

Abstract
Myeloid differentiation protein 1 (MD1) has been implicated in numerous pathophysiological processes, including immune regulation, obesity, insulin resistance, and inflammation. However, the role of MD1 in cardiac remodelling remains incompletely understood. We used MD1-knockout (KO) mice and their wild-type littermates to determine the functional significance of MD1 in the regulation of aortic banding (AB)-induced left ventricular (LV) structural and electrical remodelling and its underlying mechanisms. After 4 weeks of AB, MD1-KO hearts showed substantial aggravation of LV hypertrophy, fibrosis, LV dilation and dysfunction, and electrical remodelling, which resulted in overt heart failure and increased electrophysiological instability. Moreover, MD1-KO-AB cardiomyocytes showed increased diastolic sarcoplasmic reticulum (SR) Ca2+ leak, reduced Ca2+ transient amplitude and SR Ca2+ content, decreased SR Ca2+-ATPase2 expression, and increased phospholamban and Na+/Ca2+-exchanger 1 protein expression. Mechanistically, the adverse effects of MD1 deletion on LV remodelling were related to hyperactivated CaMKII signalling and increased impairment of intracellular Ca2+ homeostasis, whereas the increased electrophysiological instability was partly attributed to exaggerated prolongation of cardiac repolarisation, decreased action potential duration alternans threshold, and increased diastolic SR Ca2+ leak. Therefore, our study on MD1 could provide new therapeutic strategies for preventing/treating heart failure.
Introduction
Hypertension is the single most critical risk factor for heart failure (HF)1. High blood pressure is widely recognised to induce left ventricular (LV) hypertrophy and lead initially to ventricular wall thickening and stiffening, a process which is compensatory and adaptive in nature. However, sustained pressure overload contributes to maladaptive LV remodelling, progressive LV dilatation, and cardiac dysfunction2–4, and this results in arrhythmias and HF3, 5, 6, a major underlying cause of increased cardiovascular morbidity and mortality5, 7. Despite being the focus of substantial research effort in recent decades, the precise pathogenesis of maladaptive LV remodelling and the mechanisms that determine how long-standing hypertrophy ultimately progresses to HF remain unclear8. Therefore, enhanced understanding of the factors and mechanisms that modulate pathological LV remodelling could lead to novel strategies for the treatment of HF.
Evidence gathered over the past two decades has shown that Toll-like receptor 4 (TLR4) signalling is involved in several aspects of the cardiac pathological process, such as cardiac remodelling, ischaemia/reperfusion injury, hypertension, and atherosclerosis9–13. Upon stimulation, TLR4 signalling ultimately activates numerous signalling pathways14, 15, including the MAPK pathway, NF-κB pathway, and PI3K/Akt pathway. Furthermore, a link between Ca2+/calmodulin-dependent kinase II (CaMKII) signalling and TLR4 signalling has been clearly demonstrated16–18. More importantly, therapies targeting against TLR4 have shown effectiveness in attenuating murine cardiac remodelling caused by pressure overload19–21. Furthermore, a recent study demonstrated that stimulation of TLR4 in rat ventricular cardiomyocytes in vitro promoted an electrical remodelling that led to action potential duration (APD) prolongation associated with delayed afterdepolarisation and triggered activity22.
The aforementioned data led us to investigate whether a naturally occurring molecule can inhibit LV remodelling and reduce the susceptibility to ventricular tachycardia (VT) during chronic pressure overload by blocking TLR4 signalling. We speculated that a favourable candidate molecule might be myeloid differentiation protein 1 (MD1), an endogenous negative modulator of TLR4 signalling23. MD1 is expressed predominantly in B cells, macrophages, dendritic cells, and other immune cells23, 24. MD1 forms a complex with radioprotective protein 105 (RP105), which is abundantly present in heart tissue. MD1-RP105 complex can directly interact with the MD2-TLR4 complex by a lateral binding, acting as physiological negative regulators of TLR4 signalling25. Furthermore, recent evidence suggests that MD1-RP105 complex is associated with several pathophysiological processes, including immune regulation, obesity, insulin resistance, and inflammation26–28. However, to the best of our knowledge, no previous study has reported whether MD1 regulates sustained pressure overload-induced LV structural and electrical remodelling.
Here, using loss-of-function approach, we discovered that pressure overload-induced LV remodelling was aggravated in MD1-knockout (KO) mice, which led to overt HF and increased electrophysiological instability. Whereas these adverse effects of MD1 deletion on LV remodelling are related to the hyperactivation of CaMKII signalling and an exaggerated impairment of intracellular Ca2+ homeostasis, the increased electrophysiological instability is at least partly due to an increased prolongation of cardiac repolarisation, enhanced reduction of the APD alternans threshold, and exaggerated increase of diastolic sarcoplasmic reticulum (SR) Ca2+ leak.
Results
MD1 expression is down-regulated in the left ventricles of DCM patients and in failing left ventricles in mice
To explore the correlation between MD1 and LV remodelling, we first examined MD1 expression in the failing left ventricles. Western blot analysis revealed that MD1 protein levels were significantly lower in the heart samples of DCM patients than in those of normal donors (Fig. 1A). Moreover, the diminished MD1 protein levels were accompanied with increased mRNA levels of brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) and decreased ejection fraction (EF) values (Fig. 1B,C). Similarly, MD1 protein levels, EF values, and fractional shortening (FS) values in wild-type (WT) mice at 4 weeks after aortic banding (AB) were significantly reduced as compared with the corresponding values in the sham-operated group (Fig. 1D–F). These results suggest that MD1 might be involved in LV remodelling.
Figure 1.
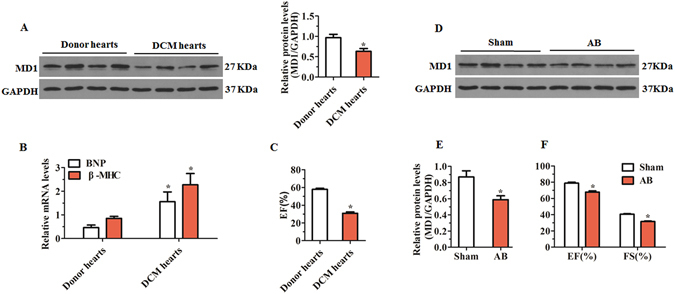
MD1 expression in left ventricles of human and mouse failing heart. (A) Representative western blots of MD1 in heart samples from normal donors (n = 7) and DCM patients (n = 8). (B) qRT-PCR analyses of BNP and β-MHC mRNA in LV tissues of normal donors (n = 7) and DCM patients (n = 8). (C) EF values of normal (n = 7) and DCM hearts (n = 8). (D,E) Representative western blots of MD1 and (F) EF/FS values in WT mouse left ventricles at 4 weeks after sham or AB operation (n = 4). *P < 0.05 vs. normal donors or shams.
Deletion of MD1 exacerbates pressure overload-induced LV structural remodelling
The absence of MD1 in the MD1-KO mouse heart was confirmed through Western blotting (Fig. 2A). Under basal conditions, MD1-KO mice showed no alterations in cardiac phenotype (data not shown). However, at 4 weeks after AB, MD1-KO mice exhibited a marked deterioration of LV hypertrophy as compared with their WT littermates, which was confirmed by the measurement in the KO mice of a relatively larger cardiomyocyte cross-sectional area (CSA), revealed by haematoxylin-eosin (H&E) staining (Fig. 2B,C), and higher ratios of heart weight (HW)/body weight (BW), HW/tibia length (TL), and lung weight (LW)/TL (Fig. 2D–F; Supplementary Table S1). Consistently, the mRNA levels of foetal genes (BNP, β-MHC) were markedly higher in the LV tissues of MD1-KO mice than in those of WT mice (Fig. 2G). Furthermore, MD1-KO mice exhibited exaggerated LV dilation and dysfunction, as shown by measurements of the following echocardiographic and haemodynamic parameters: LV end-diastolic diameter (LVEDD), LV end-systolic diameter (LVESD), LVEF, LVFS, LV end-systolic pressure (LVESP), and the maximum and minimum rates of LV pressure development (dp/dt max and dp/dt min, respectively) (Fig. 2H; Supplementary Table S1 and Fig. S2). Cardiac fibrosis, a major feature of maladaptive cardiac remodelling, was more prominent in MD1-KO-AB mice than in WT-AB mice (Fig. 2I,J). Collectively, these data indicate that MD1 loss exacerbates pressure overload-induced maladaptive LV structural remodelling.
Figure 2.
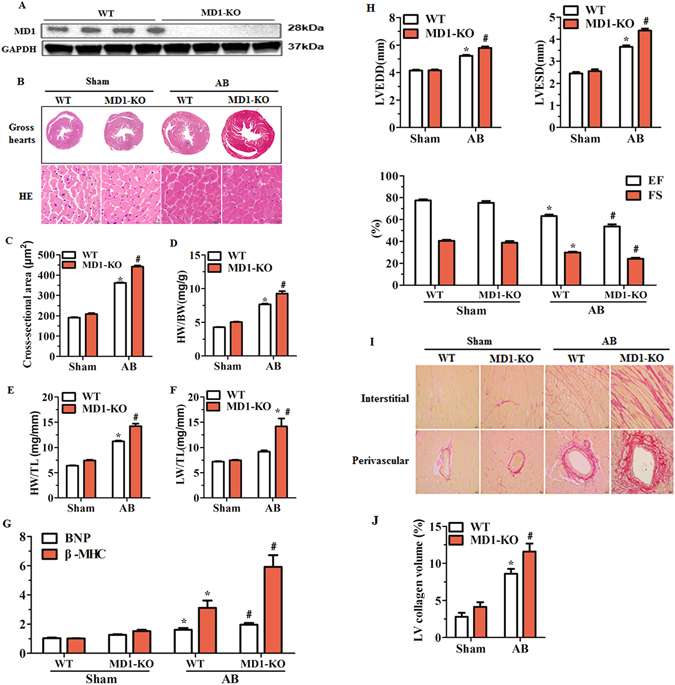
Loss of MD1 aggravates pressure overload-induced LV structural remodelling. (A) Representative western blots of MD1 expression in LV tissues from WT and MD1-KO mice (n = 6). (B) Gross hearts and H&E staining performed at 4 weeks after surgery (n = 7–8). (C) Statistical results of LV myocyte cross-sectional areas (n = 200+ cells). (D–F) HW/BW, HW/TL, and LW/TL values of the indicated groups (n = 13–14). (G) mRNA levels of the hypertrophy markers BNP and β-MHC in WT and MD1-KO left ventricles at 4 weeks after surgery, determined using qRT-PCR (n = 5). (H) Echocardiographic results of the indicated groups (n = 13–14). (I) PSR staining of histological sections prepared from LV samples of WT and MD1-KO mice at 4 weeks after surgery (n = 7–8). (J) Fibrotic areas from histological sections quantified using an image-analysis system (n = 26–28 fields). *P < 0.025 vs. WT-Sham, # P < 0.05 vs. WT-AB.
Absence of MD1 alters surface electrocardiogram (ECG) parameters during chronic pressure overload
Surface ECG (lead II) recordings were performed on mice under light anaesthesia. Mice from the groups indicated in Fig. 3 showed similar electrocardiographic PR intervals, but MD1-KO-AB mice showed shorter RR intervals and longer QRS intervals than in WT-Sham mice (Fig. 3A,B; Supplementary Table S2). The QRS intervals of MD1-KO-AB mice were significantly prolonged when compared with those of WT-AB mice (Fig. 3A,B; Supplementary Table S2). As compared with WT-Sham mice, both WT-AB and MD1-KO-AB mice exhibited prolonged QTc (corrected QT) intervals, but this prolongation was significantly higher in the KO than in the WT mice (Fig. 3A,B; Supplementary Table S2).
Figure 3.
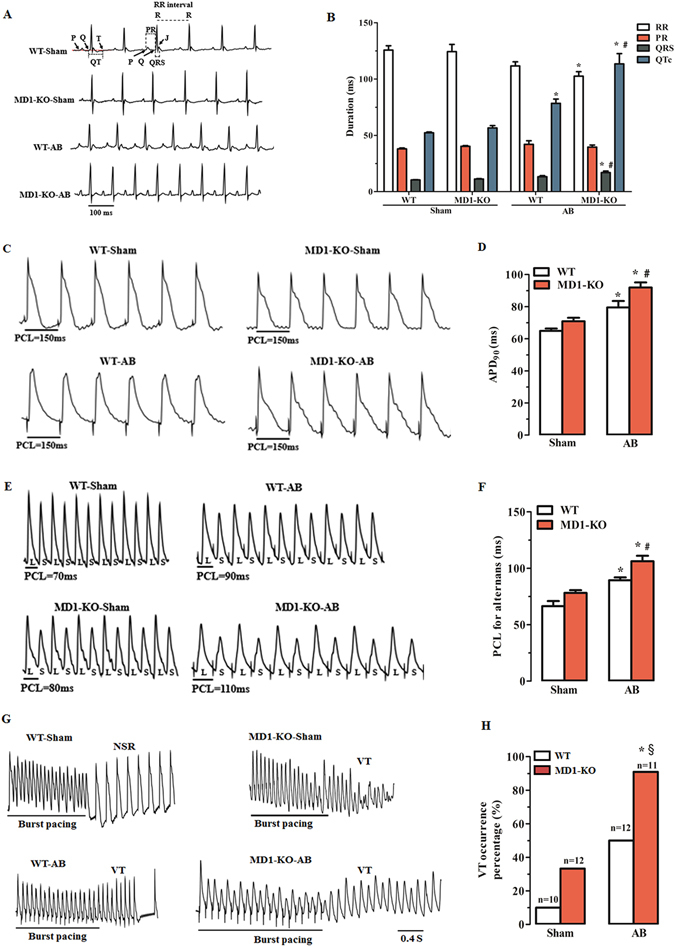
Deletion of MD1 alters surface ECG parameters and exacerbates pressure overload-induced LV electrical remodelling. (A) Examples of surface ECG recordings and (B) summary of surface ECG parameters from WT and MD1-KO mice at 4 weeks after surgery (n = 7–8). (C) Representative MAP recordings at a PCL of 150 ms and (D) statistical analysis of APD90 from Langendorff-perfused WT and MD1-KO hearts at 4 weeks after surgery (n = 8–9). (E) Representative MAP recordings of APD alternans and (F) statistical analysis of the threshold interval for APD alternans from the indicated groups (n = 7–8). (G) Examples of MAP recordings after burst pacing and (H) summary of VT inducibility rates in Langendorff-perfused WT and MD1-KO hearts at 4 weeks after surgery (n is indicated above the bar graphs). PCL = pacing cycle length; L = longer APD; S = shorter APD; NSR = normal sinus rhythm. *P < 0.03 vs. WT-Sham, § P < 0.01 vs. MD1-KO-Sham, # P < 0.05 vs. WT-AB.
Disruption of MD1 exacerbates pressure overload-induced LV electrical remodelling
Langendorff-perfused hearts and the monophasic action potential (MAP) recording technique were used to determine the changes in three electrophysiological parameters: APD90, threshold interval for APD alternans, and VT inducibility rate. After 4 weeks of AB, APD90 was markedly prolonged in both WT-AB and MD1-KO-AB mice, but the prolongation was considerably greater in the KO mice than in the WT mice (Fig. 3C,D; Supplementary Table S2). Among the four groups tested here, the changes in the threshold interval for APD alternans (Fig. 3E,F; Supplementary Table S2) were similar to the changes in APD90. The VT inducibility rate was potentially higher in MD1-KO-AB mice than in WT-AB mice (90.9% vs. 50%, Fig. 3G,H), although there is no significant difference (P = 0.069).
MD1 regulates TLR4 signalling and CaMKII signalling in response to pressure overload
We applied Western blotting to investigate the effects of MD1 loss on TLR4 signalling and CaMKII signalling after 4 weeks of AB. Our results showed that a 4-week course of AB significantly decreased RP105 expression, and increased MD2 and TLR4 expression in both WT and MD1-KO LV tissues (Fig. 4A). However, the changes of RP105, MD2 and TLR4 expression were more pronounced in MD1-KO-AB mice (P < 0.05 vs. WT-AB; Fig. 4A), suggesting a hyperactivation of TLR4 signalling. Similarly, the CaMKII signalling was activated to a higher level in MD1-KO-AB mice than in WT-AB mice; this was revealed by the increased levels of oxidised-CaMKII (ox-CaMKII), total CaMKII expression, and phosphorylation of CaMKIIβ + γ + δ in the KO mice (Fig. 4B).
Figure 4.
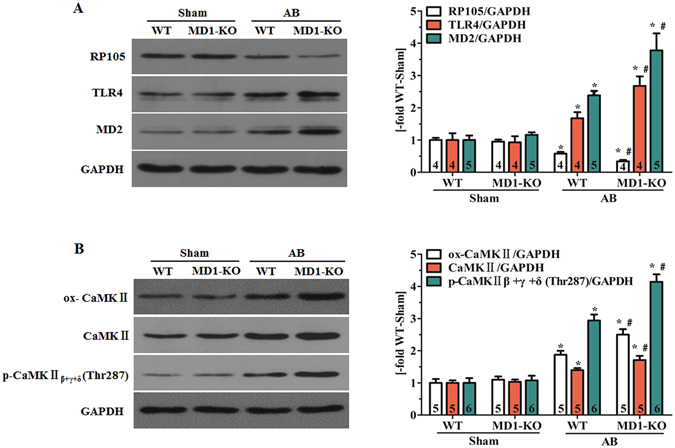
MD1 regulates TLR4 signalling and CaMKII signalling in response to pressure overload. Western blot analysis of WT and MD1-KO left-ventricle samples at 4 weeks after surgery for (A) RP105, MD2, and TLR4 expression; (B) ox-CaMKII and total CaMKII expression, and phosphorylation of CaMKIIβ + γ + δ at Thr287. Left: representative immunoblots; right: quantitative results. Data were normalised to GAPDH. Numbers of mice per group are shown inside bars. *P < 0.05 vs. WT-Sham, # P < 0.05 vs. WT-AB.
MD1 modulates intracellular Ca2+ handling in response to pressure overload
At baseline, the sham-operated WT and MD1-KO mice displayed similar Ca2+ transients in LV myocytes (Fig. 5A,C,D). However, the peak amplitude of caffeine-induced Ca2+ transients was lower in MD1-KO-Sham mice than WT-Sham mice (P < 0.05; Fig. 5A,C). After 4 weeks of AB, the LV myocytes of MD1-KO-AB mice exhibited a significantly reduced peak amplitude of Ca2+ transients and further reduced SR Ca2+ content (P < 0.05 vs. WT-AB; Fig. 5A,C), as well as prolonged time to peak [Ca2+]i transient amplitude and duration of [Ca2+]i transient decay (P < 0.03 vs. WT-AB; Fig. 5A,D).
Figure 5.
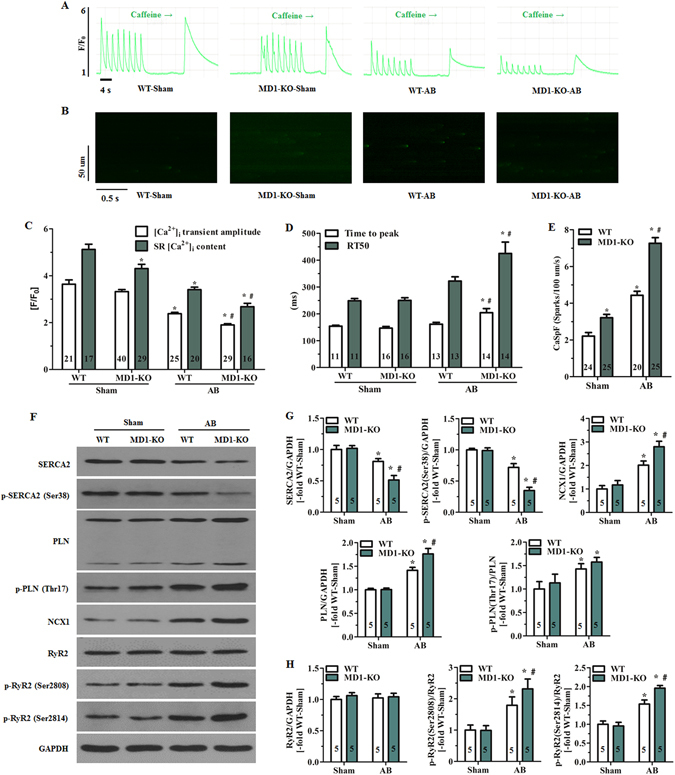
MD1 modulates intracellular Ca2+ handling in response to pressure overload. (A) Representative recordings (F/F0) of steady-state Ca2+ transients followed by rapid caffeine administration and (B) confocal line-scan images of mouse LV myocytes. Cells were isolated from WT and MD1-KO mice at 4 weeks after surgery. (C) Statistical analysis of systolic Ca2+ transient amplitude and SR Ca2+ content in the indicated groups (5–6 mice/group). (D) Time to peak [Ca2+]i transient amplitude and duration of [Ca2+]i transient decay (4–5 mice/group). RT50 = half-maximal relaxation time. (E) Quantification of CaSpF in the indicated groups (3–4 mice/group). (F) Original immunoblots of major Ca2+ handling proteins (including total and phosphorylated proteins). Statistical analysis of WT and MD1-KO heart samples at 4 weeks after surgery for (G) expression of total SERCA2, total PLN, and NCX1, phosphorylation of SERCA2 at Ser38, and PLN phosphorylation at Thr17 normalised to total PLN; (H) total RyR2 expression, and RyR2 phosphorylation at Ser2808 and Ser2814 normalised to total RyR2. Data were normalised to GAPDH. Numbers within columns indicate the number of LV myocytes (C–E) and heart samples (G,H). *P < 0.03 vs. WT-Sham, # P < 0.05 vs. WT-AB.
We also examined whether MD1 loss increases RyR2-mediated diastolic SR Ca2+ leak, measured as Ca2+ sparks (Fig. 5B), in mouse LV myocytes. The mean Ca2+ spark frequency (CaSpF) values revealed that diastolic SR Ca2+ leak was markedly higher in MD1-KO-Sham myocytes than in WT-Sham myocytes (3.21 ± 0.18 vs. 2.21 ± 0.19 sparks/100 μm/s, P < 0.05; Fig. 5E). Moreover, at 4 weeks after AB, the CaSpF increase was greater in MD1-KO mice than in WT mice (7.26 ± 0.31 vs. 4.43 ± 0.22 sparks/100 μm/s, P < 0.001; Fig. 5E).
Under baseline conditions, the expression and phosphorylation of major Ca2+ handling proteins (Fig. 5F–H), and the expression of Cav1.2, calsequestrin 2 (CASQ2), and calcineurin A (Supplementary Fig. S3) were similar in WT and MD1-KO hearts. After 4 weeks of AB, SR Ca2+ ATPase 2 (SERCA2) expression and SERCA2 phosphorylation were drastically decreased, whereas the expression of Na+/Ca2+ exchanger 1 (NCX1) and phospholamban (PLN) were significantly increased in both MD1-KO and WT hearts, although these changes were considerably greater in MD1-KO hearts than in WT hearts (Fig. 5F,G). Moreover, PLN phosphorylation was increased, markedly and to a similar extent, in both MD1-KO-AB and WT-AB hearts (Fig. 5F,G). Phosphorylation of cardiac ryanodine receptor (RyR2) (Fig. 5F,H) and the expression of CASQ2 and calcineurin A (Supplementary Fig. S3) were markedly increased, whereas Cav1.2 expression (Supplementary Fig. S3) was significantly decreased in both WT and MD1-KO hearts by a 4-week course of AB, although these changes were significantly greater in MD1-KO-AB hearts (P < 0.05 vs. WT-AB).
MD1 regulates the expression of K+ and Na+ channels in failing hearts
The K+ and Na+ channels are also associated with electrical remodelling during HF, so we investigated the expression of some K+ and Na+ channels in the mouse left-ventricle samples. Our western blotting results demonstrated that after 4 weeks of AB, both WT and MD1-KO mice showed a significant reduction in protein levels of KCNH2 (Fig. 6A), KCNE1 and KCNQ1 (Fig. 6A), Kv4.2 and Kv4.3 (Fig. 6B), and Nav1.5 (Fig. 6B), which generate IKr, IKs, Ito, and INa currents, respectively. However, this reduction was more pronounced in MD1-KO-AB mice than in WT-AB mice.
Figure 6.
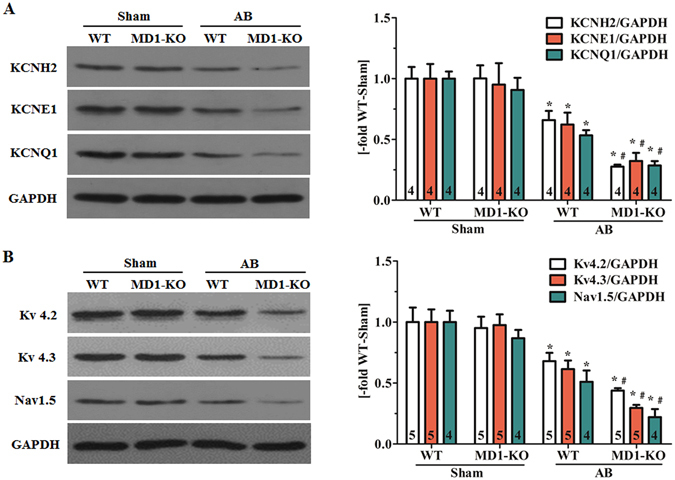
MD1 regulates the expression of K+ and Na+ channels in failing hearts. Immunoblot analysis of WT and MD1-KO left-ventricle samples at 4 weeks after surgery for (A) KCNH2, KCNE1 and KCNQ1 expression; (B) Kv4.2, Kv4.3 and Nav1.5 expression. Left: representative original immunoblots; right: quantitative results. Data were normalized to GAPDH. Numbers of mice per group are shown inside bars. *P < 0.02 vs. WT-Sham, # P < 0.05 vs. WT-AB.
Discussion
Here, we examined the role of MD1 in pressure overload-induced LV structural and electrical remodelling by using loss-of-function strategy. Expression of MD1 is decreased in LV tissues of DCM patients and failing mouse hearts. Deletion of MD1 exaggerated LV pathological hypertrophy, fibrosis, and LV dilation and dysfunction in mice in response to persistent pressure overload, which resulted in overt HF. Moreover, in agreement with recent work22, we discovered that TLR4 activation resulting from a loss of MD1 expression promoted an aggravation of LV electrical remodelling, which enhanced electrophysiological instability and tended to increase the susceptibility to VT in pressure-overloaded mice. To our knowledge, this is the first report to demonstrate a critical role of MD1 in pressure overload-induced LV structural and electrical remodelling. It should be noted, however, that the real function of MD1 in the development of HF in clinical setting needs to be further determined due to the limitation of experimental conditions.
MD1 protects against pressure overload-induced LV remodelling, but the precise molecular mechanisms remain unclear. Previous studies have suggested that TLR4 activation can increase the intracellular concentration of reactive oxygen species16–18, which results in CaMKII oxidation and sustained activation of CaMKII signalling in stressed myocardial tissues16, 18, 29. Abundant evidence indicates that CaMKII is involved in cardiac remodelling and HF. Transgenic overexpression of CaMKIIδ in mouse hearts was shown to alter Ca2+ handling through several mechanisms and cause cardiac remodelling and diseases30, 31. Conversely, CaMKIIδ deletion protects against adverse ventricular remodelling after chronic pressure overload by suppressing the CaMKII/histone deacetylase signalling pathway32, 33, and limits the progression to HF by altering the expression of Ca2+-handling proteins32. In this study, we found that loss of MD1 led to a more pronounced activation of TLR4 signalling in mouse LVs after 4 weeks of AB. Furthermore, we found that MD1 deletion further enhanced the CaMKII signalling by CaMKII oxidation in response to chronic pressure overload. These data suggest that MD1 loss exacerbates pressure overload-triggered LV remodelling, at least partly, through hyperactivation of the CaMKII signalling.
Altered Ca2+ homeostasis in cardiomyocytes is a proximal trigger of stress-induced maladaptive cardiac remodelling8, whereas defective intracellular Ca2+ homeostasis is a central cause of cardiac dysfunction and arrhythmias in failing hearts29. RyR2 hyperphosphorylation by CaMKII causes a diastolic SR Ca2+ leak, which combines with reduced SERCA2-dependent Ca2+ uptake, contribute to a reduction in SR Ca2+ content and an intracellular Ca2+ overload and thus lead to HF34, 35. Our study suggests that MD1 loss increases SR Ca2+ leak, and decreases SR Ca2+ content and Ca2+ transient amplitudes in failing LV myocytes, but this wasn’t the case with the reduction of peak L-type Ca2+ channel current (ICa,L) density (Supplementary Fig. S4A–C). The reduced SR Ca2+ content, a major cause for the severe cardiac dysfunction in MD1-KO-AB mice, was associated with increased SR Ca2+ leak and diminished SERCA2-dependent Ca2+ uptake, and was likely associated with increased expression of NCX1. Unexpectedly, the expression of CASQ2 was also increased. We speculate that this might represent a compensatory response to the increased SR Ca2+ leak: it might contribute to RyR2 deactivation and refractoriness early after Ca2+ release and to the reduced SR Ca2+ buffering36. In agreement with recent studies37–39, our study also suggested that exaggerated RyR2 dysfunction led to a hyperactivation of calcineurin/NFAT signalling pathway, which has been implicated in stress-induced pathological remodelling of the heart6, 8.
Prolongation of cardiac repolarisation is widely recognised to increase the risk of malignant ventricular arrhythmias (VAs) and sudden cardiac death40. QTc interval is a cardiac repolarisation marker and it corresponds to APD at the cellular scale; when QTc interval and APD are prolonged, they become pro-arrhythmic factors40. Consistent with these data, MD1-KO-AB hearts demonstrated prolonged QTc interval and APD, and a tendency of increasing the susceptibility to VT when compared with WT-AB hearts in this study. Moreover, our result agrees with the findings of a recent study suggesting that activation of cardiac TLR4 by lipopolysaccharide markedly increases APD22. Cardiac repolarization is mainly controlled by K+ currents, and CaMKII overactivity can impair K+ channel function and thereby prolong APD29, 41. Therefore, we examined the effect of MD1 deletion on protein expression of KCNH2, KCNE1 and KCNQ1, Kv4.2 and Kv4.3, which generate Ikr, Iks and Ito currents, respectively. Our results suggest that the APD prolongation observed in isolated MD1-KO-AB hearts might partly be through regulating cardiac K+ channels. However, the direct effect of MD1 deletion on Ikr, Iks and Ito currents in failing hearts needs to be further determined.
T-wave alternans (TWA) is a risk marker of cardiac repolarisation that has been associated with VAs40, 42, and interventions that eliminate TWA might prevent VT and sudden death42. TWA corresponds to APD alternans at the cellular level40, 43. Therefore, any factor that decreases the APD alternans threshold might induce VT. Our results showed that MD1 loss decreased the APD alternans threshold in isolated pressure-overload hearts, which suggests that reduction of the APD alternans threshold is one of the mechanisms underlying the MD1 deletion-elicited arrhythmogenic potential in response to pressure overload. A recent literature review revealed that increased SR Ca2+ leak through RyR2 channels and/or decreased SERCA pump activity contributes to alternans43. Consistent with this review’s finding, our study has shown that SERCA2 expression and phosphorylation levels were lower but RyR2 phosphorylation was higher in MD1-KO-AB mice than in WT-AB mice. In addition, alternans can be caused by instabilities originating from voltage and is influenced by INa in electrically remodelled diseased hearts43. INa is affected by CaMKII activity29, 41. Although INa might be reduced as Nav1.5 expression was demonstrated to be markedly decreased in MD1-KO-AB mice in this study, but the activation and kinetic properties of INa remain to be explored in future work.
RyR2 hyperphosphorylation due to CaMKII overactivity is widely accepted to lead to increases in diastolic SR Ca2+ leak, which can cause Ca2+ waves and activate an arrhythmogenic inward Na+/Ca2+ exchange current, causing delayed afterdepolarisations (DADs) and triggering arrhythmias29, 44–46. Here, MD1 loss enhanced the increase in CaMKII activity and CaMKII-mediated RyR2 phosphorylation in response to chronic pressure overload. Moreover, the LV myocytes isolated from MD1-KO-AB mice exhibited an increased arrhythmogenic potential in vitro as compared with WT-AB mice, including enhanced diastolic SR Ca2+ leak, reduced SERCA2 pump activity, and increased NCX1 expression. Growing evidence indicates that increased SR Ca2+ loading is necessary for enhanced RyR2 Ca2+ leak to produce arrhythmia47, but no direct evidence is available to demonstrate that spontaneous Ca2+ sparks arise only when a specific SR cistern becomes ‘overloaded’ and that a lower-than-physiological concentration of Ca2+ in the SR does not affect Ca2+ spark production48. Collectively, these data indicate that the enhanced diastolic SR Ca2+ leak is likely sufficient to contribute to the electrophysiological instability, although the occurrence of spontaneous Ca2+ waves, and DADs needs to be further investigated.
This work has limitations in the study of cardiomyocyte Ca2+ handling. (1) The analytical results of ICa,L inactivation (Supplementary Fig. S4D,E) was not consistent with previous reports that ICa,L decay is slowed in HF49–51. Fitting ICa,L decay using a double exponential equation to calculate the fast and slow decay of ICa,L would be better to analyse ICa,L decay52, and to know whether at the end of the stimulation pulse there is some residual current. (2) The increased NCX1 expression as a factor for the decreased SR Ca2+ content in MD1-KO-AB mice was not confirmed. Although enhanced NCX activity could decrease SR Ca2+ content in HF29, 35, 53, and increased NCX expression was proved to be associated with the decreased SR Ca2+ content in right atrial myocytes isolated from the patients with chronic atrial fibrillation54, additional analysis are required to calculate the contribution of NCX to Ca2+ removal. The way to do that is to fit exponential decay both on the systolic and caffeine transients and calculate rate constants of decay, as previously described54, 55. Due to technical problems and insufficient experience, we couldn’t completely solve these in a short time.
In summary, the present study indicates that MD1 loss exacerbates LV structural and electrical remodelling in response to chronic pressure overload, resulting in overt HF and increased electrophysiological instability. Mechanistically, the adverse effects of MD1 deletion on LV remodelling are associated with hyperactivated CaMKII signalling and increased impairment of intracellular Ca2+ homeostasis. The increased electrophysiological instability is partly due to aggravated electrical remodelling, including prolonged cardiac repolarisation, decreased APD alternans threshold, and increased diastolic SR Ca2+ leak. These findings suggest that endogenous MD1 protects against LV structural and electrical remodelling during long-standing pressure overload. Therefore, MD1 upregulation in the heart might serve as a new therapeutic strategy for the treatment of HF.
Methods
Human heart samples
Samples of human failing hearts were collected from the left ventricles of DCM patients undergoing heart transplants. Control samples were obtained from the left ventricles of normal heart donors who had died as a result of accidents, and whose hearts were not used for transplantation due to non-cardiac reasons. After excision, all specimens were immediately flash-frozen in liquid nitrogen and stored at −80 °C for biochemical analysis. Written informed consent was obtained from the family of prospective heart donors. The study was approved by Renmin Hospital of Wuhan University Review Board and was conducted in accordance with the Declaration of Helsinki.
Mice
Male MD1-KO mice (C57BL/6 background, purchased from RIKEN, RBRC02386) and their WT littermates aged 8–10 weeks were used for experiments. All animals were housed in a 12/12-h light/dark cycle and provided food and water ad libitum. Mice were genotyped by means of PCR performed using the protocol provided by RIKEN (Supplementary Methods and Fig. S1). All animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 1996) and approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University.
Animal model
All surgeries and subsequent analyses were performed in a blinded fashion for all groups. AB surgery and sham operation were performed as described previously56, 57. Doppler analysis was performed to ensure the induction of adequate constriction of the aorta. LV internal diameter and wall thickness were evaluated through echocardiography at various times after surgery. At the end of these procedures, some of the mice of the tested groups were sacrificed using an overdose of pentobarbital sodium (150 mg/kg, intraperitoneal injection), and ratios of HW/BW (mg/g), HW/TL (mg/mm), and LW/TL (mg/mm) of the sacrificed mice from the different groups were assessed. The LV tissues from each heart were dissected, snap-frozen in liquid nitrogen, and stored at −80 °C for biochemical studies.
Pressure-volume relationship measurements and echocardiography
We performed pressure-volume relationship measurements and echocardiography as described previously56, 57 to obtain these values: LVEF, LVFS, LVEDD, LVESD, LVESP, interventricular septum diameter in diastole and systole (IVSd and IVSs, respectively), LV posterior wall diameter in diastole and systole (LVPWd and LVPWs, respectively), and dp/dt max and dp/dt min.
Histological analysis
Hearts were excised and washed with saline solution, arrested in diastole with 10% KCl, fixed in 4% paraformaldehyde solution, and embedded in paraffin. The paraffin-embedded hearts were sectioned transversely at the level of the LV papillary muscles. Several slices (4–5 mm thick) of heart were prepared and stained with H&E for morphometric analysis or picrosirius red (PSR) for evaluation of myocardial fibrosis. All micrographs were acquired using a high-resolution optical microscope. Image-Pro Plus 6.0 software was used to determine the CSA of cardiomyocytes and the volume of LV collagen deposition.
Quantitative real-time PCR (qRT-PCR)
Total RNA from human or mouse LV tissues was extracted and reverse transcribed to generate cDNAs; qRT-PCR was performed by using an ABI-PRISM 7900 Sequence Detection System with the SYBR Color qPCR Master Mix (Vazyme Biotech Co, Nanjing, China). Gene expression was normalised relative to the housekeeping gene GAPDH, and data were analysed according to the 2−∆∆Ct method58. The method details are described in Supplementary Methods.
Western blotting
Total proteins were extracted from frozen LV samples, and a BCA Protein Assay Kit (Beyotime Biotechnology, P0010) was used to determine protein concentrations. Proteins (40 μg) were separated using SDS-PAGE and transferred onto polyvinylidene difluoride membranes, which were then incubated with primary antibodies overnight at 4 °C and subsequently with secondary antibodies for 2 h at room temperature. Chemiluminescent detection was performed using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, NCI5079). Additional details are presented in Supplementary Methods.
Surface ECG recording and analysis
Mice were lightly anaesthetised using inhaled isoflurane (1.5% isoflurane in 98% O2). Surface electrodes were placed subcutaneously and the surface-lead ECG (lead II) was recorded. Data were analysed off-line using LabChart 7 Pro (AD Instruments). To correct for heart rate, the QTc interval was calculated using Bazett’s formula59: QTc = QT/(RR/100)1/2. Supplementary Methods contains additional details.
Preparation of Langendorff-perfused hearts and MAP recording
Langendorff-perfused hearts were prepared according to published methods60. Epicardial MAP was recorded from the LV anterior free wall by using a custom-made MAP electrode, constructed from two 0.25-mm Teflon-coated silver wires (99.99% purity). The paired platinum stimulating electrode was positioned on the basal surface of the right ventricle, and it delivered regular pacing. Method details are described in Supplementary Methods.
Protocols used for electrical stimulation of isolated hearts
S1-S1 pacing was used for determining APD90 and APD alternans threshold; pacing was performed with a series of pulse trains at a regular pacing cycle length (PCL) for 30 s, and then interrupted for at least 30 s to minimise pacing memory effects. The PCL was shortened from 150 to 100 ms in 10-ms steps and then to 50 ms in 5-ms steps. The APD90 at the PCL of 150 ms was defied as the average 90% repolarisation time for 6–8 successive MAPs. Alternans was judged to have occurred when alternate APDs differed by 5% over at least 10 beats61. The APDs were measured at 90% repolarisation and the longest PCL-induced APD90 alternans was defined as the APD alternans threshold interval. To test for VT inducibility, burst pacing at cycle lengths of 100–50 ms or 2:1 capture occurrence was used62, 63. Unlike in previous studies, we applied a pacing train of 20 stimuli. VT was defined as the occurrence of at least 4 consecutive ventricular waveforms after the last paced beat.
Preparation of mouse LV myocytes
Single LV myocytes were isolated from the mice by using collagenase type II digestion. Freshly isolated LV myocytes were stored at room temperature until use. Only rod-shaped myocytes showing clear striations were studied, and experiments were performed at room temperature (20 °C–25 °C) within 6 h after cell isolation. Experimental details are presented in Supplementary Methods.
Patch-clamp recording
Whole-cell membrane currents were obtained and assessed by using an EPC-9 patch-clamp amplifier (HEKA Electronik, Lambrecht, Germany) in the whole-cell mode and Pulse/Pulsefit software (HEKA Electronik). Supplementary Methods contains the experimental details.
Measurements of Ca2+ transients and SR Ca2+ content
Isolated cardiomyocytes were loaded with the Ca2+ indicator Fluo-4 AM (10 μmol/L; Molecular Probes, Invitrogen, F14217) in Tyrode’s solution (mmol/L: NaCl 135; KCl 5.4; CaCl2 1.8; MgCl2 1; NaH2PO4 0.33; HEPES 10; glucose 10; pH adjusted to 7.35 with NaOH) at 37 °C for 30 min; cells were suspended every 5 min during incubation. The loaded cells were washed for 15 min for de-esterification, superfused for 10 min with dye-free Tyrode’s solution, and then transferred to a chamber equipped with a pair of parallel platinum electrodes. The chamber was placed on a Leica AF6000 fluorescence microscope (Leica Microsystems Inc, Germany). Systolic Ca2+ transients were recorded in steady-state conditions under constant field stimulation (8 pulses at 0.5 Hz, 10 V). We evaluated the average time to peak Ca2+ transient amplitude and duration of Ca2+ transient decay of a tested cardiomyocyte by measuring 4 consecutive and well performed Ca2+ transients during field stimulation. To assess SR Ca2+ content, the amplitude of caffeine-induced Ca2+ transients was measured; at 4 s after the stimulation was stopped, 10 mmol/L caffeine was applied directly onto the cells, which led to an immediate and complete SR Ca2+ release. The fluorescence intensity of Fluo-4 (measured at 526 nm upon excitation at 488 nm) was recorded and the Ca2+ transient amplitude (F/F0) and duration were assessed, both by using the Leica AF6000 Modular system.
Ca2+ spark measurements
Loaded cardiomyocytes were prepared as described above and transferred to the recording chamber, which was placed on a Leica TCS SP8 STED 3X laser-scanning confocal microscope (Leica Microsystems Inc.). Fluo-4 was excited using a 488-nm argon-ion laser and the emitted fluorescence was measured at 526 nm. Diastolic Ca2+ sparks of the loaded cardiomyocytes were recorded at resting conditions after repetitive field stimulation (8 pulses at 0.5 Hz, 10 V). Ca2+ sparks were analysed using SparkMaster and Image J64. The mean CaSpF of each recorded cell was expressed as sparks/100 μm/s.
Statistical analysis
Statistical analysis was performed using SPSS or GraphPad Prism software. Continuous variables are shown as means ± SEM and were evaluated with Student’s 2-tailed unpaired t test or two-way ANOVA followed by Bonferroni post hoc test. Categorical data are expressed as percentages and were analysed using Fisher exact test. P < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81570306, No. 81270249, and No. 81570459), and the Hubei Provincial commonweal Foundation of China (No. 2013BCB013).
Author Contributions
J.P. conceived the study, performed the experiments, acquired and analyzed the data, and drafted the manuscript. Y.L. designed the study, revised the manuscript, and provided a part of the research funds. X.X. performed some experiments, acquired the data. C.H. revised the manuscript. Y.M., Z.W. and J.Y. provided the samples and materials. Y.T., B.K. and W.L. offered helpful discussion in the study. T.W. provided technical assistance. H.H. designed the overall study, analyzed the data, and revised the manuscript. All authors have reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Jianye Peng and Yu Liu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05379-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet (London, England) 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 3.Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- 4.Houser SR, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circulation research. 2012;111:131–150. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- 5.McMurray JJ, Pfeffer MA. Heart failure. Lancet (London, England) 2005;365:1877–1889. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- 6.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. The Journal of clinical investigation. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Go AS, et al. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hill JA, Olson EN. Cardiac plasticity. The New England journal of medicine. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 9.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nature clinical practice. Cardiovascular medicine. 2007;4:444–454. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 10.Topkara VK, et al. Therapeutic targeting of innate immunity in the failing heart. Journal of molecular and cellular cardiology. 2011;51:594–599. doi: 10.1016/j.yjmcc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarthy CG, et al. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. American journal of physiology. Heart and circulatory physiology. 2014;306:H184–196. doi: 10.1152/ajpheart.00328.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nature reviews. Cardiology. 2011;8:292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 13.Zimmer S, Grebe A, Latz E. Danger signaling in atherosclerosis. Circulation research. 2015;116:323–340. doi: 10.1161/CIRCRESAHA.116.301135. [DOI] [PubMed] [Google Scholar]
- 14.Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nature reviews. Immunology. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh MV, et al. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. Journal of molecular and cellular cardiology. 2012;52:1135–1144. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, et al. High-mobility group box 1 (HMGB1) impaired cardiac excitation-contraction coupling by enhancing the sarcoplasmic reticulum (SR) Ca(2+) leak through TLR4-ROS signaling in cardiomyocytes. Journal of molecular and cellular cardiology. 2014;74:260–273. doi: 10.1016/j.yjmcc.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Singh MV, Anderson ME. Is CaMKII a link between inflammation and hypertrophy in heart? Journal of molecular medicine (Berlin, Germany) 2011;89:537–543. doi: 10.1007/s00109-011-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Echem C, et al. Anti-toll like receptor 4 (TLR4) therapy diminishes cardiac remodeling regardless of changes in blood pressure in spontaneously hypertensive rats (SHR) International journal of cardiology. 2015;187:243–245. doi: 10.1016/j.ijcard.2015.03.190. [DOI] [PubMed] [Google Scholar]
- 20.Ehrentraut H, et al. The toll-like receptor 4-antagonist eritoran reduces murine cardiac hypertrophy. European journal of heart failure. 2011;13:602–610. doi: 10.1093/eurjhf/hfr035. [DOI] [PubMed] [Google Scholar]
- 21.Dange RB, et al. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovascular research. 2014;103:17–27. doi: 10.1093/cvr/cvu067. [DOI] [PubMed] [Google Scholar]
- 22.Monnerat-Cahli G, et al. Toll-like receptor 4 activation promotes cardiac arrhythmias by decreasing the transient outward potassium current (Ito) through an IRF3-dependent and MyD88-independent pathway. Journal of molecular and cellular cardiology. 2014;76:116–125. doi: 10.1016/j.yjmcc.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Divanovic S, et al. Inhibition of TLR-4/MD-2 signaling by RP105/MD-1. Journal of endotoxin research. 2005;11:363–368. doi: 10.1177/09680519050110061201. [DOI] [PubMed] [Google Scholar]
- 24.Miyake K, et al. Mouse MD-1, a molecule that is physically associated with RP105 and positively regulates its expression. Journal of immunology (Baltimore, Md.: 1950) 1998;161:1348–1353. [PubMed] [Google Scholar]
- 25.Ohto U, Miyake K, Shimizu T. Crystal structures of mouse and human RP105/MD-1 complexes reveal unique dimer organization of the toll-like receptor family. Journal of molecular biology. 2011;413:815–825. doi: 10.1016/j.jmb.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe Y, et al. The radioprotective 105/MD-1 complex contributes to diet-induced obesity and adipose tissue inflammation. Diabetes. 2012;61:1199–1209. doi: 10.2337/db11-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagai Y, Watanabe Y, Takatsu K. The TLR family protein RP105/MD-1 complex: A new player in obesity and adipose tissue inflammation. Adipocyte. 2013;2:61–66. doi: 10.4161/adip.22929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Candel S, et al. Md1 and Rp105 regulate innate immunity and viral resistance in zebrafish. Developmental and comparative immunology. 2015;50:155–165. doi: 10.1016/j.dci.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 29.Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circulation research. 2013;113:690–708. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang T, et al. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. The Journal of biological chemistry. 2002;277:1261–1267. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang T, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circulation research. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 32.Ling H, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. The Journal of clinical investigation. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Backs J, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braunwald E. The war against heart failure: the Lancet lecture. Lancet (London, England) 2015;385:812–824. doi: 10.1016/S0140-6736(14)61889-4. [DOI] [PubMed] [Google Scholar]
- 35.Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling–targets for heart failure therapy. Nature reviews. Cardiology. 2012;9:717–733. doi: 10.1038/nrcardio.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu B, et al. Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovascular research. 2015;108:299–311. doi: 10.1093/cvr/cvv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou Y, et al. Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertension (Dallas, Tex.: 1979) 2011;58:1099–1110. doi: 10.1161/HYPERTENSIONAHA.111.173500. [DOI] [PubMed] [Google Scholar]
- 38.Sedej S, et al. Subclinical abnormalities in sarcoplasmic reticulum Ca(2+) release promote eccentric myocardial remodeling and pump failure death in response to pressure overload. Journal of the American College of Cardiology. 2014;63:1569–1579. doi: 10.1016/j.jacc.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 39.van Oort RJ, et al. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension (Dallas, Tex.: 1979) 2010;55:932–938. doi: 10.1161/HYPERTENSIONAHA.109.146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tse, G. & Yan, B. P. Traditional and novel electrocardiographic conduction and repolarization markers of sudden cardiac death. Europace: European pacing, arrhythmias, and cardiac electrophysiology: journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology, doi:10.1093/europace/euw280 (2016). [DOI] [PubMed]
- 41.Mustroph, J., Neef, S. & Maier, L. S. CaMKII as a target for arrhythmia suppression. Pharmacology & therapeutics, doi:10.1016/j.pharmthera.2016.10.006 (2016). [DOI] [PubMed]
- 42.Rosenbaum DS, et al. Electrical alternans and vulnerability to ventricular arrhythmias. The New England journal of medicine. 1994;330:235–241. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- 43.Qu Z, Weiss JN. Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annual review of physiology. 2015;77:29–55. doi: 10.1146/annurev-physiol-021014-071622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobrev, D. & Wehrens, X. H. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circulation research114, 1311–1319; discussion 1319, doi:10.1161/circresaha.114.300568 (2014). [DOI] [PMC free article] [PubMed]
- 45.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annual review of physiology. 2014;76:107–127. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- 46.Belevych AE, Radwanski PB, Carnes CA, Gyorke S. ‘Ryanopathy’: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovascular research. 2013;98:240–247. doi: 10.1093/cvr/cvt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovascular research. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 48.Cheng H, Lederer WJ. Calcium sparks. Physiological reviews. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 49.Nass RD, Aiba T, Tomaselli GF, Akar FG. Mechanisms of disease: ion channel remodeling in the failing ventricle. Nature clinical practice. Cardiovascular medicine. 2008;5:196–207. doi: 10.1038/ncpcardio1130. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Hill JA. Electrophysiological remodeling in heart failure. Journal of molecular and cellular cardiology. 2010;48:619–632. doi: 10.1016/j.yjmcc.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiological reviews. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 52.Boczek NJ, et al. Identification and Functional Characterization of a Novel CACNA1C-Mediated Cardiac Disorder Characterized by Prolonged QT Intervals With Hypertrophic Cardiomyopathy, Congenital Heart Defects, and Sudden Cardiac Death. Circulation. Arrhythmia and electrophysiology. 2015;8:1122–1132. doi: 10.1161/CIRCEP.115.002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. The Journal of clinical investigation. 2013;123:46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voigt N, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Voigt N, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156. doi: 10.1161/CIRCULATIONAHA.113.006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, et al. Toll-interacting protein (Tollip) negatively regulates pressure overload-induced ventricular hypertrophy in mice. Cardiovascular research. 2014;101:87–96. doi: 10.1093/cvr/cvt232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, et al. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13818–13823. doi: 10.1073/pnas.1008397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods (San Diego, Calif.) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Z, et al. Functional roles of Cav1.3(alpha1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 2005;112:1936–1944. doi: 10.1161/CIRCULATIONAHA.105.540070. [DOI] [PubMed] [Google Scholar]
- 60.Qin M, et al. Absence of Rgs5 prolongs cardiac repolarization and predisposes to ventricular tachyarrhythmia in mice. Journal of molecular and cellular cardiology. 2012;53:880–890. doi: 10.1016/j.yjmcc.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 61.Martin CA, Grace AA, Huang CL. Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. American journal of physiology. Heart and circulatory physiology. 2011;300:H605–616. doi: 10.1152/ajpheart.00824.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sovari AA, et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. Journal of the American College of Cardiology. 2011;58:2332–2339. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sovari AA, et al. Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circulation. Arrhythmia and electrophysiology. 2013;6:623–631. doi: 10.1161/CIRCEP.112.976787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. American journal of physiology. Cell physiology. 2007;293:C1073–1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.