

Abstract
Purpose
Succinate dehydrogenase subunit B (SDHB) gene mutations are associated with an aggressive clinical disease course of pheochromocytoma/paraganglioma (PHEO/PGL). Limited information is available concerning PHEO/PGL penetrance among SDHB mutation carriers with regards to primary tumor location, specific mutation type, and gender. We assessed PHEO/PGL penetrance in SDHB mutation carriers and described the clinical presentation and disease course.
Methods
Asymptomatic relatives (N=611) of 103 index patients were tested for SDHB mutations. Mutation carriers (N=328) were offered PHEO/PGL screening, of which 241 participated and were included in penetrance analysis. For additional disease outcome analysis, the 103 index patients and 40 screened individuals who developed PHEO/PGL were included. Clinical data was collected between October 2004 and June 2016.
Results
Forty (16.60%) of the 241 screened individuals developed PHEO/PGL during the study. The penetrance estimate in this population was 49.80% (95% CI 29 – 74.9) at 85 years. A significantly higher age-related penetrance of disease was observed in males compared to females, with 50% penetrance achieved at age 74 vs. not reached. Age-related penetrance analysis demonstrated 4 mutations (Ile127Ser, IVS1+1G>T, Exon 1 deletion, Arg90X) presenting with a slower rate of disease development (50% penetrance ages, respectively: not achieved, 70 years, 63 years, 61 years) compared to Arg46X and Val140Phe mutations (50% penetrance at 38 years).
Conclusions
Here, we found a higher estimated penetrance compared to several other studies, and a striking difference in age-related penetrance between male and female SDHB mutation carriers with no association between mutation and gender or tumor location.
Keywords: SDHB, Pheochromocytoma, Paraganglioma, Penetrance, Sex differences
Introduction
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are neuroendocrine tumors arising from adrenal and extra-adrenal chromaffin tissues, respectively. PGLs can arise from both the sympathetic and parasympathetic nervous systems. They are preferentially found arising along the sympathetic chain in the chest, abdomen, and pelvis, or along the parasympathetic chain in the head and neck. PHEOs/PGLs are known to be catecholamine producing tumors. Although rarely, they can present as biochemically silent (Pacak et al. 2007; Timmers et al. 2008). Thirty to forty percent of patients carry a germline mutation in one of the PHEO/PGL susceptibility genes (Erlic et al. 2009; Gimenez-Roqueplo et al. 2012; Neumann et al. 2002), including von Hippel-Lindau (VHL) tumor suppressor gene; RET proto-oncogene in multiple endocrine neoplasia type 2 (MEN2); neurofibromatosis type 1 (NF1); TMEM127; Myc-associated factor X (MAX), the A, B, C, and D subunits of the mitochondrial succinate dehydrogenase complex (SDHA, SDHB, SDHC, SDHD), succinate dehydrogenase assembly factor 2 (SDHAF2), and in rare cases, HIF prolyl hydroxylase 1 and 2 (PHD1/EGLN2, PHD2/EGLN1), fumarate hydratase (FH), hypoxia-inducible factor 2α (HIF2A/EPAS1), and malate dehydrogenase 2 (MDH2) genes. Additionally, 10 – 39% of tumors were found to harbor somatic mutations in genes such as NF1, VHL, RET, MAX, HIF2A/EPAS1, KIF1Bβ, and ATRX (Jochmanova et al. 2015; Fishbein et al. 2017).
SDHB mutations are currently associated with the most aggressive clinical presentation, course, and lethality. Several published studies have described the clinical presentation, aggressiveness, unique biochemical and imaging phenotypes, and therapeutic challenges, including failures, of SDHB-related PHEOs/PGLs (Amar et al. 2007; Ayala-Ramirez et al. 2012; Benn et al. 2006; Benn et al. 2015; Brouwers et al. 2006; Eisenhofer et al. 2012; Ellis et al. 2014; Fonte et al. 2012; Gimenez-Roqueplo et al. 2003; Hadoux et al. 2014; King et al. 2011; Srirangalingam et al. 2008; Timmers et al. 2009; Timmers et al. 2007a). These studies should be carefully considered in the context of the penetrance of these tumors. Increased knowledge of SDHB mutation penetrance supports focused early intervention, follow-up, and improved medical outcomes for patients and their relatives.
In 2006, Benn et al. published a landmark study looking at the clinical presentation and penetrance of PHEO/PGL in an international population of SDHB and SDHD genes mutation carriers. Benn et al. (2006) reported an age-related penetrance of 45% for 82 SDHB mutation carriers by age 40 years. Since their report, several investigators have demonstrated a similar penetrance. Ricketts et al. (2010) reported a 52% penetrance of the disease by age 60 in a group of 295 SDHB mutation carriers. Srirangalingam et al. (2008) observed an overall penetrance of 50% in a group of 32 SDHB mutation carriers. However, after excluding index patients, the overall penetrance decreased to 24%. In 2004, Neumann et al., demonstrated a much higher penetrance of 77% by 50 years of age (Neumann et al. 2004). Overall, studies aimed at evaluating the penetrance of disease in families with specific SDHB mutations, have found lower penetrance estimates. Solis et al. (2009) identified a family with 41 mutation carriers with a large exon 1 deletion and only 11 members diagnosed with PGL. The estimated disease penetrance in this study was 35% by age 40. Hes et al. (2010) reported an estimated penetrance of 26% at 48 years of age in a family with a c.423+1G>A SDHB mutation. In both of these family-based studies, index patients were included in the penetrance calculations. The studies from Neumann, Benn, and Ricketts, which used standard Kaplan-Meier methods to estimate penetrance, likewise fell into a similar problem. Schiavi et al. (2010) correctly indicated that these studies could have overestimated the average penetrance because no compensations were made for the ascertainment of mutation carriers. In their own penetrance study, Schiavi et al. (2010) adjusted their calculations for ascertainment bias and reported an average penetrance of the disease at 50 years of age to be 13% and the average lifetime penetrance (to 80 years of age) to be 30%, which was similar to that reported by Srirangalingam et al. (2008) after excluding index patients. Jafri et al. (2013) also showed a significantly lower penetrance of PHEO/PGL in SDHB mutation carriers after excluding index patients from their analysis. A recent study from Rijken et al. (2016) included SDHB exon 3 deletion carriers from a single family and showed a penetrance of 9% at 50 years of age and 21% at age 70 after correcting for ascertainment bias by excluding index patients from the penetrance calculations.
Despite their discordance, previous studies have accelerated the determination of: (a) the best diagnostic tactics and therapeutic decisions for SDHB-related PHEO/PGL; (b) time-dependent screening of asymptomatic carriers for earlier detection and treatment; and (c) discovery of previously unrecognized SDHB mutations, which could be specifically linked to development of these tumors with unique clinical presentations. Despite this progress, a complete understanding of SDHB-related PHEO/PGL is lacking – especially in terms of its mutation-specific penetrance, differences between males and females, and potential associations among specific mutations, clinical presentation, and outcome. Thus, further knowledge of the penetrance of these tumors may lead to earlier detection of tumors in carriers, development of better treatment options, a greater understanding of SDHB-related PHEO/PGL as a whole, and more directed genetic counseling that would enhance its impact in this very aggressive, and often lethal, disease.
Here, we present data analysis of 344 SDHB mutation carriers on the penetrance of SDHB-related PHEO/PGL linked to sex, age, and specific mutation types, as well as an analysis of the clinical presentation of those with disease.
Patients and Methods
Patients
Between October 2004 and June 2016, family members of index patients who presented to the National Institutes of Health (NIH) for known SDHB related PHEO/PGL, were offered SDHB genetic testing and screening. Penetrance calculations only included family members of index patients. Disease clinical presentation and mutation analyses comprised index patients and family members who developed PHEO/PGL.
Genetic testing was either performed by the NIH in collaboration with the Mayo Clinic in Rochester, MN, or through outside facilities who forwarded the genetic results to the NIH. Screening involved biochemical analysis with plasma and/or urine metanephrines and catecholamines, as well as full body imaging with computer tomography (CT) or magnetic resonance imaging (MRI), including the neck. Index and screened patients diagnosed with PHEO/PGL were either followed at the NIH semiannually or annually. SDHB mutation carriers negative for PHEO/PGL at the time of screening were advised to follow-up with their physicians and to seek medical attention if any symptoms of PHEO/PGL should appear.
This study was approved by the Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development at the NIH and all patients gave written informed consent prior to any performed testing.
Clinical Data
Demographic and clinical information was obtained from medical records. Demographic data included year of birth and gender. For screened individuals, clinical data included plasma and/or urine metanephrines and catecholamine levels, imaging studies results, age at initial screening, SDHB genetic status reports, and occurrence of tumors other than PHEO/PGL. In those diagnosed with PHEO/PGL, additional data involved age at diagnosis, primary tumor location, age at diagnosis of recurrent and/or metastatic disease, and age at death for deceased patients. Diagnosis of PHEO/PGL was made either histologically (when possible), or by a combination of positive biochemistry and imaging studies.
Statistical Methods
Penetrance is defined as the percentage of individuals with a given genotype who exhibit the phenotype associated with that genotype (Griffiths et al. 2000; Khoury et al. 1993). In disease penetrance studies related to gene mutations, the studied population should involve all known gene mutation carriers to prevent false high penetrance of the disease. To prevent overestimation, adjustments for ascertainment bias were made by excluding all index patients from penetrance calculations.
Categorical variables were summarized as frequency counts and percentages. Continuous variables were summarized as the median and range. Penetrance estimates and associated 95% confidence intervals (CIs) at various ages were computed using standard survival methods, including Kaplan-Meier curves and Greenwood’s formula for the associated standard error and CI. Estimates of median values were based on interpolating values from the Kaplan-Meier estimates. Tests to compare penetrance in groups (e.g., males vs. females; or by 3 tumor location sites) used the logrank test, which compares the groups across all time points. To account for a second factor in group comparisons, the stratified version of the logrank test was used. Analyses of the time until metastases developed, or time until death, following the initial diagnosis of PHEO/PGL also used the same survival methods. Average values of numeric quantities are reported as: mean ± 1 standard deviation. Comparisons of percentages in cross-tabulated tables to assess independence or association of factors used Fisher’s exact method for both simple 2 × 2 contingency tables and those with more rows and/or columns. This test assumes that neither the rows nor columns have an inherent ordering, as was the case in our applications (e.g., rows being males or females, columns being the top 6 mutation types). All reported P-values are two-sided.
Results
Initially we evaluated 105 (46 (43.81%) females and 59 (56.19%) males) index patients found to harbor a mutation in the SDHB gene (64 tested at NIH, 41 tested at an outside facility prior to NIH evaluation). From October 1, 2004 to June 31, 2016, 611 (326 (53.36%) females and 285 (46.64%) males) family members from 86 index patients were tested for the SDHB mutation (Figure 1). Results were not available from the remaining 19 index patients because their family members declined/were not yet tested. Two of the 86 index patients were excluded from all analyses due to a lack of available clinical data. The remaining 84 studied index patients, along with the 19 index patients without tested family members (103 index patients in total), were excluded from the PHEO/PGL penetrance calculations, but not from disease clinical presentation or mutation analyses.
Figure 1. Study Population.
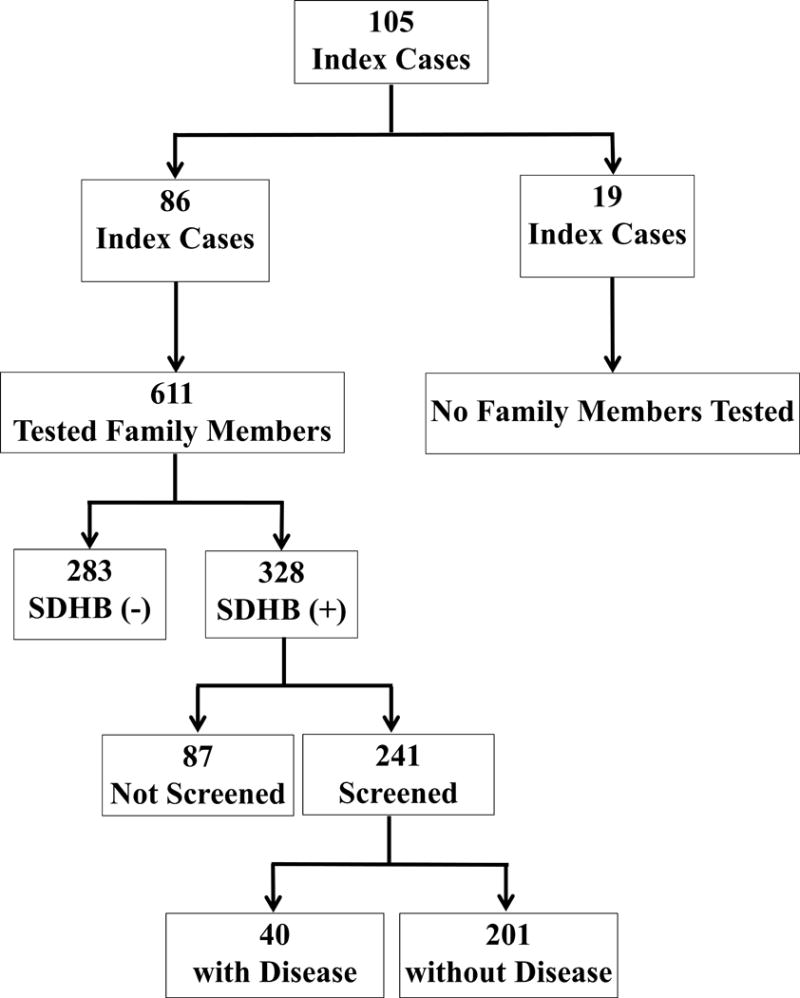
A total of 611 asymptomatic family members from 86 out of 105 index patients harboring SDHB mutations underwent genetic testing. From this cohort, 328 were found to likewise have an SDHB mutation. Screened individuals totaled 241, with 87 not screened due to refusal. Of the screened patients, 40 were diagnosed with PHEO/PGL histologically, or via a combination of positive biochemistry and imaging studies. Two out of the 105 index patients were excluded from analyses due to lack of clinical data.
Of the 611 family members, 231 had their genetic testing performed through the NIH in collaboration with the Mayo Clinic, 183 through NIH collaboration with other research institutions, and 197 at outside facilities. Results revealed that 328 individuals (from 77 families) were positive for an SDHB mutation and were offered screening for PHEO/PGL at the NIH. From the above population, 87 subjects declined screening.
The remaining 241 (130 (53.94%) females and 111 (46.06 %) males) positive individuals were included in the study. Upon initial evaluation, 14 SDHB positive family members were found to have a prior history of PHEO/PGL. Up until June 31, 2016, PHEO/PGL was diagnosed in another 26 SDHB mutation carriers. Characteristics of the 241 screened SDHB mutation carriers are summarized in Table 1. The analyses focused on clinical presentation of the disease included 143 PHEO/PGL patients: 40 diseased patients from the 241 screened family members, and 103 index patients with available clinical data. In 140 patients, the diagnosis was confirmed histologically. In 3 patients, a histologic diagnosis was not made, but the combination of positive biochemistry and imaging studies led to a diagnosis of PHEO/PGL. Three hundred and forty-four (103 index patients with available clinical data and 241 SDHB positive family members) SDHB mutation carriers were included in the mutation analysis.
Table 1.
Characteristics of 241 screened SDHB mutation carriers
| Total | Male | Female | P | |
|---|---|---|---|---|
| Screened SDHB mutation carriers (N, %) | 241 (100%) | 111 (46.06%) | 130 (53.94%) | 0.25 |
| Age of initial screening* | 38 (2 – 85) | 40 (2 – 85) | 37.5 (3 – 83) | 0.80 |
| Years of screening/follow-up at NIH* | 1 (0-14) | 2 (0 – 11) | 1 (0 – 14) | 0.11 |
| Number of screenings/follow-ups* | 2 (1-26) | 2 (1 – 26) | 2 (1 – 10) | 0.11 |
| Other tumors (N, %) | 27 (11.20%) | 15 (13.51%) | 12 (9.23%) | 0.20 |
| PHEO/PGL diagnosis | 40 (16.60%) | 27 (24.32%) | 13 (10.00%) | 0.003 |
| Age at initial diagnosis* | 36.5 (11 – 74) | 34 (11 – 74) | 44 (13 – 64) | 0.56 |
| Diagnosed at initial screening (N, %) | 23 (57.50%) | 18 (66.67%) | 5 (38.46%) | 0.17 |
| Diagnosed prior coming to NIH | 14 (35%) | 8 (29.63%) | 6 (46.15%) | 0.48 |
years, median (range)
Penetrance
A PHEO/PGL diagnosis was made in 40 (16.60%) of the 241 screened SDHB mutation carriers (Table 1). For clinical screening, ages ranged from 2 to 85 years, with a median age of 38 years; 191 SDHB mutation carriers were screened at age 20 or later, 152 at age 30 or later, 118 at age 40 or later, 76 at age 50 or later, 32 at age 60 or later, and 8 at age 70 or later. At initial screening, 14 (5.81%) SDHB mutation carriers were found to have a history of PHEO/PGL and 23 (9.54%) out of 241 SDHB mutation carriers were diagnosed with PHEO/PGL (Table 1). Three (1.25%) more SDHB mutation carriers developed PHEO/PGL within 5 years after initial screening. The estimated overall penetrance of PHEO/PGL in this population of patients was 49.8% by age 85 years. The age-related penetrance for all 241 subjects with clinical data was 3.3% by age 20 (95% CI: 1.6 – 6.9), 12.4% by age 40 (95 % CI: 8.3 – 18.3), 26.4% by age 60 (95% CI: 19.0 – 35.8), and 48.8% by age 80 (95% CI: 29.0 – 74.9) (Figure 2A). The estimated 50% penetrance for this group of SDHB mutation carriers was not reached by 80 years of age (95% CI: 64 – ∞ years).
Figure 2. Kaplan-Meier Penetrance Curves.
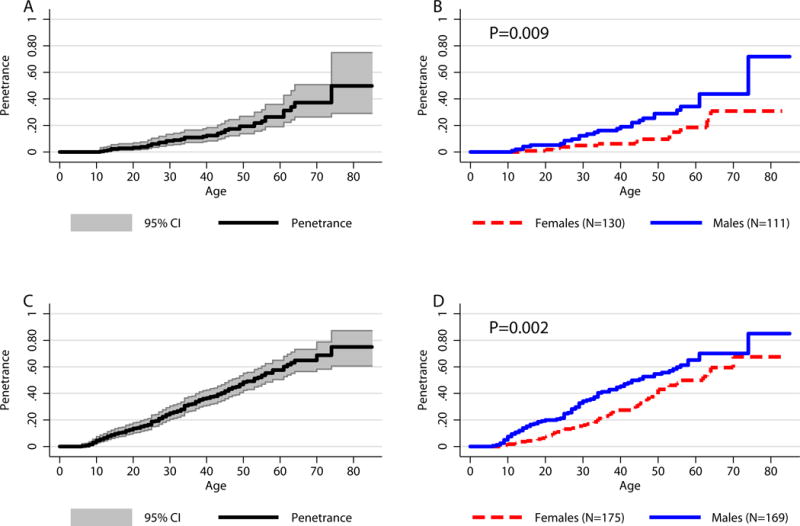
A) Overall age-related penetrance for the 241 SDHB mutation carriers screened for the disease. B) Age-related penetrance in the 241 SDHB mutation carriers separated by sex. C) Overall age-related penetrance for the 241 SDHB mutation carriers screened for the disease and 103 index patients (N=344). D) Age-related penetrance in the 241 SDHB mutation carriers and 103 index patients (N=344) separated by sex. C) and D) represent penetrance estimates if analysis was not corrected for ascertainment bias.
Upper gray line represents estimated upper bound of the age-related penetrance. Lower gray line represents estimated lower bound of the age-related penetrance.
In the studied group of SDHB mutation carriers, 24.32% (27/111) of males and 10% (13/130) of females developed PHEO/PGL. The age range of detection of PHEO/PGL was 11–74 years (median 36.5 years), with 7 patients diagnosed between the ages of 11 and 20, 9 patients between the ages of 21 and 30, 6 patients between the ages of 31 and 40, 8 patients between the ages of 41 and 50, 5 patients between the ages of 51 and 60, and 5 patients diagnosed after the age of 61.
The age-related penetrance values for the 111 screened males were: 5.2% at age 20 (95% CI: 2.2 – 12.0), 19.0% at age 40 (95% CI: 12.0 – 29.3), 34.3% at age 60 (95% CI: 23.7 – 47.8), and 71.8% at age 75 (95% CI: 34.0 – 97.9). For the 130 screened females, the age-related penetrance values were: 1.8% at age 20 (95% CI: 0.5 – 7.1), 6.2% at age 40 (95% CI: 2.8 – 13.4), 18.5% at age 60 (95% CI: 9.8 – 33.2), and 30.8% at age 75 (95% CI: 16.3 – 53.5) (Figure 2B). The estimated 50% penetrance was 74 years (95% CI: 61 – ∞ years) in males and was not achieved by 80 years of age in females (95% CI: 64 – ∞ years) (P=0.0088).
For comparison, when subjects from the screened population are combined with the 103 index patients, i.e. no compensations were made for the ascertainment of mutation carriers, the portion of patients who developed PHEO/PGL during the duration of our study is much higher – 41.57% (143 diseased/344 screened+index patients). Accordingly, the age related penetrance of the disease would change: 4.6% at age 10 (95% CI: 2.8 – 7.5), 13.6% at age 20 (95% CI: 10.3 – 17.9), 36.5% at age 40 (95% CI: 31.0 – 42.5), 57.6% at age 60 (95% CI: 50.4 – 65.0), and 75% at age 75 (95% CI: 29.0 – 74.9) (Figure 2C). The estimated overall penetrance changes to 75% by age 75 and the age at 50% penetrance changed to 53 years (95% CI: 47 – 58 years). The age at 50% penetrance for males was 45 years (95% CI: 37 – 55 years) and for females, 62 years (95% CI: 50 – 70 years) (P=0.002). The age related penetrance of PHEO/PGL in males and females would change accordingly (Figure 2D). Additionally, the age range for clinical diagnosis of PHEO/PGL in this group was 6 – 74 years, with 15 patients diagnosed between the ages of 6 and 10, 28 patients between the ages of 11 and 20, 32 patients between the ages of 21 and 30, 27 patients between the ages of 31 and 40, 23 patients between the ages of 41 and 50, 14 patients between the ages of 51 and 60 years, and 7 patients diagnosed at or after the age of 61.
Mutation Analysis
Mutation analysis was performed on a cohort of 344 SDHB mutation carriers (103 index patients + 241 screened family members). Forty-eight different mutations (Table 2) were found. Six mutations (Arg90X, Ile127Ser, Exon 1 deletion, IVS1+1G>T, Val140Phe, Arg46X), identified in the largest number of patients (184; 99 females, 85 males), were analyzed separately. Age-related penetrance analysis did not demonstrate a strongly significant stratification of risk within the six mutation groups (P=0.072). Although non-significant, we observed a slower rate of disease development (50% penetrance ages, respectively: not achieved, 70 years, 63 years, 61 years) in 4 mutations (Ile127Ser, IVS1+1G>T, Exon 1 Deletion, Arg90X) when compared to patients with Val140Phe or Arg46X mutations (50% penetrance ages, respectively: 38 years, 38 years) (Figure 3A). The two sexes were fairly equally represented among the 6 mutation groups studied, with no statistical evidence that mutation types are related to the sex of the patient (P=0.83, Fisher exact test for 2×6 contingency table). However, as noted earlier, we observed males having an earlier onset of the disease (compare curves in Figure 3A to those in Figure 3B); and the age-related penetrance differences among the 6 mutation groups in males (P=0.039, Figure 3B) appeared possibly greater than those in females (P=0.114, Figure 3B). There appears to be no association between mutation type and location of primary tumor (P=0.82).
Table 2.
Germline mutations in the SDHB gene in the index cases
| Exon/Intron | SDHB mutation (cDNA nucleotide) | SDHB mutation/protein change | Type | Pathogenicity* | Index cases | Carriers+ | Carriers with disease | Types of PHEO/PGL in families |
|---|---|---|---|---|---|---|---|---|
| 1 – 8 | c.1 – c.843+del | Exon 1–8 deletion | large deletion | AF | 2 | 2 | 1 | EA-S |
| 1 | c.1-16418_73-5173del | Exon 1 deletion | large deletion | AF | 4 | 27 | 8 | EA-S, A, EA-P |
| 1 | c.1A>T | p.Met1Leu | missense | AF | 1 | 0 | 0 | EA-P |
| 1 | c.26T>A | p.Leu9X | nonsense | AF | 1 | 2 | 0 | EA-S |
| 1 | c.32G>A | p.Arg11His | missense | VUS | 1 | 1 | 0 | A |
| IVS 1 | c.72+1G>T | IVS1+1G>T | splice site | AF | 8 | 19 | 1 | EA-S, EA-P, A |
| IVS 1 | c.73-9A>G | IVS1–9A>G | splice site | VUS | 1 | 0 | 1 | EA-S |
| 2 | c.79C>A | p.Arg27X | nonsense | AF | 1 | 12 | 0 | EA-S |
| 2 | c.136C>T | p.Arg46X | nonsense | AF | 10 | 10 | 0 | EA-S, A, EA-P |
| 2 | c.137G>A | p.Arg46Gln | missense | AF | 4 | 14 | 2 | EA-S |
| 2 | c.183T>G | p.Tyr61X | nonsense | AF | 1 | 1 | 0 | A |
| 3 | c.268C>T | p.Arg90X | nonsense | AF | 5 | 43 | 9 | EA-S, EA-P |
| 3 | c.271A>T | p.Arg91X | nonsense | AF | 1 | 0 | 0 | EA-S |
| 3 | c.274T>C | p.Ser92Pro | missense | VUS | 1 | 0 | 0 | EA-S |
| 3 | c.275C>A | p.Ser92X | nonsense | 1 | 0 | 0 | EA-S | |
| 3 | c.277T>C | p.Cys93Arg | missense | likely AF | 1 | 0 | 0 | A |
| 3 | c.286G>A | p.Gly96Ser | missense | likely AF | 1 | 1 | 0 | EA-P |
| IVS 3 | c.286+1G>A | IVS3+1G>A | splice site | likely AF | 3 | 2 | 0 | EA-S, EA-P |
| IVS 3 | c.286+2T>A | IVS3+2T>A | splice site | AF | 3 | 0 | N/A | EA-S |
| IVS 3 | c.287-1G>C | IVS3-1G>C | splice site | likely AF | 2 | 6 | 1 | EA-S, EA-P |
| IVS 3 | c.287-3C>G | IVS3-3C>G | splice site | AF | 1 | 0 | N/A | EA-P |
| 4 | c.287G>A | p.Gly96Asp | missense | likely AF | 1 | 0 | 0 | EA-S |
| 4 | c.330_331del | p.Leu111SerfsX7 | frameshift | AF | 1 | 3 | 1 | A |
| 4 | c.343C>T | p.Arg115X | nonsense | AF | 1 | 0 | 0 | EA-S |
| 4 | c.348_352het_del GATTG | p.Ile117HisfsX5 | frameshift | VUS | 1 | 1 | 0 | A |
| 4 | c.369_370insA | p.Val124SerfsX39 | frameshift | AF | 1 | 0 | 0 | EA-S |
| 4 | c.380T>G | p.Ile127Ser | missense | likely AF | 5 | 28 | 5 | EA-S, A, EA, P |
| 4 | c.392delC | p.Pro131HisfsX5 | frameshift | AF | 1 | 0 | 0 | A, EA-S |
| 4 | c.418G>T | p.Val140Phe | missense | AF | 9 | 16 | 5 | EA-S, EA-P, A |
| 5 | c.445-447delCAinsGGTATCT | p.Gln149LeufsX159 | frameshift | VUS | 1 | 1 | 0 | EA-S |
| 5 | c.445C>T | p.Gln149X | nonsense | AF | 1 | 0 | 0 | A |
| 5 | c.487T>C | p.Ser163Pro | missense | VUS | 1 | 0 | 0 | EA-S |
| 5 | c.490C>T | p.Gln164X | nonsense | AF | 1 | 2 | 0 | EA-S |
| 5 | c.540G>A | p.Leu180Leu | splice site | VUS | 1 | 1 | 1 | EA-S |
| IVS 5 | c.541-2A>G | IVS5-2A>G | splice site | AF | 3 | 5 | 3 | A, EA-S, EA-P |
| 6 | c.574T>C | p.Cys192Arg | missense | AF | 1 | 0 | 0 | EA-S |
| 6 | c.575G>A | p.Cys192Tyr | missense | AF | 1 | 6 | 0 | EA-P |
| 6 | c.587G>A | p.Cys196Tyr | missense | AF | 3 | 1 | 1 | EA-S, A |
| 6 | c.590C>G | p.Pro197Arg | missense | AF | 1 | 1 | 0 | EA-S |
| 6 | c.600G>T | p.Trp200Cys | missense | AF | 2 | 3 | 0 | EA-S |
| 6 | c.642G>C | p.Gln214His | missense | VUS | 1 | 0 | 0 | A |
| IVS 6 | c.642+1G>A | IVS6+1G>A | splice site | AF | 2 | 4 | 1 | EA-S |
| 7 | c.683_684delAG | p.Glu228GlyfsX27 | frameshift | AF | 1 | 0 | 0 | EA-S |
| 7 | c.688C>T | p.Arg230Cys | missense | AF | 3 | 5 | 1 | EA-S, EA-P |
| 7 | c.689G>A | p.Arg230His | missense | AF | 3 | 9 | 0 | A, EA-S, EA-P |
| 7 | c.725G>A | p.Arg242His | missense | AF | 1 | 0 | 0 | EA-S |
| 7 | c.746G>T | p.Cys249Phe | missense | VUS | 1 | 0 | 0 | A |
| 7 | c.761insC | p.Pro254fsX255 | frameshift | VUS | 1 | 0 | 0 | EA-S |
As reported by LOVD and NCBI ClinVar databases
Number of carriers out of 241 included in the study, excluding the index cases
Abbreviations: A, adrenal; AF, affects function; EA-S, extra-adrenal sympathetic; EA-P, extra-adrenal parasympathetic; SNV, Single nucleotide variant; VUS, variant of unknown significance
Figure 3. Kaplan-Meier Penetrance Curve Related to Mutation Type.
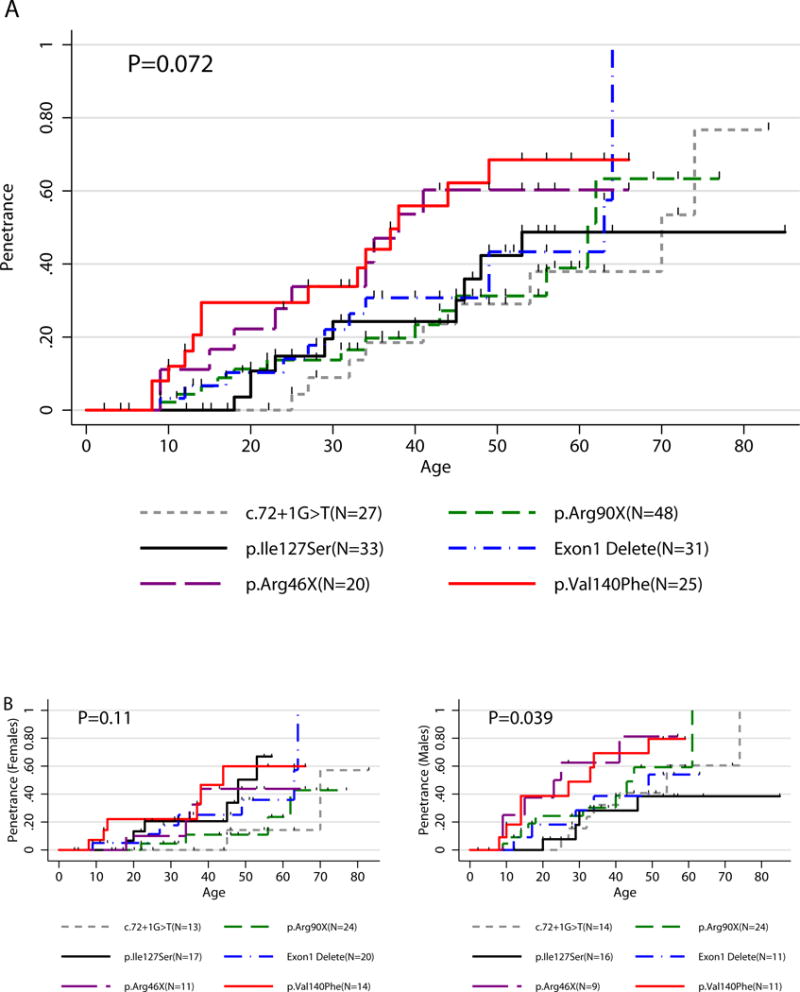
A) Age related penetrance of PHEO/PGL in SDHB mutation carriers (N=184/344) with 6 most common mutation types in SDHB gene. B) Age related penetrance of PHEO/PGL in SDHB mutation carriers (N=184/344) with 6 most common mutation types in SDHB gene separated by sex.
Clinical Presentation of PHEO/PGL
Clinical presentation of PHEO/PGL was evaluated in 103 index patients and 40 SDHB mutation carriers diagnosed with PHEO/PGL during screening. Their characteristics are summarized in Table 3.
Table 3.
Characteristics of 143 patients diagnosed with PHEO/PGL
| All PHEO/PGL patients (N=143) | Index patients (N=103) | Family members with PHEO/PGL (N=40) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Male | Female | P | Total | Male | Female | P | Total | Male | Female | P | |
| Patients (N, %) | 143 (100%) | 85 (59.44%) | 58 (40.56%) | 0.029 | 103 (100%) | 58 (56.31%) | 45 (43.69%) | 0.24 | 40 (100%) | 27 (67.50%) | 13 (32.50%) | 0.038 |
| Age of initial diagnosis* | 29 (6 – 74) | 27 (6 – 74) | 34 (8 – 70) | 0.022 | 28 (6 – 70) | 25.0 (6 – 58) | 34 (8 – 70) | 0.005 | 36.5 (11 – 74) | 34 (11 – 74) | 44 (13 – 64) | 0.56 |
| Primary tumor location | 0.28 | 0.34 | 0.54 | |||||||||
| Adrenal (N, %) | 20 (13.99%) | 10 (11.77%) | 10 (17.24%) | – | 16 (15.53%) | 8 (13.79%) | 8 (17.78%) | – | 4 (10%) | 2 (7.41%) | 2 (15.38%) | – |
| Extra-adrenal sympathetic (N, %) | 97 (67.83%) | 62 (72.94%) | 35 (60.35%) | – | 74 (71.84%) | 45 (77.59%) | 29 (64.44%) | – | 23 (57.5%) | 17 (62.96%) | 6 (46.15%) | – |
| Extra-adrenal parasympathetic (N, %) | 25 (17.48%) | 13 (15.29%) | 12 (20.69%) | – | 12 (11.65%) | 5 (8.62%) | 7 (15.56%) | – | 13 (32.50%) | 8 (29.63%) | 5 (38.46%) | – |
| Adrenal and extra-adrenal sympathetic (N, %) | 1 (0.70%) | 0 | 1 (1.72%) | – | 1 (0.97%) | 0 | 1 (2.22) | – | 0 | 0 | 0 | – |
| Recurrence (N, %) | 7 (4.90%) | 4 (4.71%) | 3 (5.17%) | 1.00 | 6 (5.83%) | 4 (6.90%) | 2 (4.44%) | 0.69 | 1 (2.50%) | 0 | 1 (7.69%) | 0.33 |
| Age at recurrence* | 26 (13 – 54) | 22 (13 – 54) | 48 (17 – 54) | 0.59 | 22 (13 – 54) | 22 (13 – 54) | 35.5 (17–54) | 0.81 | 48 | n/a | 48 | n/a |
| Diagnosis to recurrence interval* | 3 (1 – 8) | 2 (1 – 8) | 3 (2 – 4) | 0.59 | 2.5 (1 – 8) | 2 (1 – 8) | 3 (2–4) | 0.64 | 3 | n/a | 3 | n/a |
| Metastatic disease (N, %) | 85 (59.44%) | 51 (60.00%) | 34 (58.62%) | 1.00 | 78 (75.73%) | 46 (79.31%) | 32 (71.11%) | 0.36 | 7 (17.50%) | 5 (18.52%) | 2 (15.38%) | 1.00 |
| Age of metastasis diagnosis* | 32 (8 – 78) | 29 (10 – 58) | 37 (8 – 78) | 0.054 | 30.5 (8 – 70) | 28.5 (10 – 58) | 35 (8 – 70) | 0.069 | 43 (24 – 78) | 34 (24 – 50) | 60.5 (43 – 78) | 0.25 |
| Diagnosis to metastasis interval* | 3 (0 – 25) | 2 (0 – 25) | 3 (0 – 21) | 0.90 | 3 (0 – 25) | 2.5 (0 – 25) | 3 (0 – 15) | 0.60 | 2 (0 – 21) | 1 (0 – 4) | 16 (11 – 21) | 0.051 |
| Synchronous metastasis | 25 (29.41%) | 14 (27.45%) | 11 (32.35%) | 0.64 | 23 (29.49%) | 12 (26.09%) | 11 (34.38%) | 0.46 | 2 (28.57%) | 2 (40.00%) | 0 (0.00%) | 1.00 |
| Metachronous metastasis | 60 (70.59%) | 37 (72.55%) | 23 (67.65%) | – | 55 (70.51%) | 34 (73.91%) | 21 (65.62%) | – | 5 (71.43%) | 3 (60.00%) | 2 (100.00%) | – |
| Deceased (N, %) | 39 (27.27%) | 19 (22.35%) | 20 (34.48%) | 0.13 | 36 (34.95%) | 18 (31.03%) | 18 (40.00%) | 0.41 | 3 (7.50%) | 1 (3.70%) | 2 (15.38%) | 0.24 |
| Diagnosis to death interval* | 9 (1 – 28) | 8 (1 – 28) | 9 (1 – 28) | 0.53 | 8.5 (1 – 28) | 8.5 (1 – 28) | 8.5 (1 – 23) | 0.80 | 14 (8 – 28) | 8 | 21 (17 – 28) | 0.22 |
| Other tumors (N, %) | 9 (6.29%) | 4 (4.71%) | 5 (8.62%) | 0.49 | 3 (2.91%) | 1 (1.72%) | 2 (4.44%) | 0.58 | 6 (15.00%) | 3 (11.11%) | 3 (23.08%) | 0.37 |
| Years from diagnosis to last follow-up/death* | 7 (0 – 56) | 7 (1 – 56) | 6 (0 – 29) | 0.70 | 7 (1 – 56) | 7.5 (1 – 56) | 7 (1 – 23) | 0.50 | 5 (0 – 43) | 5 (1 – 43) | 5 (0 – 29) | 0.70 |
| Years of follow-up at NIH* | 4 (0 – 16) | 4 (0 – 14) | 3 (0 – 16) | 0.60 | 4 (0 – 16) | 4 (0 – 14) | 3 (0 – 16) | 0.95 | 4 (0 – 14) | 4 (1 – 11) | 4 (0 – 14) | 0.47 |
| Number of visits to NIH* | 4.5 (1 – 42) | 4 (1 – 32) | 5 (1 – 42) | 0.89 | 5 (1 – 42) | 4 (1 – 32) | 5 (1 – 42) | 0.95 | 3.5 (1 – 26) | 4 (1 – 26) | 3 (1 – 8) | 0.37 |
years, median (range)
Tumor characteristics
Of the 143 patients diagnosed with PHEO/PGL, 25 (17.48%) primary tumors were located in the head and neck, 20 (13.99%) in the adrenal gland, and 98 (67.83%) in an extra-adrenal location along the sympathetic chain (Table 3). At the time of diagnosis, one patient was found to have two primary tumors – one in the retroperitoneum and one in the adrenal gland. This patient was excluded from penetrance analysis based on tumor location. In comparing the penetrance of this patient population based on the location of the primary tumor, we found that patients with extra-adrenal parasympathetic tumors had significantly later age-related penetrance as compared to patients with adrenal or extra-adrenal sympathetic tumors (Figure 4A; P=0.0051 comparing 3 locations). The 50% penetrance in patients with primary adrenal tumors was 29 years (95% CI: 15 – 46 years), 27 years in patients with primary extra-adrenal sympathetic tumors (95% CI: 23 – 30 years), and 42 years in patients with primary extra-adrenal parasympathetic tumors (95% CI: 33 – 47 years).
Figure 4. Kaplan-Meier Penetrance Curves Related to Clinical Presentation.
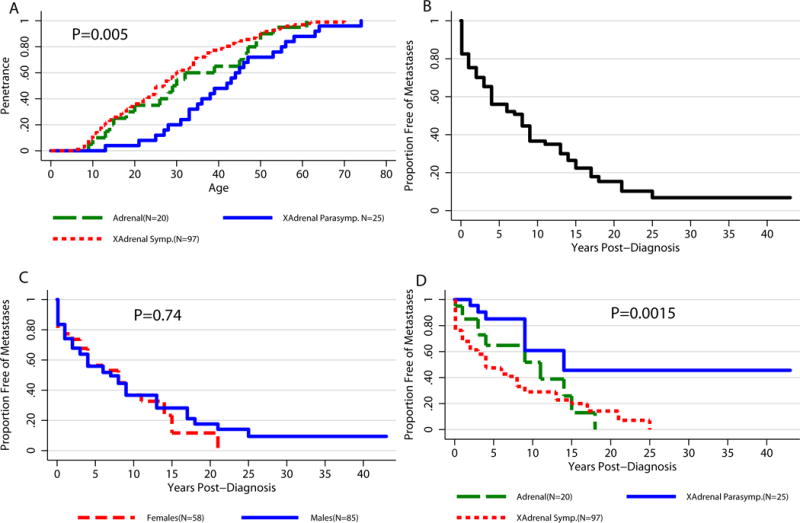
A) Age-related penetrance for three primary tumor locations – adrenal, extra-adrenal sympathetic, extra-adrenal parasympathetic (N=143) B) Time to metastatic disease development after diagnosis. 50% of the 143 individuals with PHEO/PGL will be diagnosed with metastatic disease within 8 years from initial diagnosis. C) Proportion free-of metastasis post diagnosis of PHEO/PGL (N=143; P=0.74). D) Proportion free-of metastasis post diagnosis of PHEO/PGL by primary tumor location (N=143; P=0.0015).
Abbreviations: XAdrenal Symp., extra-adrenal sympathetic; XAdrenal Parasymp., extra-adrenal parasympathetic
Metastatic disease
Eighty-five patients (59.44% of the 143 patients with disease) developed metastatic disease (defined as PHEOs/PGLs in non-chromaffin tissue: bones, lungs, liver, and/or lymph nodes) (Linnoila et al. 1990, DeLellis et al. 2004) (Table 3). The median time between initial diagnosis of PHEO/PGL and diagnosis of metastatic disease was 3 years (Figure 4B). Based on the data, we estimated that among all 143 patients with the disease, 50% will develop metastatic disease within 8 years of diagnosis. The estimated interval for the time to metastasis development is very similar in males and females, at 7 and 8 years, respectively (P=0.74) (Figure 4C). Similarly, time from diagnosis to metastases did not differ significantly among the 6 mutation groups studied (P=0.15). However, the 3 groups of patients differentiated by primary tumor location did have significant differences in time to development of metastatic disease (P=0.0015). Patients presenting with a primary extra-adrenal sympathetic tumor showed significantly faster metastatic disease development when compared to patients with primary extra-adrenal parasympathetic tumors (P=0.0004) (Figure 4D). Patients with primary adrenal tumors showed faster metastasis development than patients with primary extra-adrenal parasympathetic tumors (P=0.043), but there was no statistical difference when compared to patients with primary extra-adrenal sympathetic tumors (P=0.36).
However, when looking at the separate groups of index patients (N=103) and screened family members diagnosed with PHEO/PGL (N=40), the risk of metastatic disease differs significantly. Metastatic disease was diagnosed in 7 (17.5%) out of 40 screened family members diagnosed with PHEO/GPL compared to 78 (75.73%) out of 103 index patients (P<0.0001; Table 3). Among the index patients, 50% will develop metastatic disease within 4 years of diagnosis, whereas 50% of family members diagnosed with PHEO/PGL will develop metastases within 21 year of diagnosis (P<0.0001; Figure 5).
Figure 5.
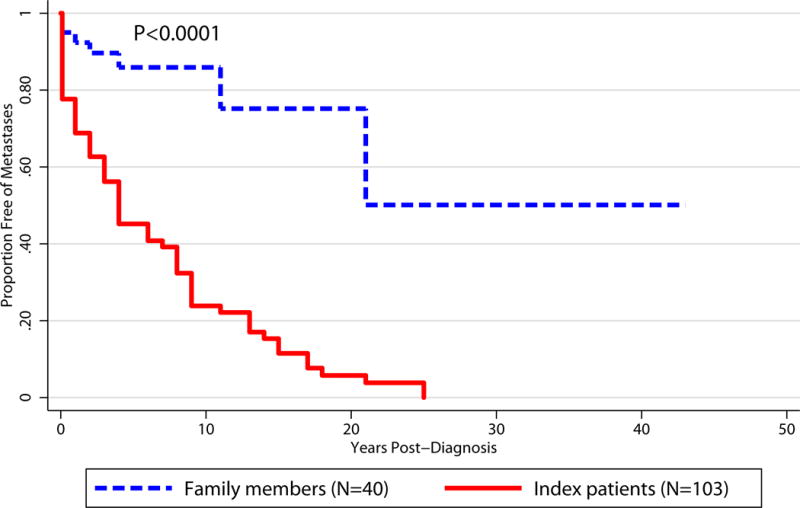
Kaplan-Meier Curves for Development of Metastases Post-Diagnosis: Family Members vs. Index Patients
Mortality
At the conclusion of this study, 39 patients (19 male, 20 female) with diagnosed PHEO/PGL had died (Table 1). From these 39 patients, the median number of years between initial diagnosis and death was 9. For the 105 remaining patients, the median follow-up time since initial diagnosis is 6 years. The Kaplan-Meier estimate of the median time to death after diagnosis, based on all 143 patients with disease, is 20 years (95% CI: 17 – 26 years). All but one patient was diagnosed with metastatic disease prior to death. This patient, although not officially diagnosed with metastatic disease, had multiple soft tissue tumors with likely metastatic lesions. However, PHEO/PGL-specific imaging studies could not be performed. Post-diagnosis survival appears to be moderately better among males compared to females (of those with the disease, 34.48% of females have died vs. 22.35% of males). The median time interval between initial diagnosis and death is 9 years shorter in females than in males, with 17 and 26 years, respectively (P=0.023, 95% CI: 9 – 19 years and 20 – not reached, respectively). The 39 deceased patients presented with 26 different mutations in the SDHB gene, with multiple patients presenting with the following mutations: Arg46X (6 patients), IVS1+1G>A (5 patients), exon 1 deletion (4 patients), and IVS6+1G>A (2 patients).
Presence of other tumors
Thirty (8.72%) out of the 344 SDHB mutation carriers also presented with other tumor types. Of those diagnosed with PHEO/PGL (N=143), 9 (6.29%) mutation carriers presented with another tumor type (Table 3). Two of these patients presented with clear cell renal carcinoma and 2 with breast carcinoma. Melanoma, schwannoma, lung chondroma, prostate, and endometrial adenocarcinoma were each present in a single patient.
Among the remaining 201 SDHB mutation carriers who screened negatively for PHEO/PGL, 21 (10.45%) subjects developed other tumors. Four SDHB mutation carriers were diagnosed with two different types of tumors (oncocytoma and unspecified eye tumor, prostate cancer and basal cell carcinoma, schwannoma with fibroadenomas, and uterine fibroids with myomas). The remaining 17 SDHB mutation carriers presented with clear cell renal carcinoma (N=4), breast cancer (N=3), prostate cancer (N=3), papillary thyroid carcinoma (N=2), neuroblastoma (N=2), melanoma, (N=1), seminoma (N=1), stomach cancer (N=1), basal cell carcinoma (N=1), and uterine fibroids (N=1).
Discussion
With the arrival of more targeted and wide-spread genetic testing for PHEO/PGL, we have gained a clearer understanding of the clinical presentations associated with many of the PHEO/PGL related genes, which has allowed us to make advances in designing genetically-targeted diagnostics and therapeutics. Germline mutations in the SDHB gene account for approximately 10% of PHEOs/PGLs (Bjorklund et al. 2016) and together with MAX and FH mutations, are associated with more aggressive disease than other susceptibility genes mutations. This warrants a dedicated guideline for different screening and evaluation protocols based on varying mutations.
In the present study, we sought to build on our current knowledge base of SDHB-related PHEO/PGL by looking at its penetrance in a population of 344 SDHB mutation carriers. We observed age-related penetrance of 12.4% by age 40 after excluding the 103 index patients (i.e. correcting for ascertainment bias), which is in agreement with the results reported in studies using the same statistical approach (Rijken et al. 2016; Schiavi et al. 2010). Our estimate for overall penetrance (49.8%) is higher than reported in previous studies, which also corrected their estimates for ascertainment bias (21 – 30%) (Hes et al. 2010; Rijken et al. 2016; Schiavi et al. 2010), but lower than in studies that did not corrected for ascertainment bias (65 – 100%) (Neumann et al. 2004; Benn et al. 2006; Solis et al. 2009; Ricketts et al. 2010). However, without correcting for ascertainment bias, the age-related penetrance and overall penetrance would be much higher (36.5% by age 40 and 75%, respectively), which is similar to the first studies on penetrance of SDHB-related PHEO/PGL (Benn et al. 2006; Ricketts et al. 2010; Solis et al. 2009). Therefore, these results demonstrate the importance of excluding index patients from penetrance calculations to avoid overestimation of the disease penetrance.
Previously published results, especially single family-based studies, indicate potential differences in penetrance amongst patients with different mutations within the SDHB gene (Hes et al. 2010; Rijken et al. 2016; Solis et al. 2009). Thus, we analyzed the penetrance and clinical features of the patients harboring the six most represented SDHB mutations in this PHEO/PGL population: Arg90X, Ile127Ser, Exon 1 deletion, IVS1+1G>T, Val140Phe, Arg46X. Solis et al. (2009) reported age-related penetrance of PHEO/PGL in members of a single family harboring gross Exon 1 deletion to be 35% by age 40 and 50% penetrance at age 65 (estimated from figure). In our subset of SDHB Exon 1 deletion carriers, the estimated age-related penetrance was similar – 50% at age 63 (95% CI: 34 – ∞) (Figure 3A). None of the SDHB mutation carriers included in our study harbored the mutations c.423+1G>A or Exon 3 deletion, which were reported in two other family based studies (Hes et al. 2010; Rijken et al. 2016). When comparing the six mutation groups, we observed a tendency for earlier disease development in Val140Phe and Arg46X SDHB mutation carriers (Figure 3A). However, we acknowledge that some of the SDHB mutation carriers harboring the same type of mutation were from the same families. Thus, the influence of modifier genes or environmental factors could explain the different penetrance or phenotype of the disease. For example, in SDHD mutation-related PHEOs/PGLs, altitude has been found to be a phenotypic modifier (Astrom et al. 2003), and recently, two candidate 11p15 tumor modifier genes were described, which may be involved in PHEO/PGL development (Hoekstra et al. 2016). Additional studies, with larger patient samples, are needed to truly determine if different mutations in the SDHB gene are related to contrasting clinical presentations with regard to primary tumor location or metastatic disease development, or, possibly, differences across gender.
Among the presented cohort of SDHB mutation carriers, we found a striking difference in penetrance of PHEO/PGL between males and females, which has not been previously reported. Males showed an earlier progression to disease when compared to females, with a 50% penetrance at 74 years and never reached, respectively (P=0.0088) (Figure 2B). Males also presented with initial diagnosis and diagnosis of metastatic disease at younger ages than females (Table 1). However, rates of metastatic disease and time from initial diagnosis to diagnosis of metastatic disease were similar between the two sexes. Based on the results, males show slightly better post-diagnosis survival than females. This slightly higher proportion of deaths in female’s post-diagnosis, and almost identical time interval from diagnosis to death, largely offsets the earlier onset of disease demonstrated in males. Thus, the early onset of disease in males showed little or no evidence of being associated with accelerated fatal outcome among all 143 followed patients. More studies are necessary, not only to validate these findings in other patient populations, but to also discover why SDHB-related PHEO/PGL occurs later in life in the female population. While the study of male/female differences in disease presentation and penetrance should be repeated in other populations of patients, we should also consider why there are such differences in the present patient cohort.
This study confirms previously described genotype-phenotype correlations found in SDHB-related PHEO/PGL (Benn et al. 2006; Srirangalingam et al. 2008; Timmers et al. 2007b). The majority of the present patient cohort presented with primary tumors in extra-adrenal sympathetic location, with lower portion presentation with primary adrenal tumors or primary head and neck tumors. Patients with primary adrenal or extra-adrenal sympathetic tumors demonstrated faster disease development compared to patients with primary extra-adrenal parasympathetic tumors. This can be, at least partially, attributed to the fact that most extra-adrenal parasympathetic tumors are non-secreting and do not present with symptoms leading to evaluation. Hence, patients with sympathetic PGLs and PHEOs are diagnosed earlier due to the presence of symptoms of catecholamine overproduction.
During the duration of this study, 59.44% patients have developed metastatic disease (Table 1). The high rate of metastatic disease development in patients with SDHB mutations was also reported in previous studies (34 – 71%) (Amar et al. 2005; Benn et al. 2006; Neumann et al. 2004; Timmers et al. 2007b). In 26 (18.18%) patients, malignant disease was present at the time of diagnosis, which is similar to previous findings (Benn et al. 2006; Timmers et al. 2007b). However, in ours as well as some of the previous studies, the high proportion of patients with metastatic disease is most probably due to a referral bias of more complicated cases to our institution. When index patients are excluded from the calculations, metastatic disease development is observed in much lower portion of patients (Figure 5). Patients presenting with primary tumors in extra-adrenal sympathetic locations were found to develop metastatic disease at a significantly faster rate than patients with extra-adrenal parasympathetic tumors. However, the small number of patients studied could have affected these results and they should be examined in other patient populations. Thirty-nine patients died by the time data was collected for this study. Given the possible differences in penetrance rates for the various mutations discussed earlier, we believe it is quite possible that certain mutations will be associated with higher rates of metastatic disease and possibly with different rates of death. However, at this time, we do not have definitive evidence for this (P=0.15 comparing the top 6 mutations for time post-disease to the development of metastases).
Since SDHB mutations are known to be involved in several cancers outside the paraganglial system, we investigated the presence of other tumors in our cohort of SDHB mutation carriers. Only a small number of SDHB mutation carriers presented with other tumors, with the most prevalent being clear cell renal carcinoma, breast adenocarcinoma, and prostate cancer. Previous studies reported SDHB mutations in patients with gastrointestinal stromal tumors, clear cell renal carcinoma, or papillary thyroid cancer (Neumann et al. 2004; Ni et al. 2008; Ricketts et al. 2008; Vanharanta et al. 2004). One of our patients presented with Carney-Stratakis syndrome, which is caused by germline mutations in SDHB, -C, or –D (Janeway et al. 2011; Pasini et al. 2008). However, associations of SDHB-related PHEO/PGL with other tumors are still not clear.
When drawing conclusions of genotype-phenotype correlations and the course of the disease, we fully recognize the limitations of our study. The small cohort of patients with each of the mutations limits our ability to make conclusions about the possible correlations with each mutation. Moreover, the NIH PHEO/PGL research initiative represents a tertiary care center. While the protocol was designed as an all-inclusive clinical program for diagnosis and clinical management of PHEO/PGL for all patients with these tumors regardless of race, age, or gender, we are aware of the potential referral bias of more complicated cases because we are able to provide tests that are not currently available at some institutions across the United States. Thus, multi-institutional and international mutation-specific studies, including in non-tertiary centers, need to be conducted in order to determine whether certain mutations are associated with specific clinical phenotypes.
Nevertheless, our data provides the basis for timing of screening and diagnostic processes and enables personalized treatment decisions. Current Endocrine Society Clinical Practice Guideline (Lenders et al. 2014) recommends employing personalized management of patients with PHEO/PGL based on the known genotype-phenotype presentations. Our results indicate that the overall penetrance of PHEO/PGL in SDHB mutation carriers is slightly higher than in previously published international reports that also corrected their results for ascertainment bias. Moreover, we have found significant differences amongst the male and female SDHB mutation carrier populations, which may result in earlier tumor development in the male population. While females demonstrated a later onset of disease when compared to males, they also showed a slightly shorter interval between diagnosis and death, indicating that more frequent follow-up of this population may be warranted. Currently, there is no consensus about recommendations for screening SDHB mutation carriers. The suggested age of when to start screening varies from 5 to 10 years of age, and the recommended frequency of screening is annual biochemical testing and anatomical imaging every 1 to 2 years (Benn et al. 2006; Srirangalingam et al. 2008; Neumann et al. 2009; Prodanov et al. 2009; Kirmani and Young 2014). Thus, in light of the presented results, we encourage physicians to begin screening asymptomatic SDHB mutation carriers between the ages of 5 and 7 years old with annual biochemical testing. If the initial biochemistry is negative, we suggest just one MRI prior to the 10th birthday. Based on our experience, these tumors rarely present before the age of 10, and if they do, they usually present as biochemically active. Moreover, an MRI can be a very stressful experience for young children, and imaging cannot usually be performed without sedation. After the age of 10, we recommend anatomic imaging with alternating CT/MRI once every two years. Very large, multi-institutional and international studies are certainly necessary to extend results of the present study and to confirm (or repel) the differences in age of presentation among males and females with SDHB mutations. New data on sex differences could potentially lead to changes in genetic counseling and clinical disease management.
Acknowledgments
Funding: This work was supported, in part, by the Intramural Research Program of the NIH, NICHD.
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interest.
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent: Informed consent was obtained from all individual participants included in the study.
References
- Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, Bertagna X, Schlumberger M, Jeunemaitre X, Gimenez-Roqueplo AP, Plouin PF. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–3828. doi: 10.1210/jc.2007-0709. [DOI] [PubMed] [Google Scholar]
- Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul JL, Strompf L, Schlumberger M, Bertagna X, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- Astrom K, Cohen JE, Willett-Brozick JE, Aston CE, Baysal BE. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet. 2003;113:228–237. doi: 10.1007/s00439-003-0969-6. [DOI] [PubMed] [Google Scholar]
- Ayala-Ramirez M, Feng L, Habra MA, Rich T, Dickson PV, Perrier N, Phan A, Waguespack S, Patel S, Jimenez C. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012;118:2804–2812. doi: 10.1002/cncr.26577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PL, Elston M, Gimm O, Henley D, Herman P, Murday V, Niccoli-Sire P, Pasieka JL, Rohmer V, Tucker K, Jeunemaitre X, Marsh DJ, Plouin PF, Robinson BG. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr Relat Cancer. 2015;22:T91–103. doi: 10.1530/ERC-15-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund P, Pacak K, Crona J. Precision medicine in pheochromocytoma and paraganglioma: current and future concepts. J Intern Med. 2016 doi: 10.1111/joim.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, Pacak K. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- DeLellis RA, Ronald A, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumours of endocrine organs Lyon. IARC Press; 2004. [Google Scholar]
- Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, Mannelli M, Linehan WM, Adams K, Timmers HJ, Pacak K. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48:1739–1749. doi: 10.1016/j.ejca.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, Patel D, Prodanov T, Nilubol N, Pacak K, Kebebew E. The presence of SDHB mutations should modify surgical indications for carotid body paragangliomas. Ann Surg. 2014;260:158–162. doi: 10.1097/SLA.0000000000000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, Mussig K, Muresan M, Schaffler A, Reisch N, Schott M, Fassnacht M, Opocher G, Klose S, Fottner C, Forrer F, Plockinger U, Petersenn S, Zabolotny D, Kollukch O, Yaremchuk S, Januszewicz A, Walz MK, Eng C, Neumann HP. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15:6378–6385. doi: 10.1158/1078-0432.CCR-09-1237. [DOI] [PubMed] [Google Scholar]
- Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA, Ghayee HK, Else T, Ling S, Jefferys SR, de Cubas AA, Wenz B, Korpershoek E, Amelio AL, Makowski L, Rathmell WK, Gimenez-Roqueplo AP, Giordano TJ, Asa SL, Tischler AS, Cancer Genome Atlas Research Network. Pacak K, Nathanson KL, Wilkerson MD. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell. 2017;31:181–193. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonte JS, Robles JF, Chen CC, Reynolds J, Whatley M, Ling A, Mercado-Asis LB, Adams KT, Martucci V, Fojo T, Pacak K. False-negative 123I-MIBG SPECT is most commonly found in SDHB-related pheochromocytoma or paraganglioma with high frequency to develop metastatic disease. Endocr Relat Cancer. 2012;19:83–93. doi: 10.1530/ERC-11-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Dahia PL, Robledo M. An Update on the Genetics of Paraganglioma, Pheochromocytoma, and Associated Hereditary Syndromes. Horm Metab Res. 2012;44:328–333. doi: 10.1055/s-0031-1301302. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- Griffiths AJF, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM. An Introduction to Genetic Analysis. W. H. Freeman; New York: 2000. [Google Scholar]
- Hadoux J, Favier J, Scoazec JY, Leboulleux S, Al Ghuzlan A, Caramella C, Deandreis D, Borget I, Loriot C, Chougnet C, Letouze E, Young J, Amar L, Bertherat J, Libe R, Dumont F, Deschamps F, Schlumberger M, Gimenez-Roqueplo AP, Baudin E. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer. 2014;135:2711–2720. doi: 10.1002/ijc.28913. [DOI] [PubMed] [Google Scholar]
- Hes FJ, Weiss MM, Woortman SA, de Miranda NF, van Bunderen PA, Bonsing BA, Stokkel MP, Morreau H, Romijn JA, Jansen JC, Vriends AH, Bayley JP, Corssmit EP. Low penetrance of a SDHB mutation in a large Dutch paraganglioma family. BMC Med Genet. 2010;11:92. doi: 10.1186/1471-2350-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra AS, Addie RD, Ras C, Seifar RM, Ruivenkamp CA, Briaire-de Bruijn IH, Hes FJ, Jansen JC, Corssmit EPM, Corver WE, Morreau H, Bovée JVMG, Bayley J-P, Devilee P. Parent-of-origin tumourigenesis is mediated by an essential imprinted modifier in SDHD-linked paragangliomas: SLC22A18 and CDKN1C are candidate tumour modifiers. Hum Mol Genet. 2016;25:3715–3728. doi: 10.1093/hmg/ddw218. [DOI] [PubMed] [Google Scholar]
- Jafri M, Whitworth J, Rattenberry E, Vialard L, Kilby G, Kumar AV, Izatt L, Lalloo F, Brennan P, Cook J, Morrison PJ, Canham N, Armstrong R, Brewer C, Tomkins S, Donaldson A, Barwell J, Cole TR, Atkinson AB, Aylwin S, Ball SG, Srirangalingam U, Chew SL, Evans DG, Hodgson SV, Irving R, Woodward E, Macdonald F, Maher ER. Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin Endocrinol (Oxf) 2013;78:898–906. doi: 10.1111/cen.12074. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, Pediatric NIH, Wild-Type GC, O’Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochmanova I, Zhuang Z, Pacak K. Pheochromocytoma: Gasping for Air. Horm Cancer. 2015;6:191–205. doi: 10.1007/s12672-015-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, Beaty TH, Cohen BH. Fundamentals of Genetic Epidemiology. Oxford University Press; New York: 1993. [Google Scholar]
- King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, Wesley R, Lodish M, Raygada M, Gimenez-Roqueplo AP, McCormack S, Eisenhofer G, Milosevic D, Kebebew E, Stratakis CA, Pacak K. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011;29:4137–4142. doi: 10.1200/JCO.2011.34.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmani S, Young WF. Hereditary paraganglioma-pheochromocytoma syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Gene Reviews. University of Washington; Seattle: 2014. [Google Scholar]
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF, Jr, Endocrine S. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915–1942. doi: 10.1210/jc.2014-1498. [DOI] [PubMed] [Google Scholar]
- Linnoila RI, Keiser HR, Steinberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol. 1990;21:1168–1180. doi: 10.1016/0046-8177(90)90155-x. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Eng C. The approach to the patient with paraganglioma. J Clin Endocrinol Metab. 2009;94:2677–2683. doi: 10.1210/jc.2009-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak K, Lenders JWM, Eisenhofer G. Pheochromocytoma Diagnosis, Localization, and Treatment. Blackwell Publishing Inc; Malden, MA, USA: 2007. [Google Scholar]
- Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E, Chompret A, Ellison JW, Briere JJ, Rustin P, Gimenez-Roqueplo AP, Eng C, Carney JA, Stratakis CA. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- Prodanov T, Havekes B, Nathanson KL, Adams KT, Pacak K. Malignant paraganglioma associated with succinate dehydrogenase subunit B in an 8-year-old child: the age of first screening? Pediatr Nephrol. 2009;24:1239–1242. doi: 10.1007/s00467-008-1111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, Cole TR, Armstrong R, Kumar VK, Morrison PJ, Atkinson AB, Douglas F, Ball SG, Cook J, Srirangalingam U, Killick P, Kirby G, Aylwin S, Woodward ER, Evans DG, Hodgson SV, Murday V, Chew SL, Connell JM, Blundell TL, Macdonald F, Maher ER. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- Rijken JA, Niemeijer ND, Corssmit EP, Jonker MA, Leemans CR, Menko FH, Hensen EF. Low penetrance of paraganglioma and pheochromocytoma in an extended kindred with a germline SDHB exon 3 deletion. Clin Genet. 2016;89:128–132. doi: 10.1111/cge.12591. [DOI] [PubMed] [Google Scholar]
- Schiavi F, Milne RL, Anda E, Blay P, Castellano M, Opocher G, Robledo M, Cascon A. Are we overestimating the penetrance of mutations in SDHB? Hum Mutat. 2010;31:761–762. doi: 10.1002/humu.21269. [DOI] [PubMed] [Google Scholar]
- Solis DC, Burnichon N, Timmers HJ, Raygada MJ, Kozupa A, Merino MJ, Makey D, Adams KT, Venisse A, Gimenez-Roqueplo AP, Pacak K. Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin Genet. 2009;75:354–363. doi: 10.1111/j.1399-0004.2009.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srirangalingam U, Walker L, Khoo B, MacDonald F, Gardner D, Wilkin TJ, Skelly RH, George E, Spooner D, Monson JP, Grossman AB, Akker SA, Pollard PJ, Plowman N, Avril N, Berney DM, Burrin JM, Reznek RH, Kumar VK, Maher ER, Chew SL. Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH-B) gene mutation carriers. Clin Endocrinol (Oxf) 2008 doi: 10.1111/j.1365-2265.2008.03274.x. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, Pacak K. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2009;16:391–400. doi: 10.1677/ERC-08-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers HJ, Kozupa A, Chen CC, Carrasquillo JA, Ling A, Eisenhofer G, Adams KT, Solis D, Lenders JW, Pacak K. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol. 2007a;25:2262–2269. doi: 10.1200/JCO.2006.09.6297. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007b;92:779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Pacak K, Huynh TT, Abu-Asab M, Tsokos M, Merino MJ, Baysal BE, Adams KT, Eisenhofer G. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J Clin Endocrinol Metab. 2008;93:4826–4832. doi: 10.1210/jc.2008-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]