. doi: 10.1002/ajmg.1320420422. [DOI] [PubMed] [Google Scholar]
- Colby SL, Ortman JM. Projections of the size and composition of the U.S. population: 2014 to 2060. Current Population Reports. 2014:25–1143. [Google Scholar]
- Crow YJ, Tolmie JL, Howatson AG, Patrick WJ, Stephenson JB. Batten disease in the west of Scotland 1974-1995 including five cases of the juvenile form with granular osmiophilic deposits. Neuropediatrics. 1997;28:140–4. doi: 10.1055/s-2007-973690. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, Kim JH, Rosenthal EA, Kim DS, National Heart L, Blood Institute Grand Opportunity Exome Sequencing, P. Tabor HK, Bamshad MJ, Motulsky AG, Scott CR, Pritchard CC, Walsh T, Burke W, Raskind WH, Byers P, Hisama FM, Nickerson DA, Jarvik GP. Actionable, pathogenic incidental findings in 1,000 participants' exomes. Am J Hum Genet. 2013;93:631–40. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleder M, Franc J, Kraus J, Nevsimalova S, Sixtova K, Zeman J. Neuronal ceroid lipofuscinosis in the Czech Republic: analysis of 57 cases. Report of the ‘Prague NCL group’. Eur J Paediatr Neurol. 1997;1:109–14. doi: 10.1016/s1090-3798(97)80041-4. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium. Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O'Donnell-Luria A, Ware J, Hill A, Cummings B, Tukiainen T, Birnbaum D, Kosmicki J, Duncan L, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Cooper D, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki M, Levy Moonshine A, Natarajan P, Orozco L, Peloso G, Poplin R, Rivas M, Ruano-Rubio V, Ruderfer M, Shakir K, Stenson P, Stevens C, Thomas B, Tiao G, Tusie-Luna M, Weisburd B, Won H, Yu D, Altshuler D, Ardissino D, Boehnke M, Danesh J, Elosua R, Florez J, Gabriel S, Getz G, Hultman C, Kathiresan S, Laakso M, McCarroll S, McCarthy M, McGovern D, McPherson R, Neale B, Palotie A, Purcell S, Saleheen D, Scharf J, Sklar P, Sullivan P, Tuomilehto J, Watkins H, Wilson J, Daly M, MacArthur D. Analysis of protein-coding genetic variation in 60,706 humans. 2015 doi: 10.1038/nature19057. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Wu Y, Narzisi G, O'Rawe JA, Barron LT, Rosenbaum J, Ronemus M, Iossifov I, Schatz MC, Lyon GJ. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89. doi: 10.1186/s13073-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33:42–63. doi: 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, McLean CY, Tung JY, Yu LP, Gambetti P, Blevins J, Zhang S, Cohen Y, Chen W, Yamada M, Hamaguchi T, Sanjo N, Mizusawa H, Nakamura Y, Kitamoto T, Collins SJ, Boyd A, Will RG, Knight R, Ponto C, Zerr I, Kraus TF, Eigenbrod S, Giese A, Calero M, de Pedro-Cuesta J, Haik S, Laplanche JL, Bouaziz-Amar E, Brandel JP, Capellari S, Parchi P, Poleggi A, Ladogana A, O'Donnell-Luria AH, Karczewski KJ, Marshall JL, Boehnke M, Laakso M, Mohlke KL, Kahler A, Chambert K, McCarroll S, Sullivan PF, Hultman CM, Purcell SM, Sklar P, van der Lee SJ, Rozemuller A, Jansen C, Hofman A, Kraaij R, van Rooij JG, Ikram MA, Uitterlinden AG, van Duijn CM, Exome Aggregation C, Daly MJ, MacArthur DG. Quantifying prion disease penetrance using large population control cohorts. Science translational medicine. 2016;8:322ra9. doi: 10.1126/scitranslmed.aad5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison HM, O'Rawe AM, Taschner PE, Sandkuijl LA, Santavuori P, de Vos N, Breuning MH, Mole SE, Gardiner RM, Jarvela IE. Batten disease gene, CLN3: linkage disequilibrium mapping in the Finnish population, and analysis of European haplotypes. Am J Hum Genet. 1995;56:654–62. [PMC free article] [PubMed] [Google Scholar]
- Mole SE, Williams RE, Goebel HH. The neuronal ceroid lipofuscinoses (Batten disease) 2. Oxford University Press; Oxford: 2011. [Google Scholar]
- Moore SJ, Buckley DJ, MacMillan A, Marshall HD, Steele L, Ray PN, Nawaz Z, Baskin B, Frecker M, Carr SM, Ives E, Parfrey PS. The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland. Clin Genet. 2008;74:213–22. doi: 10.1111/j.1399-0004.2008.01054.x. [DOI] [PubMed] [Google Scholar]
- Munroe PB, Mitchison HM, O'Rawe AM, Anderson JW, Boustany RM, Lerner TJ, Taschner PE, de Vos N, Breuning MH, Gardiner RM, Mole SE. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet. 1997;61:310–6. doi: 10.1086/514846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntais C, Polycarpou A, Ioannidis JP. Meta-analysis of the association of the cathepsin D Ala224Val gene polymorphism with the risk of Alzheimer's disease: a HuGE gene-disease association review. Am J Epidemiol. 2004;159:527–36. doi: 10.1093/aje/kwh069. [DOI] [PubMed] [Google Scholar]
- Ostergaard JR, Hertz JM. Juvenile neuronal ceroid lipofuscinosis. Ugeskr Laeger. 1998;160:3895–900. [PubMed] [Google Scholar]
- Papassotiropoulos A, Lewis HD, Bagli M, Jessen F, Ptok U, Schulte A, Shearman MS, Heun R. Cerebrospinal fluid levels of beta-amyloid(42) in patients with Alzheimer's disease are related to the exon 2 polymorphism of the cathepsin D gene. Neuroreport. 2002;13:1291–4. doi: 10.1097/00001756-200207190-00015. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider JA, Rider DL. Thirty years of Batten disease research: present status and future goals. Mol Genet Metab. 1999;66:231–3. doi: 10.1006/mgme.1999.2827. [DOI] [PubMed] [Google Scholar]
- Ropers HH, Wienker T. Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders. Eur J Med Genet. 2015;58:715–8. doi: 10.1016/j.ejmg.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Santavuori P, Haltia M, Rapola J. Infantile type of so-called neuronal ceroid-lipofuscinosis. Dev Med Child Neurol. 1974;16:644–53. doi: 10.1111/j.1469-8749.1974.tb04183.x. [DOI] [PubMed] [Google Scholar]
- Santorelli FM, Garavaglia B, Cardona F, Nardocci N, Bernardina BD, Sartori S, Suppiej A, Bertini E, Claps D, Battini R, Biancheri R, Filocamo M, Pezzini F, Simonati A. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J Rare Dis. 2013;8:19. doi: 10.1186/1750-1172-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–45. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower-Brooks S, Pullarkat RK, Palmer DN, Lerner TJ, Boustany RM, Uldall P, Siakotos AN, Donnelly RJ, Lobel P. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am J Hum Genet. 1999;64:1511–23. doi: 10.1086/302427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90:1102–7. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner PE, Franken PF, van Berkel L, Breuning MH. Genetic heterogeneity of neuronal ceroid lipofuscinosis in The Netherlands. Mol Genet Metab. 1999;66:339–43. doi: 10.1006/mgme.1999.2810. [DOI] [PubMed] [Google Scholar]
- Teixeira C, Guimaraes A, Bessa C, Ferreira MJ, Lopes L, Pinto E, Pinto R, Boustany RM, Sa Miranda MC, Ribeiro MG. Clinicopathological and molecular characterization of neuronal ceroid lipofuscinosis in the Portuguese population. J Neurol. 2003;250:661–7. doi: 10.1007/s00415-003-1050-z. [DOI] [PubMed] [Google Scholar]
- Uvebrant P, Hagberg B. Neuronal ceroid lipofuscinoses in Scandinavia. Epidemiology and clinical pictures. Neuropediatrics. 1997;28:6–8. doi: 10.1055/s-2007-973654. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong E. Clinical Genetics Has a Big Problem That's Affecting People's Lives. The Atlantic 2016 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.