

Abstract
Used lubricating oil (ULO) is a widespread contaminant, particularly throughout tropical regions, and may be a candidate for bioremediation. However, little is known about the biodegradation potential or basic microbial ecology of ULO-contaminated soils. This study aims to determine the effects of used lubricating oil (ULO) on bacterial community structure and diversity. Using a combination of culture-based (agar plate counts) and molecular techniques (16S rRNA gene sequencing and DGGE), we investigated changes in soil bacterial communities from three different ULO-contaminated soils collected from motorcycle mechanical workshops (soil A, B, and C). We further explored the relationship between bacterial community structure, physiochemical soil parameters, and ULO composition in three ULO-contaminated soils. Results indicated that the three investigated soils had different community structures, which may be a result of the different ULO characteristics and physiochemical soil parameters of each site. Soil C had the highest ULO concentration and also the greatest diversity and richness of bacteria, which may be a result of higher nutrient retention, organic matter and cation exchange capacity (CEC), as well as freshness of oil compared to the other soils. In soils A and B, Proteobacteria (esp. Gammaproteobacteria) dominated the bacterial community, and in soil C, Actinobacteria and Firmicutes dominated. The genus Enterobacter, a member of the class Gammaproteobacteria, is known to include ULO-degraders, and this genus was the only one found in all three soils, suggesting that it could play a key role in the in situ degradation of ULO-contaminated tropical Thai soils. This study provides insights into our understanding of soil microbial richness, diversity, composition and structure in tropical ULO-contaminated soils, and may be useful for the development of strategies to improve bioremediation.
Keywords: Illumina MiSeq, PCR-DGGE, Bacterial community, Used lubricating oil
Introduction
Lubricating oils (LO) are manufactured in various formulations for different applications. Large amount of LO are used in engines of motorized vehicles (Koma et al. 2001), and there are more than one billion motor vehicles around the world (Sperling and Gordon 2009). Used lubricating oils (ULOs) are produced after being subjected to high temperature and high mechanical strain in car engine processes (Bhattacharya and Biswas 2014). ULO is a brown-to-black liquid and is highly viscous due to contamination with dirt, water, salt, metals, incomplete products of combustion, and other materials during the operation of equipment (Ali et al. 2010). ULO is a complex mixture of different chemicals, including linear and branched paraffins, cyclic alkanes, polycyclic aromatic hydrocarbons (PAHs), lubricative additives, and heavy metals (Kim et al. 2003; Aucélio et al. 2007; Dike et al. 2013). These chemicals can be hazardous to human health, and include known mutagens and carcinogens (Haritash and Kaushik 2009). ULO generated from mechanical workshops is becoming a major environmental problem in Thailand (Naladta and Milintawisamai 2011; Pimda and Bunnag 2012; Saimmai et al. 2012) due to inaccessibility of proper disposal facilities and improper disposal.
Several remediation technologies for removal of contaminants, such as hydrocarbons and complex chemical mixtures, from the environment have been developed. Bioremediation is one such method and uses microorganisms and/or plants to transform or reduce environmental contaminant concentrations (Anyasi and Atagana 2011). Successful bioremediation treatments are dependent on multiple factors including the type and concentration of the contaminant, indigenous microbial population, temperature, oxygen, nutrients, and soil characteristics (Khan et al. 2004). Phenotypic and genotypic characterizations of indigenous microorganisms can be valuable for assessing the potential efficacy of bioremediation processes at contaminated sites (Kao et al. 2010).
Assessing the diversity of bacteria that degrade hydrocarbons and associated degradation products in contaminated sites is an important step prior to the implementation of bioremediation projects. As only a small fraction (<1%) of microbial communities can be cultured in the laboratory, culture-independent techniques are necessary to gain a more accurate representation of the total community present at any site (Stewart 2012). Through the use of culture-independent molecular techniques, researchers can more fully characterize the microbial community structure and function across a wide arrange of sites. DNA fingerprinting techniques, such as denaturing gradient gel electrophoresis (DGGE), can provide a snapshot of genetic diversity from mixed microbial samples, however fingerprinting techniques are still not sufficient to fully characterize microbial community structure. High-throughput sequencing (HTS) technologies, such as the Illumina MiSeq, has allowed for more in-depth investigations into microbial community diversity and function (Caporaso et al. 2012; Fadrosh et al. 2014). Using HTS, researchers can more fully analyze the microbial population structure, functional gene content, and metabolic potential.
This study seeks to understand the relationship between bacterial community structure, physiochemical soil parameters, and the ULO composition in three ULO-contaminated soils collected from motorcycle mechanic shops near Prince of Songkla University, Thailand. Both culturing (heterotrophic bacteria and ULO-degrading bacteria) and a combination of culture-independent techniques (DGGE and Illumina MiSeq) were used to estimate bacterial diversity and to monitor the effect of ULO on bacterial community structure.
Materials and Methods
Soil samples
ULO-contaminated soils were obtained from three motorcycle mechanical workshops around Prince of Songkla University, Hat Yai, Songkhla, Thailand. Soil A was located the university campus (6.9983N, 100.4924E) and other soils are located along Thungree Rroad near the university (Soil B at 7.0007N, 100.4964E and Soil C at 7.0045N, 100.4950E). Three replicates soil samples of approximately 500 g each were collected at each site. Individual soil samples were collected from different sites within approximately 1.5 m of each other to a depth of 15 cm by digging with a sterile trowel. Three replicates soils were mixed thoroughly, and then were sieved through a screen with 4 mm sterile mesh to remove gravel, stones and plant debris. The homogenized soil samples were stored in sterilized containers placed in a dark and dry area at room temperature (29±2°C) to prevent photooxidation.
Soil physiochemical analyses
Air-dried soil samples were primarily characterized for moisture content determination (Brady 1974). Soil texture, pH, CEC, NO3−, NH4+, P, K and organic matter were analyzed at the University of Alaska Fairbanks, Palmer Center for Sustainable Living (Jackson 1958; Day 1965; Holmgren et al. 1977; Bremner 1982; Peech 1982; Michaelson et al. 1987).
Enumeration of heterotrophic bacteria and used lubricating oil-degrading bacteria
Within 24 hours of collection, 10 g of homogenized soil from each site was transferred to sterile glass bottles containing 90 ml of 0.85% sterile saline (NaCl) solution. A dilution series (to 10−8) of the suspension was made in 0.85% sterile saline (NaCl) solution. One hundred microliters from each of the dilutions in the series was evenly distributed over both the surface of nutrient agar plates incubated at room temperature (29±2°C) overnight for heterotrophic total viable counts, onto the mineral salt medium agar (MSM) containing 1% of ULO obtained after use in a motorcycle engine for 1 year as the sole carbon source. Plates with ULO as the sole carbon source were incubated for 7 days at room temperature prior to performing culturable ULO-degrading bacterial counts (Jain et al. 2010). Average mean counts of colonies from triplicate plates were recorded and used for the calculation of colony forming units (log CFU g−1).
Hydrocarbon analysis
In order to quantify ULO in each of the collected soils, soils were extracted using a Soxhlet apparatus with 150 ml of n-hexane for 4 h. The organic phase was concentrated by rotary evaporator and dissolved in 1 ml of pure hexane. The extracts were purified with alumina cartridges (Waters Corporation, USA). Aliphatic hydrocarbons in these concentrated and purified extracts were determined by GC-MS chromatography. The GC-MS analysis was performed using a gas chromatograph-mass spectrometer, trace GC Ultra/ISQ MS (Thermo Scientific Inc., USA). The capillary column used was TR-5MS, 30 m × 0.25 mm × 0.25 μm. Helium was used as the carrier gas, at a flow rate of 1 ml min−1. The temperatures of the GC system were the following: injection temperature 240°C; transfer line temperature 280°C; oven initial temperature 50°C 1.5 min; ramp to 180°C at 30°C min−1; ramp to 275°C at 10°C min−1. For qualitative analysis, the MS detector was operated in acquisition scan mode (35–500 amu) (Dąbrowska et al. 2003).
DNA extraction
Bacterial community DNA extraction from agar plate washes
Bacterial colonies from both the nutrient agar (NA, NB, NC) and MSM agar (MA, MB, MC) plates were collected using the agar plate wash method after culturing and counting (Ercolini et al. 2001). For the best dilution (30–300 colonies) of each sample, all colonies present on the surface of the plate (NA and MSM agar) were washed with 2 ml of 0.85% sterile saline (NaCl) solution, harvested with a sterile pipette and stored by freezing at −20 °C. One milliliter of plate wash for each medium was used for the DNA extraction using a phenol-chloroform isoamyl alcohol DNA extraction method (Shahriar et al. 2011). The DNA solution was stored at −20°C until use.
Total soil DNA extraction
Total soil DNA of three soil samples (SA, SB, SC) were extracted and purified using the PowerSoil DNA isolation kit (MO BIO Laboratories, Inc., USA) for PCR-DGGE analysis and the FastDNA Spin kit (MP Biomedicals, Ohio, USA) for MiSeq analysis according to the manufacturer’s instructions. The DNA solution was stored at −20°C until used. The purity and concentration of DNA were evaluated by measuring absorbance at 260 and 280 nm using a NanoDrop ND-1000 Spectrophotometer (Thermo Fischer Scientific, USA), and DNA extracts were stored at −20°C until further analysis.
PCR-DGGE analysis of ULO-contaminated soil
PCR amplification of the 16S rDNA
The variable V3 region of 16S rRNA was PCR-amplified from DNA extracts from both the soil and agar washes with primers targeting the conserved regions of the 16S rRNA gene. PCR universal primers 341F (5′-CCT ACG GGA GGC AGC AG-3′) and 518R (5′-ATT ACC GCG GCT GCT GG-3′) were used to amplify the V3 region of bacterial 16S rRNA connected to GC clamp (5′-CGC CCG CCG CGC CCC GCG CCC GGC CCG CCG CCC CCG CCC C-3rime;) attached to the forward primer (Muyzer and Smalla 1998).
All PCR reactions were performed using Emerald Amp GT PCR Master Mix (Takara, Japan). PCR volumes of 50 μl contained 25 μl of PCR Master Mix, 5 μl of each primer (2.0 μM final concentration), 14.8 μl of water (Takara, Japan), and 0.2 μl of DNA template (500 ng). Amplification was performed using TC-5000 Series PCR Gradient Thermal Cyclers (Techne, United Kingdom) as follows: 95°C for 3 min; followed by 30 cycles of 94°C for 0.5 min; 55°C for 1 min and 72°C for 1 min; with a final elongation of 72°C for 10 min. PCR products were gel checked using 1.5% agarose gel in 1×TAE buffer at 100V for 26 min. After electrophoresis, the gel was stained with SYBR safe DNA gel stain (Invitrogen, USA) for 20 min and DNA bands were visualized (photographed) with UVI gel documentation (Uvtec, UK) (Röling et al. 2002).
DGGE profiling and DNA sequencing
DNA concentrations of purified PCR products were determined by the absorbance values obtained at 260 and 280 nm, using a spectrophotometer. Equal concentrations of purified PCR products (40 μg) were then loaded onto 1 mm thick vertical gels containing 10% polyacrylamide (w/v) (37.5:1 acrylamide: bisacrylamide). Each gel contained a linear gradient of the denaturants urea and formamide, increasing from 30% at the top of gel to 60% at the bottom was applied to separate 16S rDNA fragments (100% denaturant is defined as 7 M urea and 40% (v/v) deionized formamide). The gels were prepared in 1×TAE buffer (40 mM Tris-acetate and 1 mM Na-EDTA, pH 8.0) at 60°C and 60V for 12 h using the D-Code™ Universal Mutation Detection System (BioRad, Hercules, CA, USA) (Röling et al. 2002). Gels were stained in 1×TAE buffer containing SYBR safe DNA gel stain and photographed. All bands in DGGE profiles were cut out of gels with a sterile scalpel and placed in microfuge tubes containing 50 μl of sterilized milli-Q water. The DNA of each band was allowed to diffuse into the water at 4°C overnight. Ten microliters of the solution was used as the DNA template in a PCR reaction using the same primer as previous, without the GC clamp. The reaction was carried out for 30 cycles of 95°C for 0.5 min, 50°C for 1 min and 72°C for 1 min. The PCR products were purified with HiYield™ Gel/PCR DNA Fragments Extraction Kit (RBC Bioscience, Taiwan) then sent for sequencing using the BigDye® Terminator v3.1 cycle sequencing kit (1st BASE, Malaysia). Nucleotide sequences were compared with the Ribosomal Database Project (RDP) naïve Bayesian Classifier tool (Wang et al. 2007). The RDP classifier tool was used to classify all sequences to the genus level. The RDP SeqMatch tool compared with representative sequences from individual operational taxonomic units (OTUs) to the RDP database, and a match was detected at 0.34–1.00 similarity (S_ab) score.
MiSeq Illumina sequencing analysis of ULO-contaminated soil
Total soil DNA extracts were submitted to the Research Technology Support Facility, Michigan State University for PCR optimization and MiSeq Illumina sequencing analyses. The 16S rRNA gene was amplified from soil genomic DNA samples using Illumina fusion primers as described by Caporaso et al. (2011). PCR output for all samples was normalized using a Life Technologies SequalPrep Normalization plate. The normalized products were pooled. After Ampure clean up, quality control and quantitation pool samples were loaded on a standard v2 MiSeq flow cell and sequenced in a 2×250bp format using custom V4 sequencing and index primers and a MiSeq 500 cycle reagent cartridge (v2). Base calling was done by Illumina Real Time Analysis (RTA) v1.18.54 and output of RTA demultiplexed converted to FastQ with Illumina Bcl2fastq v1.8.4. Amplicon sequence data from the Illumina MiSeq were processed using the mother software package version v. 1.32.0 (Schloss et al. 2009) and following the standardized operating procedure. Operational taxonomic units (OTUs) were defined at 3% dissimilarity.
Nucleotide sequence accession numbers
The nucleotide sequences obtained from PCR-DGGE have been deposited in the GenBank database under accession numbers AB934284 to AB934342. All Illumina MiSeq sequence data is publicly available on MG-RAST (ID no. 4687128.3, 4687129.3, 4687130.3) (http://metagenomics.anl.gov/).
Statistical analyses
Alpha diversity measures, including Chao richness estimators, the inverse Simpson diversity index, non-parametric Shannon diversity index and rarefaction curves, were also calculated in mothur. Relationships among bacterial community structures in the three different samples were examined using Bray-Curtis similarity measure in cluster analysis (Bray et al. 1957).
Results
Soil and used lubricating oil characteristics
The physicochemical properties are summarized in Table 1. Soil A was classified as loamy sand, and soil B and C were classified as sandy loam based on the relative proportions of sand, silt and clay. The major nutrient ions such as ammonium (NH4+), nitrate (NO3−), phosphorus (P) and potassium (K) levels in three soils investigated were 3–20 ppm, 1–101 ppm, 14–138 ppm and 18–95 ppm, respectively. The organic matter content and CEC of soil A, B and C were 6–12 % and 18–95 meq/100 g soil, respectively.
Table 1.
Physico-chemical and microbiological characteristics of soil samples A, B and C
| Sample | pH | Moisture content (%) | NH4+ (ppm) | NO3− (ppm) | P (ppm) | K (ppm) | CEC (meq/100 g soil) | Sand (%) | Silt (%) | Clay (%) | C (%) | Heterotrophic total viable counts (log CFU g−1) | ULO degrading bacteria counts (log CFU g−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 5.55 | 4.6 | 10 | <1 | 14 | 18 | 0.98 | 87.2 | 4.2 | 8.6 | 6 | 7.57 | 7.16 |
| B | 6.02 | 11.9 | 20 | <1 | 138 | 67 | 4.68 | 65.2 | 22.2 | 12.6 | 8 | 7.34 | 6.48 |
| C | 5.31 | 4.8 | 3 | 101 | 41 | 95 | 4.76 | 67.2 | 17.2 | 15.6 | 12 | 5.04 | 5.97 |
The ULO extracted from three soil samples was characterized by the presence of hydrocarbons ranging from C13 to C28. Peaks of C13, C21 and C27 were not observed in soil A and peak of C28 only found in soil C (Fig. 1).
Fig. 1.

Gas chromatogram of ULO extracted from three soil samples (A, B, C)
Microbial community characteristics
Results from plate counts showed that the ULO-degrading bacterial counts of soil A, B and C were 7.16, 6.48 and 5.97 log CFU g−1, respectively (Table 1). The heterotrophic total viable counts of soils A, B and C were 7.57, 7.34 and 5.04 log CFU g−1, respectively. However, soils A and B had relatively high densities of culturable ULO-degrading bacteria at 7.16 and 6.48 log CFU g−1, respectively, than soil C which had a higher abundance of ULO-degraders than heterotrophs (5.97 log CFU g−1 and 5.04 log CFU g−1, respectively).
Bacterial community structure and composition determined by PCR-DGGE and MiSeq
Genetic diversity in ULO-contaminated soils was investigated by DGGE analysis of 16S rRNA amplicons. A total of 59 bands were detected in the DGGE profiles from direct soil analyses and plate washes from nutrient agar and ULO-utilizing cultures from the three soils (Fig. 2), and the taxonomic affiliations of the bands are shown in Table 2. The DGGE patterns appeared to be different for the different sources of DNA in each soil. Twenty bands of ULO-degraders from ULO-containing MSM agar plate washes were present in MA, MB and MC lanes on DGGE profiles. These bands were affiliated with 7 different genera of ULO-degraders in all three soils. Members of the genus Enterobacter sp. were present in all of soil samples. Pseudomonas and Pantoea were found in both of soil A and B. Citrobacter sp. and Rahnella sp. were found in soils A and B, respectively. Acinetobacter sp. and Brevibacterium sp. were found as ULO-degraders only in soil C. Twenty-six sequences of heterotrophic bacteria from three ULO-contaminated soils were affiliated with 26 different genera. Five different heterotrophic bacterial genera dominated in soil A (NA) including Citrobacter sp., Enterobacter sp., Marinobacter sp., Acinetobacter sp., and Pseudomonas sp. Heterotrophic bacteria in soil B (NB) were affiliated with 6 genera with Pseudomonas sp., Pseudoalteromonas sp., Rahnella sp., Cronobacter sp., Citrobacter sp., and Enterobacter sp. The genera Lysinibacillus sp., Bacillus sp., and Pantoea sp. were found in soil C (NC). Pseudomonas sp., Citrobacter sp., Rahnella sp. and Enterobacter sp. were found in both culture-based analyses of soil A and B.
Fig. 2.

DGGE fingerprints of the bacterial 16S rRNA gene V3 region amplified from soil A, B, C. The DNA extracted from soil directly (SA, SB, SC), agar plate washes from nutrient ager (NA, NB, NC) and MSM agar (MA, MB, MC). The individual DNA bands selected for sequencing are numbered 1–59. Ladder M: M1 - Bacillus sp., M2 - Enterobacter cancerogenus, M3 - Enterobacter sp., M4 - Pseudomonas putida
Table 2.
16S rDNA-V3 sequence similarities to closest relatives of DNA recovered from the respective bands in DGGE gels
| Class | Genus | Soil A | Soil B | Soil C | Accession umber for the nearest match | |||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Band(s) | S_ab score | Band(s) | S_ab score | Band(s) | S_ab score | |||
| Actinobacteria | Dietzia | - | - | - | - | 9,11 | 0.93–1.00 | AY211169 |
| Brevibacterium | - | - | - | - | 58,59 | 0.83–0.88 | AJ746164 | |
| uncultured bacterium | - | - | - | - | 10 | 0.84 | AY593453 | |
| Bacilli | Bacillus | - | - | - | - | 35, 36,37,38 | 0.80–0.84 | AM778699, DQ147593 |
| Lysinibacillus | - | - | - | - | 33,34 | 0.64–0.84 | KC465744 | |
| Anaerolineae | uncultured bacterium | - | - | 5 | 0.46 | - | - | JN434428 |
| Alphaproteobacteria | Rhizobium | - | - | 6 | 0.32 | - | - | JN688942 |
| uncultured bacterium | - | - | 7 | 0.48 | - | - | EU808178 | |
| Betaproteobacteria | Comamonas | - | - | 31 | 0.76 | - | - | AM503546 |
| Bordetella | - | - | - | - | 12 | 0.79 | FN691469 | |
| uncultured beta-Proteobacterium | 2 | 0.42 | - | - | - | - | AF063633 | |
| Gammaproteobacteria | Enterobacter | 15,18,20,45,48 | 0.74–0.89 | 4,23,29,32,53 | 0.47–0.84 | 13,57 | 0.77–0.91 | EU260145, GU271945 |
| Cronobacter | - | - | 27,30 | 0.79–0.83 | - | - | JQ936997 | |
| Citrobacter | 14,19,40,46,47 | 0.84–0.96 | 28 | 0.78 | - | - | FJ009572 | |
| Pantoea | 44 | 0.69 | 55 | 0.89 | 39 | 0.51 | AY315450 | |
| Marinobacter | 16 | 0.46 | - | - | - | - | GQ131648 | |
| Enterobacteriaceae bacterium | 1 | 0.34 | - | - | - | - | FJ348017 | |
| Rahnella | - | - | 26,51,52,54 | 0.60–0.86 | - | - | KJ997758 | |
| Pseudoalteromonas | - | - | 24, 25 | 0.42–0.48 | - | - | FJ357649 | |
| Acinetobacter | 3,17 | 0.67–0.85 | - | - | 56 | 0.80 | KM108483 | |
| Pseudomonas | 21,41,42,43 | 0.44–0.88 | 8,22,49,50 | 0.34–0.97 | - | - | FM161360, EF427772 | |
Bold of the band number are the bacterial strain from direct soil DNA
S_ab score of 1.0 is the perfect match
Bacterial 16S rRNA amplicons from total soil DNA extractions produced 13 bands on DGGE profiles. Of these 13 bands, 3 bands from soil A (SA), 5 bands from soil B (SB) and 5 bands from soil C (SC) were detected. From extracts of soil A, 3 different bacterial genera dominated, including Acinetobacter sp., Enterobacteriaceae bacterium, and uncultured Betaproteobacteria. Overall, tThe microbial community associated from soil B was affiliated with 4 genera, namely, Enterobacter sp., Rhizobium sp., Pseudomonas sp., uncultured Alphaproteobacteria, and Uncultured Anaerolineae bacterium. The bacterial genera detected from soil C were Dietzia sp., Bordetella sp., Enterobacter sp., and uncultured Actinobacteria. Acinetobacter sp. and Pseudomonas sp. were detected with culturing and culture-independent methods in soil A and B, respectively. Only Enterobacter sp. was found in soil B and C with culture-dependent and culture-independent methods.
The 59 classifiable sequences from DGGE analysis were affiliated with 23 different genera. The greatest bacterial richness was found in soil C (Fig. 3A). The bacterial taxa detected in soil C were affiliated with three main groups: Proteobacteria (Gammaproteobacteria and Betaproteobacteria), Firmicutes (Bacilli) and Actinobacteria. Proteobacteria were dominant in soil A and B, while Firmicutes were dominant in soil C. Overall, Gammaproteobacteria and Betaproteobacteria were the most abundant in all soils.
Fig. 3.

Bacterial community composition and relative abundances at phyla level, based on the classification of partial 16S rDNA sequences of bacteria from of three ULO-contaminated soils (A, B, C) derived from PCR-DGGE (A) and MiSeq (B). Shown are the percentages of the classified sequences
A total of 316,606 high-quality partial 16S rDNA sequences were obtained by MiSeq analysis, including 165,375 from soil A, 75,965 from soil B and 75,266 from soil C. The diversity estimation of those sequences were calculated in MOTHUR and after sequences clustered into 3,449 OTUs (97% similarity). The diversity of the total individual communities was measured by the Inverse Simpson or Shannon indices (Table 3). The result showed that the diversity was higher in soil C than soil A and B. The OTU richness estimate (Sobs), showed that soil B possessed greater species richness than the soil A and C, which is consistent with rarefaction curves that compare the richness observed in different soil samples (Fig. 4). In contrast, the Chao richness index indicated that soil C had higher richness than the other soils.
Table 3.
Alpha-diversity indices based on MiSeq Illumina sequencing data from soil samples
| Group | Nseqsa | Sobsb | Invsimpsonc | Invsimpson_lcid | Invsimpson_hcie | npShannonf | Chaog | Chao_lci | Chao_hci |
|---|---|---|---|---|---|---|---|---|---|
| Soil A | 165,375 | 1,100 | 15.73 | 15.60 | 15.86 | 3.63 | 2207.33 | 1964.46 | 2518.42 |
| Soil B | 75,965 | 1,452 | 4.79 | 4.73 | 4.86 | 3.03 | 1757.64 | 1693.92 | 1838.14 |
| Soil C | 75,266 | 897 | 21.92 | 21.54 | 22.33 | 4.41 | 2658.63 | 2153.30 | 3367.23 |
| Total | 316,606 | 3,449 |
Calculated by MOTHUR at the 0.03 distance level.
Nseqs = number of sequences in the sample
Sobs = number of observed OTUs
InvSimpson = inversed Simpson’s index
Low confidence interval.
High confidence interval
npShannon = non-parametric Shannon index
Chao = richness index
Fig. 4.
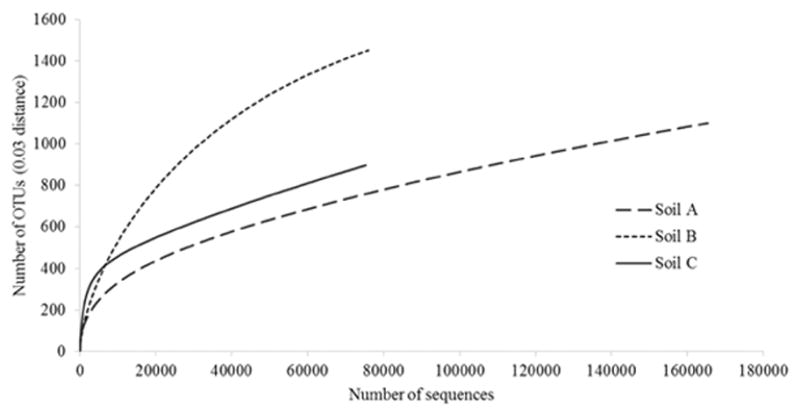
Rarefaction curves of V4 region of the bacterial 16S rRNA gene from three ULO-contaminated soils (A, B, C) obtained from MiSeq amplicon sequencing
Microbial community composition of the various samples was assessed by relative abundances at the phylum level and reads grouped into twenty-four different phyla: Acidobacteria, Actinobacteria, Armatimonadetes, Bacteroidetes, Caldiserica, Chlamydiae, Chloroflexi, Deinococcus-Thermus, Firmicutes, Fusobacteria, Gemmatimonadetes, Planctomycetes, Proteobacteria, Spirochaetes, Synergistetes, Thermotogae, Tenericutes, unclassified, Verrucomicrobia, candidate division TM7, candidate division OD1, candidate division WS3, candidate division BRC1, and candidate division OP11 (Fig. 3B). Of these, the phylum Proteobacteria was the most abundant phylum in soil A (67.14%) and B (72.08%), and Actinobacteria was dominant in soil C (48.68%). Candidate divisions and unclassified bacteria were only found in soil A.
Only OTUs belonging to Pseudomonas sp. were detected using culturing and both culture-independent methods (PCR-DGGE and MiSeq) in soil B. Species of Bacillus sp. and Brevibacterium sp. were represented in culture-dependent and -independent methods in soil C. Members of the genus Dietzia sp. and Bordetella sp. were also found using both culture-independent techniques in soil C. The similarity of microbial communities among the 3 samples was analyzed with a cluster analysis at the OTU level (Fig. 5A, 5B). Based on OTU presence and abundance, soil A and soil B clustered together, but soil C was distinct from those other soils in both PCR-DGGE and MiSeq.
Fig. 5.

Cluster analysis of bacterial community structure based on OTU presence and abundance in ULO-contaminated soils (A, B, C) derived from PCR-DGGE (A) and MiSeq (B). Bray-Curtis dissimilarity was used to determine clusters
Discussion
In this study, molecular and culture-based techniques were used to investigate the diversity of bacteria potentially involved in ULO biodegradation in contaminated soils. Our findings showed that the three contaminated soils possessed substantial biodegradation potential, as ULO-degrading bacterial counts ranged from 5.97–7.16 log CFU g−1 in investigated soils. Many indigenous hydrocarbon-degrading bacteria can quickly respond to the presence of hydrocarbons and are able to perform biodegradation (Das and Chandran 2011).
Strains related to Enterobacter sp. were isolated as ULO-degrading strains in all three soil samples, indicating they could play an important role in hydrocarbon degradation as many Enterobacter sp. have been reported to possess hydrocarbon-degrading capabilities (Jain et al. 2010; Khorasani et al. 2013). For example, Enterobacter cloacae has been associated with the degradation of hexadecane, naphthalene, anthracene and phenanthrene in minimal medium broth with PAHs mixture and using the 2, 6-DCPIP assay (Bučková et al. 2013).
The ULO extracted from three soil samples was characterized by the presence of hydrocarbons ranging from C13 to C28. These hydrocarbon profiles indicate that the ULO in soil C is less weathered, based on the variety and higher amount of low molecular weight aliphatic hydrocarbons components compared to soils A and B (Fig. 1C). Soils A and B have profiles consistent with weathered ULO, indicating that they have been contaminated longer than soil C. Aliphatic hydrocarbons lower than C13 were not observed in any of the samples, potentially due to physiochemical processes such as photo-oxidation, volatilization and evaporation (Ezekoye et al. 2015). Some compounds typically present in ULOs were undetected in all samples such as C21, which might be due to partial decomposition of the oil during its use in the engine (Song et al. 2010).
Soil A was classified as loamy sand and was dominated by sand particles, but still contained a high percentage of silt and clay which can be important for maintaining soil moisture and nutrients. Although sandy soils tend to have lower nutrient retention, organic matter, and CEC, soil A also had the highest number of heterotrophic and ULO-degrading bacteria (Table 1). Some of the culturable bacteria in this soil were capable of rapid aerobic growth with fresh ULO as the sole carbon source and electron donor in MSM agar, as well as peptone and beef extract as the carbon and nitrogen source in NA agar, yet they may or may not be active in situ. Although some sandy soils may have poor microbial activity due to the very low quantities of nutrients in soil with low concentrations of organic material and clays, this soil texture may have allowed for increased aeration therefore enabling higher rates of hydrocarbon biodegradation (Adams et al. 2013, 2011). In contrast, soil C had the lowest numbers of culturable heterotrophic bacteria and ULO-degrading bacteria but the highest bacterial richness based on PCR-DGGE. The high richness may be due to the soil’s high nutrient retention, organic matter and CEC (Table 1), and also the larger amount and diversity of aliphatic hydrocarbons which may support the growth of diverse taxa (Fig. 1C). Our findings suggest that hydrocarbon composition, soil type, and time affect the microbial community structure after hydrocarbon contamination in soil, and these factors are important determinants of the diversity and number of hydrocarbon-degrading bacteria.
Our results with both PCR-DGGE and MiSeq techniques showed that Proteobacteria was the major bacterial phylum present in soil A and B and was a minor group in soil C. This is generally consistent with previous studies that have reported Proteobacteria as the most abundant phylum in hydrocarbon-contaminated soils, based on both culture-dependent methods and culture-independent methods (Mahjoubi et al. 2013). The same dominant bacterial groups were detected in soils with different physiochemical properties such as organic carbon content, CEC, or nutrients (Tab. 1), as previously reported by Will et al. (2010). The phyla Firmicutes, Actinobacteria and Proteobacteria were the main bacterial community members in soil C based on both PCR-DGGE and MiSeq techniques. Members of the phyla Firmicutes, Actinobacteria and Proteobacteria have also been reported to have potential for in-situ degradation of hydrocarbon compounds (Yang et al. 2014). Candidate divisions (TM7, OD1, WS3, BRC1, and OP11) and unclassified organisms were also found in this study using Illumina MiSeq. This direct molecular approach successfully detected previously uncultured organisms belonging to candidate divisions, as has been previously observed (Yang et al. 2015). All of these candidate divisions have been detected in environmental samples such as sediments and bioaerosols of a coal mine (Basak et al. 2014; Wei et al. 2015).
Newer molecular methods such as HTS of 16S rRNA amplicons can overcome some limitations of PCR-DGGE, such as detection of non-dominant taxa and deeper sequencing of complex microbial systems. While previous studies have applied HTS to characterize crude oil contaminated soils (Al-Kindi and Abed 2016), this study is the first to characterize the microbial community in tropical ULO-contaminated soil using combined fingerprinting and Illumina MiSeq techniques. The increased sequencing depth associated with HTS allows for the identification of rare bacteria and provided access to the genetic diversity of microbial assemblages in these ULO-contaminated soils. The combination of culturing and molecular techniques allowed for a broad examination of the investigated soils and helped to decrease of some methodological limitations associated with the individual technologies.
In this study we investigated the diversity and identity of ULO-degraders and the environmental factors that affect bioremediation potential and total microbial communities in ULO-contaminated soil. Culturable ULO-degrading bacteria were present in ULO-contaminated soils, indicating that these soils have ULO-degrading potential. These strains should be subjected to further testing to confirm their biodegradation abilities. ULO-degraders often are very slow to degrade oil in ULO-contaminated sites in the environment, possibly due to limitations in environmental conditions. Methods are needed to enhance the biodegradation process. This study suggests that, because indigenous ULO-degraders are present, biostimulation methods such as nutrient additions or biosurfactants may support and accelerate biodegradation rates in these and other similar contaminated soils. Biostimulation methods have been shown to increase the degradative activities of indigenous bacterial populations and reduce time required in other soils.
Conclusion
In this study, bacterial community structure differed alongside physiochemical soil parameters and ULO weathering. Soil C showed the highest richness, which may be a result of higher nutrient retention, organic matter and CEC, as well as freshness of oil compared to the other soils. Overall, our results with culture-independent and culture-dependent methods indicated that the phylum, Proteobacteria, which includes petroleum degraders, was present and abundant in all investigated soils. Enterobacter sp. were cultivated on MSM agar containing ULO as the sole carbon source from all soil samples, suggesting that this strain may be widespread in contaminated soils from this region of Thailand and potentially important to ULO biodegradation. The results, obtained through a combination of culture-dependent and independent techniques applied here, may be useful for monitoring natural attenuation and developing strategies to accelerate ULO bioremediation.
Acknowledgments
This work was supported by the grants from the Strategic Scholarships Fellowships Frontier Research Networks (specific for Southern region), Office of the Higher Education Commission, Thailand (Grant no. 020/2553) and Prince of Songkla University (Grant no. AGR590252S). Additional support was provided by NSF DEB-1257424 (MBL and MCL), the M.J. Murdock Charitable Trust, and by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103395. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- Adams RH, Kanga-Leyva K, Guzmán-Osorio FJ, Escalante-Espinosa E. Comparison of moisture management methods for the bioremediation of hydrocarbon contaminated soil. Afr J Biotechnol. 2011;10(3):394–404. doi: 10.5897/AJB09.1868. [DOI] [Google Scholar]
- Adil E. Corrective measures of denaturing gradient gel electrophoresis limitations. J Environ Sci Technol. 2015;8(1):1–12. doi: 10.3923/jest.2015.1.12. [DOI] [Google Scholar]
- Ali SMW, Ripin A, Ahmad A. Adsorption of heavy metal from recovered base oil using zeolite. J Appl Sci. 2010;10(21):2688–2692. [Google Scholar]
- Al-Kindi S, Abed RMM. Effect of biostimulation using sewage sludge, soybean meal, and wheat straw on oil degradation and bacterial community composition in a contaminated desert soil. Front Microbiol. 2016;7:1–14. doi: 10.3389/fmicb.2016.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anyasi RO, Atagana HI. Biological remediation of polychlorinated biphenyls (PCB) in the environment by microorganisms and plants. Afr J Biotechnol. 2011;10:18916–18938. doi: 10.5897/AJB10.557. [DOI] [Google Scholar]
- Aucélio RQ, de Souza RM, de Campos RC, Miekeley N, de Silveira CLP. The determination of trace metals in lubricating oils by atomic spectrometry. Spectrochim Acta B. 2007;62:952–961. [Google Scholar]
- Basak P, Majumder NS, Nag S, Bhattacharyya A, Roy D, Chakraborty A, SenGupta S, Roy A, Mukherjee A, Pattanayak R, Ghosh A, Chattopadhyay D, Bhattacharyya M. Spatiotemporal analysis of bacterial diversity in sediments of Sundarbans using parallel 16S rRNA gene tag sequencing. Microb Ecol. 2014;69(3):500–511. doi: 10.1007/s00248-014-0498-y. doi:101007/s00248-014-0498-y. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Biswas D. Enhancement of waste engine oil biodegradation by optimization of media using factorial design study. Indian J Biotechnol. 2014;13:293–300. [Google Scholar]
- Brady NC. The nature and properties of soils. 8. Macmillan Publishing Company, Inc; New York: 1974. [Google Scholar]
- Bray JR, Curtis JT. An ordination of upland forest communities of southern Wisconsin. Ecol Monogr. 1957;27(4):325–349. [Google Scholar]
- Bremner JM. Methods of soil analysis, Part 2. Soil Science Society of America; Madison, WI: 1982. Extraction of exchangeable ammonium, nitrate, and nitrite. [Google Scholar]
- Bučková M, Puškarová A, Chovanová K, Kraková L, Ferianc P, Pangallo D. A simple strategy for investigating the diversity and hydrocarbon degradation abilities of cultivable bacteria from contaminated soil. World J Microbiol Biotechnol. 2013;29:1085–1098. doi: 10.1007/s11274-013-1277-5. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS. 2011;108(suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dąbrowska H, Dabrowski L, Biziuk M, Gaca J, Namiesnik J. Solid-phase extraction clean-up of soil and sediment extracts for the determination of various types of pollutants in a single run. J Chromatogr A. 2003;1003:29–42. doi: 10.1016/S0021-9673(03)00849-5. [DOI] [PubMed] [Google Scholar]
- Das N, Chandran P. Microbial degradation of petroleum hydrocarbon contaminants: An overview. Biotechnol Res Inter. 2011:1–13. doi: 10.4061/2011/941810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day PR. Methods of soil analysis, Part 1. Soil Science Society of America; Madison, WI: 1965. Particle fractionation and particle-size analysis, hydrometer method. [Google Scholar]
- Dike BU, Okoro BC, Nwakwasi NN, Agbo KC. Remediation of used motor engine oil contaminated soil: A soil washing treatment approach. J Civil Environ Eng. 2013;3(1):1–3. doi: 10.4172/2165-784X1000129. [DOI] [Google Scholar]
- Ercolini D, Moschetti G, Blaiotta G, Coppola S. The potential of a polyphasic PCR-DGGE approach in evaluating microbial diversity of natural whey cultures for water-buffalo mozzarella cheese production: Bias of culture-dependent and culture-independent analyses. Syst Appl Microbiol. 2001;24:610–617. doi: 10.1078/0723-2020-00076. [DOI] [PubMed] [Google Scholar]
- Ezekoye CC, Amakoromo ER, Ibiene AA. Bioremediation of hydrocarbon polluted mangrove swamp soil from the Niger Delta using organic and inorganic nutrients. Br Biotechnol J. 2015;6(2):62–78. doi: 10.9734/BBJ/2015/15083. [DOI] [Google Scholar]
- Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2:1–7. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haritash AK, Kaushik CP. Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs): A review. J Hazard Mater. 2009;169:1–15. doi: 10.1016/j.jhazmat.2009.03.137. [DOI] [PubMed] [Google Scholar]
- Hernandez RR, Debenport SJ, Leewis M-CCE, Ndoye F, Nkenmogne KIE, Soumare A, Thuita M, Gueye M, Miambi E, Chapuis-Lardy L, Diedhiou I, Dick RP. The native shrub, Piliostigma reticulatum, as an ecological “resource island” for mango trees in the Sahel. Agric Ecosyst Environ. 2015;204:51–61. org/10.1016/j.agee.2015.02.009. [Google Scholar]
- Holmgren GS, Juve RL, Geschwender RC. A mechanically controlled variable rate leaching device. Soil Sci Soc Am J. 1977;41:1207–1208. [Google Scholar]
- Jackson ML. Soil chemical analysis. Prentice-Hall; Englewood Cliffs, NJ, USA: 1958. [Google Scholar]
- Jain PK, Gupta VK, Pathak HP, Lowry M, Jaroli DP. Characterization of 2T engine oil degrading indigenous bacteria, isolated from high altitude (Mussoorie), India. World J Microbiol Biotechnol. 2010;26:1419–1426. doi: 10.1007/s11274-010-0316-8. [DOI] [Google Scholar]
- Kao C-M, Chen CS, Tsa F-Y, Yang K-H, Chien C-C, Liang S-H, Yang C, Chen SC. Application of real-time PCR, DGGE fingerprinting, and culture-based method to evaluate the effectiveness of intrinsic bioremediation on the control of petroleum-hydrocarbon plume. J Hazard Mater. 2010;178:409–416. doi: 10.1016/j.jhazmat.2010.01.096. [DOI] [PubMed] [Google Scholar]
- Khan FI, Husain T, Hejazi R. An overview and analysis of site remediation technologies. J Environ Manage. 2004;71:95–122. doi: 10.1016/j.jenvman.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Khorasani AC, Mashreghi M, Yaghmaei S. Study on biodegradation of Mazut by newly isolated strain Enterobacter cloacae BBRC10061: improving and kinetic investigation. Iranian J Environ Health Sci Eng. 2013;10(2):1–7. doi: 10.1186/1735-2746-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS, Chun BH, Kim SH. Non-isothermal pyrolysis of waste automobile lubricating oil in a stirred batch reactor. Chem Eng J. 2003;93:225–231. [Google Scholar]
- Koma D, Hasumi F, Yamamoto E, Ohta T, Chung S-Y, Kubo M. Biodegradation of long-chain n-paraffins from waste oil of car engine by Acinetobacter sp. J Biosci Bioeng. 2001;91:94–96. doi: 10.1263/jbb.91.94. [DOI] [PubMed] [Google Scholar]
- Mahjoubi M, Jaouani A, Guesmi A, Amor SB, Jouini A, Cherif H, Najjari A, Boudabous A, Koubaa N, Cherif A. Hydrocarbonoclastic bacteria isolated from petroleum contaminated sites in Tunisia: isolation, identification and characterization of the biotechnological potential. New Biotechnol. 2013;30(6):723–733. doi: 10.1016/j.nbt.2013.03004. [DOI] [PubMed] [Google Scholar]
- Michaelson GJ, Ping CL, Mitchell GA. Correlation of Mehlich 3, bray 1, and ammonium acetate extractable P, K, Ca, and Mg for Alaska agricultural soils 1. Commun Soil Sci Plant Anal. 1987;18:1003–1015. [Google Scholar]
- Muyzer G, Smalla K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek. 1998;73:127–141. doi: 10.1023/a:1000669317571. [DOI] [PubMed] [Google Scholar]
- Naladta A, Milintawisamai N. Isolation of motor oil degrading-microorganism from contaminated soil. In: Svasti J, Pinitglang S, Dejkriengkraikul PL, editors. Proceedings of the third international conference on biochemistry and molecular biology; Chiang Mai, Thailand. 2011. pp. 247–250. [Google Scholar]
- Navarro-Noya Y, Hernández-Rodríguez C, Zenteno JC, Buentello-Volante B, Cancino-Díaz ME, Jan-Roblero J, Cancino-Díaz JC. 16S rRNA gene-based identification of bacteria in postoperative endophthalmitis by PCR denaturing gradient gel electrophoresis (PCR-DGGE) fingerprinting. Braz J Microbiol. 2012:283–287. doi: 10.1590/S1517-838220120001000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peech M. Methods of soil analysis, Part 1. Soil Science Society of America; Madison, WI: 1982. Hydrogen ion activity. [Google Scholar]
- Pimda W, Bunnag S. Biodegradation of used motor oil by Nostoc piscinale TISTR 8401. Afr J Microbiol Res. 2012;6(10):2367–2372. doi: 10.5897/AJMR11.1378. [DOI] [Google Scholar]
- Roling WFM, Milner MG, Jones DM, Lee K, Daniel F, Swannell RJP, Head IM. Robust hydrocarbon degradation and dynamics of bacterial communities during nutrient-enhanced oil spill bioremediation. Appl Environ Microbiol. 2002;68(11):5537–5548. doi: 10.1128/AEM.68.11.5537-5548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saimmai A, Kaewrueng J, Maneerat S. Used lubricating oil degradation and biosurfactant production by SC-9 consortia obtained from oil-contaminated soil. Ann Microbiol. 2012;4:1757–1767. doi: 10.1007/s13213-012-0434-7. [DOI] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Horn DJV, Weber CF. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahriar M, Haque R, Kabir S, Dewan I, Bhuyian MA. Effect of proteinase-K on genomic DNA extraction from gram-positive strains. Stamford J Pharm Sci. 2011;4(1):53–57. [Google Scholar]
- Song G-J, Seo Y-C, Pudasainee D, Kim I-T. Characteristics of gas and residues produced from electric arc pyrolysis of waste lubricating oil. Waste Manage. 2010;30:1230–1237. doi: 10.1016/j.wasman.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Sperling D, Gordon D. Two Billion Cars: Driving Toward Sustainability. Oxford: Oxford University Press; 2009. [Google Scholar]
- Stewart EJ. Growing unculturable bacteria. J Bacteriol. 2012;194(16):4151–4160. doi: 10.1128/JB.00345-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Grundmann S, Schmid M, Dörfler U, Roherer S, Munch JC, Hartmann A, Jiang X, Schroll R. Isolation and characterization of 1,2,4-trichlorobenzene mineralizing Bordetella sp and its bioremediation potential in soil. Chemosphere. 2007;67:896–902. doi: 10.1016/j.chemosphere.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Wei M, Yu Z, Zhang H. Molecular characterization of microbial communities in bioaerosols of a coal mine by 454 pyrosequencing and real-time PCR. J Environ Sci. 2015;30:241–251. doi: 10.1016/j.jes.2014.07.035. [DOI] [PubMed] [Google Scholar]
- Will C, Thürmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F, Daniel R. Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol. 2010;76(20):6751–6759. doi: 10.1128/AEM.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wen X, Zhao L, Shi Y, Jin H. Crude oil treatment leads to shift of bacterial communities in soils from the deep active layer and upper permafrost along the China-Russia crude oil pipeline route. PLOS ONE. 2014;9(5):1–10. doi: 10.1371/journal.pone.0096552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Li Y, Zhou B, Zhou Y, Zheng W, Tian Y, Van Nostrand JD, Wu L, He Z, Zhou J, Zheng T. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen sea, China. Sci Rep. 2015;5(8476):1–11. doi: 10.1038/srep08476. [DOI] [PMC free article] [PubMed] [Google Scholar]