

Abstract
Despite the highly oxidative environment of the phagosomal lumen, the need for maintaining redox homeostasis is a critical aspect of mycobacterial biology. The pathogens are equipped with the sophisticated thioredoxin- (Trx) and peroxiredoxin system, including TrxC and the alkyl hydroperoxide reductase subunit C (AhpC), whereby TrxC is one of the reducing partners of AhpC. Here we visualize the redox modulated dodecamer ring formation of AhpC from Mycobacterium bovis (BCG strain; MbAhpC) using electron microscopy and present novel insights into the unique N-terminal epitope (40 residues) of mycobacterial AhpC. Truncations and amino acid substitutions of residues in the unique N-terminus of MbAhpC provide insights into their structural and enzymatic roles, and into the evolutionary divergence of mycobacterial AhpC versus that of other bacteria. These structural details shed light on the epitopes and residues of TrxC which contributes to its interaction with AhpC. Since human cells lack AhpC, the unique N-terminal epitope of mycobacterial AhpC as well as the MbAhpC-TrxC interface represent an ideal drug target.
Introduction
Tuberculosis (TB) with its causative agent, Mycobacterium tuberculosis (Mtb) has claimed many lives1 ever since it has been made known to mankind from ancient times. Ranked above HIV/AIDS in causing mortality due to an infectious disease2, World Health Organization’s Global Tuberculosis Report 2016 revealed an additional 10.4 million new cases worldwide in 20152. It was also understood that the current TB research and development is still severely unfunded2 and that the emergence of resistant strains against the first line drug, isoniazid (INH), is a huge cause of concern3. Despite being exposed to numerous oxidative and nitrosative stresses when mounting a pathogenic cycle of infection and transmission, Mtb is still able to persist and proliferate through various antioxidant defense systems4, 5. These antioxidant defense systems in Mtb have led to increased drug resistance and virulence. The susceptibility of Mtb to INH results from the activation of the gene KatG, encoding the mycobacterial specific catalase-peroxidase that plays a crucial role in evading or countering the host’s respiratory burst by decomposing hydrogen peroxide (H2O2) into water and oxygen6. The deletion of KatG in Mtb has shown to increase the bacteria’s sensitivity to H2O2 7. To ensure its virulence, Mycobacterium have evolved an alternative peroxidase system including the alkyl hydroperoxide reductase subunit C (AhpC)8 (Fig. 1A). Importantly, when exposed to INH, the INH-resistant Mtb exudes a KatG-deficiency, but also enhanced expression of MtAhpC characteristic9.
Figure 1.
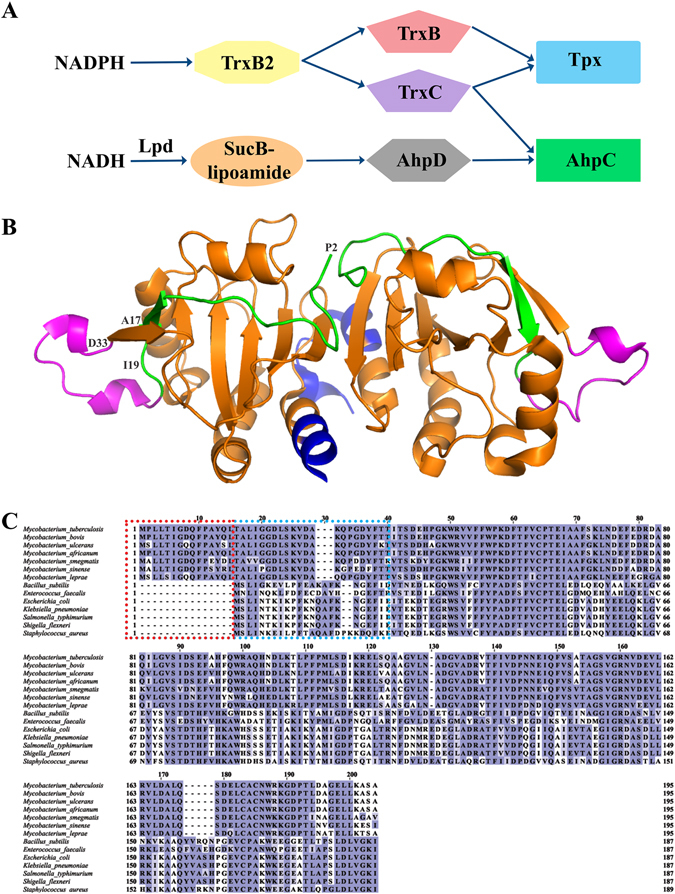
Peroxidatic catalytic pathway of Mycobacterium. (A) AhpC has various disulfide oxidoreductases such as TrxC and the proposed AhpD. In the former, AhpC has been proposed to function with TrxC in the reduction of ROS. In the latter, an orchestra of AhpD, SucB-lipoamide and LpD is necessary to reduce ROS. In both mechanisms, the reduction by either TrxC or AhpD results in the formation of a disulfide bridge along two cysteine residues of AhpC. (B) MtAhpCC176S mutant dimer structure (PDB ID: 2BMX)13. The N-terminal residues 1–15 are highlighted in green. The extra loop (amino acids 23–34) is shown in magenta, and the very C-terminal residues are colored in blue. (C) Multiple sequence alignment of AhpC from M. tuberculosis, M. bovis, M. ulcerans, M. africanum, M. smegmatis, M. sinense, M. leprae, B. subtilis, E. faecalis, E. coli, K. pneumonia, S. typhimurium, S. flexneri and S. aureus using Clustal-Omega30. The stretch of additional residues found in mycobacterial AhpC are highlighted with a red dashed box. The residues deleted for the generation of MbAhpC41–195 are highlighted with a blue dashed box.
Like most bacteria, the mycobacterial AhpC belongs to the typical 2-Cys peroxiredoxins (Prxs)5. Prxs are a crucial class of antioxidant enzymes needed to protect the cell from oxidative damages induced by reactive oxygen species (ROS). AhpCs of gram-negative bacteria described so far exist as a basic dimeric unit when it is oxidized and form a decamer-ring under reduced conditions10, 11. A peroxidatic cysteine (CP) residue in one unit of the dimeric AhpC reacts with H2O2 to form sulfenic acid. A reducing cysteine (CR) residue in the other unit of the dimer then attacks the sulfenic acid to release water5, 12. As a result, a disulfide bond is formed between the peroxidatic and reducing cysteine residues. Finally, the oxidized AhpC becomes regenerated by the NADH-dependent oxidoreductase, AhpF12. In the case of mycobacterial AhpC, a crystallographic structure of the Mtb AhpC mutant C176S (MtAhpCC176S)13 revealed that the asymmetric unit contains a dimer and a monomer. It was proposed that MtAhpCC176S would form a dodecamer-ring in solution13 instead of a decamer as described for the AhpC of gram-negative bacteria10, 11, 14. Connected with this diversity, the mycobacterial antioxidant system is not equipped with a gene encoding the oxidoreductase AhpF. Instead of AhpF, AhpC is reduced by the mycobacterial specific alkyl hydroperoxidase subunit D (AhpD)15 (Fig. 1A) or the thioredoxin TrxB and TrxC (TrxB, TrxC)16, as well as thioredoxin reductase TrxB2, although the role of the reduction through the thioredoxin system is still a debate1, 15 and its interaction epitopes are not resolved yet. It may reflect the difference of mycobacterial AhpCs to related typical 2-Cys peroxiredoxins by employing three rather than two cysteine residues involved in catalysis13.
Interestingly, the dimer structure of MtAhpCC176S mutant13 (Fig. 1B) as well as a protein sequence analysis of mycobacterial AhpCs revealed differences in the amino acid composition of AhpCs from other sources (Fig. 1C), in particular the presence of a unique stretch of 15 amino acid residues and the secondary structure of its N-terminal 40 residues. This N-terminal stretch, highlighted in green in Fig. 1B, is proposed to span one AhpC molecule and thereby bridging the two oligomeric interfaces, in which the catalytic CP and CR of the AhpC-ring are located. In addition, a crystal structure alignment with EcAhpC revealed that MtAhpCC176S forms an additional loop in the N-terminus from residues 23–34 (Fig. 1B, magenta). Elucidating the molecular mechanism and architecture of mycobacterial AhpC as well as its interaction with AhpD and TrxC in solution will shed light on the potential differences in function and regulation of the unique mycobacterial thioredoxin- and peroxiredoxin system.
Here we used 2-dimensional (2D) projection analysis of electron micrographs to visualize the dodecamer ring of reduced wild-type (wt) AhpC from M. bovis (BCG strain; MbAhpC), whose amino acid sequence is identical to Mtb, and to demonstrate the oligomeric formation of MbAhpC due to redox-modulation. Using NMR-titration experiments of labeled MbTrxC with MbAhpC, the epitope to which MbAhpC binds in MbTrxC is presented and opens the door for an ideal drug target. Truncations and amino acid substitutions of residues in of the unique N-terminus of MbAhpC provide insights into their structural and enzymatic roles.
Results
Production, purification and characterization of recombinant MbAhpC
Recombinant MbAhpC was purified in oxidized conditions by affinity- and size exclusion chromatography as described in Materials and Methods. The pure protein eluted at 16 ml on a Superdex 200 HR 10/30 column in the absence of the reducing agent DTT (oxidized condition), corresponding to a dimeric species (Fig. 2A). To confirm the observation of size exclusion chromatography (SEC), DLS experiments with MbAhpC were performed. As shown in Fig. 2B and C, the DLS profile revealed a dimeric species with an estimated molecular weight of 59.9 ± 22.6 kDa (26.2% polydispersity), and thus, confirming the SEC result described above.
Figure 2.
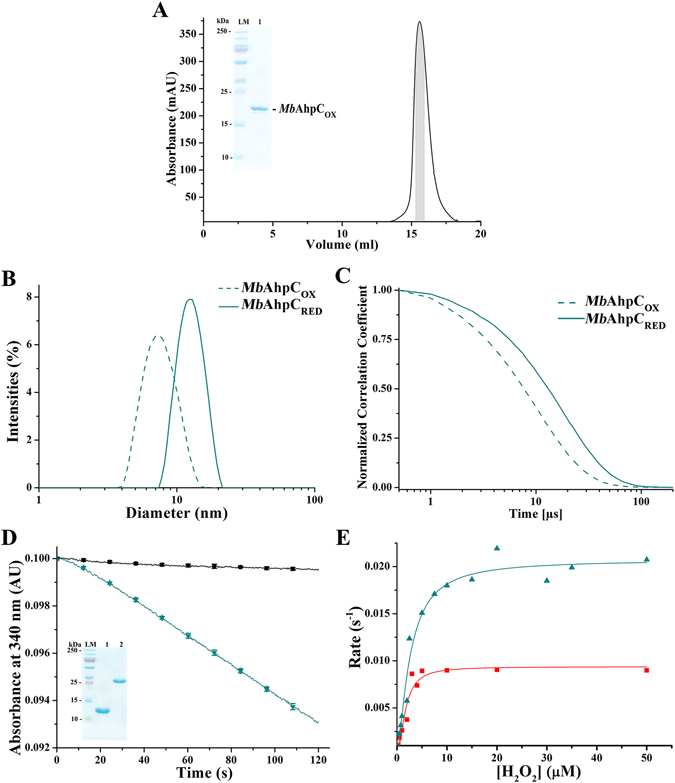
Protein characterization of MbAhpC. (A) The recombinant and oxidized MbAhpC eluted at approximately 16 ml on a Superdex 200 HR 10/30 column and showed high purity on a 17% SDS gel (inset). (B) DLS analysis of the oxidized (green dashed line) and reduced (green solid line) MbAhpC. (C) Normalized correlation function of DLS studies of MbAhpC in 50 mM Tris/HCl, pH 7.5, 200 mM NaCl. A slowed relaxation of the autocorrelation curve in reduced MbAhpC (green solid line) was observed as compared with oxidized MbAhpC (green dashed line). (D) NADPH-oxidation of MbTrxC and MbAhpC. The control experiment was performed in the absence of enzymes and H2O2 (black solid line). The utilized MbTrxC (lane 1) and MbTrxB2 (lane 2) showed high purity on a 17% SDS gel (inset) after size exclusion chromatography purification. The size of MbTrxC is 12,203 Da and MbTrxB2 is 35,643 Da. A significant drop in absorbance was observed due to the presence of wt MbAhpC (green solid line). (E) The Michaelis-Menten plot of MbAhpC and MbAhpCT5A/D8A were performed by fitting data of at least ten concentrations of H2O2 to establish the enzymatic kinetic parameters of wt MbAhpC and MbAhpCT5A/D8A. MbAhpC showed a higher enzymatic efficiency (green line and triangle symbols) as compared to MbAhpCT5A/D8A (red line and square symbols) whereby the reaction approached Vmax rapidly in the mutant as compared to wt MbAhpC.
To examine the enzymatic activity of MbAhpC, the MbTrxB2-TrxC system composed of TrxB2 and TrxC (Fig. 1A) was utilized, and both proteins were generated and purified for the peroxidase assay (see Material and Methods and inset of Fig. 2D). In this assay, NADPH-oxidation by TrxC provides the electrons via TrxB2 to MbAhpC for H2O2 reduction. As shown in Fig. 2D, MbAhpC reacted with 50 μM H2O2 and actively oxidized NADPH, as compared to the profile of the control measurement in which no MbAhpC was injected. To further characterise the enzymatic traits of MbAhpC, the Michaelis-constant (K m) of 2.57 ± 0.36 µM and the catalytic turnover number (k cat) of 0.021 ± 0.001 s−1 were determined (Fig. 2E, Table 1), resulting in a catalytic efficiency of MbAhpC (k cat /K m (H2O2)) of 8.17 × 103 M−1 s−1. In addition, the enzymatic parameters of MbAhpC with tert-butyl hydroperoxide (t-bOOH) as a substrate and NADPH as electron donor were determined with a K m of 2.23 ± 0.49 µM, a k cat value of 0.027 ± 0.003 s−1, and a k cat /K m of 12.20 × 103 M−1 s−1 (Table 1).
Table 1.
Kinetic parameters of mycobacterial AhpC with TrxC and NADPH.
| Protein | k cat ± SD (s−1) | K m ± SD (μM) | k cat /K m (M−1s−1) |
|---|---|---|---|
| H 2 O 2 as substrate | |||
| MbAhpC + MbTrxC | 0.021 ± 0.001 | 2.57 ± 0.36 | 8.17 × 103 |
| MbAhpCT5A/D8A + MbTrxC | 0.009 ± 0.001 | 1.65 ± 0.32 | 5.44 × 103 |
| t-BOOH as substrate | |||
| MbAhpC + MbTrxC | 0.0272 ± 0.003 | 2.23 ± 0.49 | 12.20 × 103 |
Ring formation of MbAhpC and its enzymatic traits under reduced conditions
In order to visualize and to define the presence of an oligomeric form of MbAhpC under reduced conditions, electron micrographs of reduced MbAhpC were collected. Images of the reduced enzyme showed considerable amounts of ring-like and rod-shaped particles, likely representing the top and side views of the ring structure, respectively (Fig. 3A). A total of 37 micrographs were subjected to two rounds of 2-dimensional (2D) classifications in RELION (Fig. 3C). Particles were sorted into 32 different classes in the first round to discard bad particles. A total of 3744 particles (eight 2D classes with clear features) selected from the first round of 2D classification were further averaged into four different classes during a second round of 2D classification to improve the image contrast. The averaged projection images show 12 defined masses, indicating that MbAhpC forms a dodecameric-ring under reducing conditions (Fig. 3D). The particles have a diameter of approximately 155 Å, and it is consistent with the reported 140 Å diameter of MtAhpCC176S mutant structure13, which reveals that six molecules (A–F) in the asymmetric unit form a half-ring conformation and a dodecameric ring is generated by its crystallographic two-fold symmetry operation (A′–F′).
Figure 3.
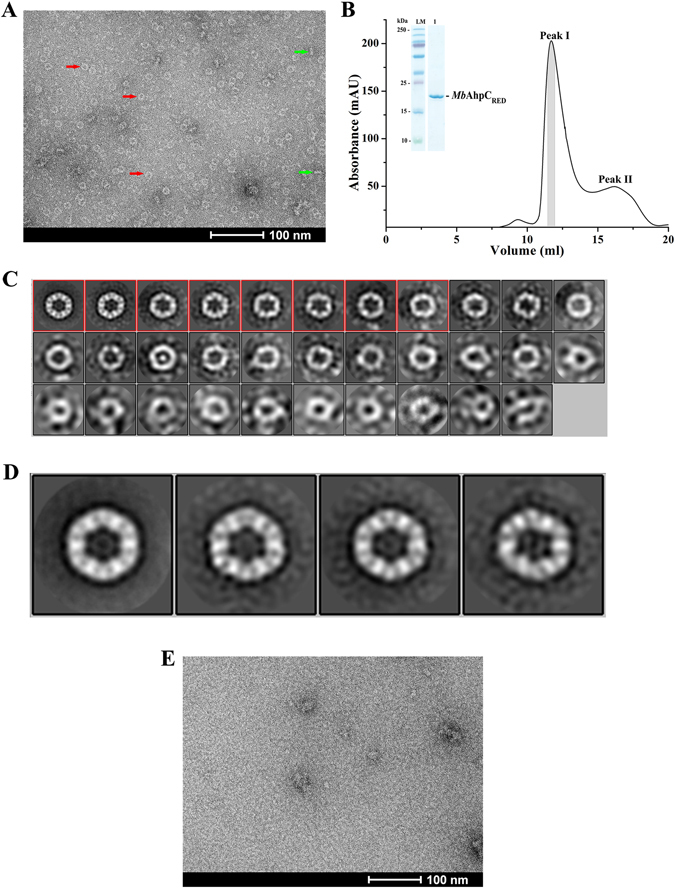
Negative stain electron micrographs of MbAhpC. (A) Micrograph of MbAhpC in its reduced form showed ring and rod shape particles, which were likely representing the top (red arrow) and side (green arrow) views of the ring structures, respectively. (B) The recombinant and reduced MbAhpC eluted with majority of the protein fractions at approximately 12 ml on a Superdex 200 HR 10/30 column and showed high purity on a 17% SDS gel (inset). (C) 2D classification of MbAhpC particles in its reduced form. Raw particles were classified in 32 classes employing 2D classification procedure of RELION image processing package. (D) Projection analysis revealed a dodecameric MbAhpC. Particles from the boxed classes in C were further classified into 4 classes to improve the quality of 2D classification. (E) The ring-shaped particles seen in the micrographs of reduced MbAhpC above were absent in the micrograph of the oxidized protein.
The SEC-elution profile of reduced (2 mM DTT) MbAhpC revealed a major peak at the column volume of 12 ml (Fig. 3B, peak I) and a minor peak at about 16 ml (Fig. 3B, peak II). As shown by the SDS-PAGE, both peak I (inset of Fig. 3B) and peak II contain MbAhpC, indicating that peak I contains an oligomeric form of MbAhpC, and peak II represents a smaller fraction of the dimeric MbAhpC. To confirm this, a DLS experiment was performed using the protein eluted at peak I. The DLS profile of the peak I fraction revealed that reduction of the recombinant protein led to the formation of a higher molecular weight species of about 226.0 ± 55.9 kDa (20.7% polydispersity) (Fig. 2B) with slower diffusion properties, and hence slower relaxation of the autocorrelation function (Fig. 2C), confirming the SEC interpretation above.
In contrast to the reduced MbAhpC, no ring-shaped particles were observed in the electron micrographs of the oxidized MbAhpC protein (Fig. 3E). This confirms the results obtained in the SEC and DLS experiments described above (Fig. 2A–C), that oxidized MbAhpC forms a dimer, and reduction of the enzyme results in a defined dodecameric ring structure of MbAhpC.
The N-terminal residues are essential for mycobacterial AhpC
Figure 1B and C reveal the unique stretch of the residues 1–40 at the very N-terminus of mycobacterial AhpC. To understand whether this unique stretch is critical for the structure and/or the enzymatic traits of the protein, a mutant with deletion of residues 1–40 was genetically engineered (MbAhpC41–195). Despite good production of the recombinant MbAhpC41–195 mutant in the induction assay, the mutant showed lower protein solubility when compared with wt MbAhpC (Fig. 4A), indicating the importance of the unique N-terminal stretch in protein solubility and stability.
Figure 4.
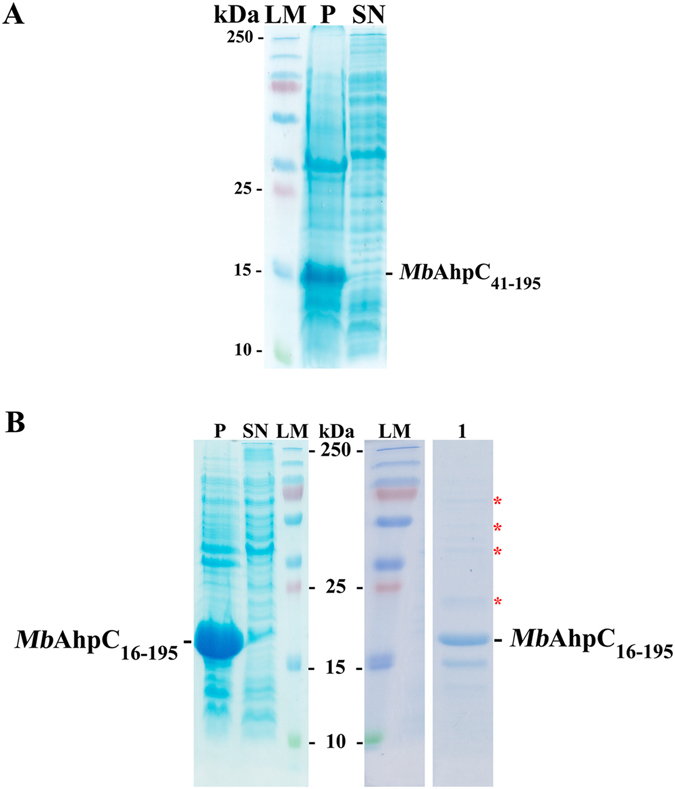
Importance of N-terminal residues. (A) Solubility assay of MbAhpC41–195 showed low solubility of the protein as reflected in the pellet, labelled as P. SN represents supernatant. (B) MbAhpC16–195 also showed a less soluble protein (left) and the presence of a laddering pattern (right) in the 125 mM imidazole fraction after affinity chromatography, indicates the presence of undefined oligomers. LM represents a molecular weight standard. Both SDS gels revealed the importance of the N-terminal residues, particularly from residues 1–15, in maintaining the solubility and oligomerization of MbAhpC.
To further determine the critical residues in the N-terminus for the stability of MbAhpC, the N-terminal deletion mutant MbAhpC16–195 was genetically engineered. High amount of recombinant MbAhpC16–195 was produced however, the amount of soluble protein derived from 6 g of cells was low (Fig. 4B). As shown in the SDS gel after Ni-NTA chromatography (Fig. 4B), ladder formation of protein bands in the SDS gel were observed, reflecting that the protein is not stable and tends to form undefined oligomers (Fig. 4B). These results suggest that the N-terminal deletions may affect the defined oligomeric state of mycobacterial AhpC and its overall stability.
The importance of the N-terminal residues 23–34 in activity and assembling of MbAhpC
As shown by the multiple sequence alignments of AhpCs from different bacteria species using the program ClustalW17, a stretch of additional residues was observed in the N-terminus of mycobacterial AhpC and predicted to form an extra loop in the N-terminus of mycobacterial AhpC (Fig. 5A). In order to understand the function of this extra loop, a construct with the deletion of the loop residues from 23 to 34 (MbAhpCΔ23–34) (Fig. 5A, red dashed box) was generated. Oxidized- and reduced (presence of 2 mM DTT) MbAhpCΔ23–34 were purified using the purification protocol following wt MbAhpC (Fig. 5B and C). Both the oxidized as well as the reduced form of MbAhpCΔ23–34 eluted at 16 ml, resembling the oxidized dimeric form of MbAhpC. Furthermore, no significant change in the hydrodynamic diameter of both the oxidized (6.5 ± 1.9 nm, 25.5% polydispersity) and reduced (5.9 ± 1.8 nm, 28.5% polydispersity) MbAhpCΔ23–34 was observed in DLS experiments (Fig. 5D). These results demonstrate that the amino acid stretch 23–34 of MbAhpC is essential for the assembling of the dodecameric ring of reduced MbAhpC. This is underlined by electron micrographs of MbAhpCΔ23–34 (Fig. 5E), in which the reduced MbAhpCΔ23–34 particles resembled those of oxidized MbAhpC (Fig. 3E). Furthermore, as reflected in Fig. 5F, the MbAhpCΔ23–34 mutant did not show significant activity in the NADPH-oxidation assay, indicating that no electron transfer occurred in the catalytic cycle and demonstrating that the N-terminal residues 23–34 are also crucial in maintaining the enzymatic activity.
Figure 5.
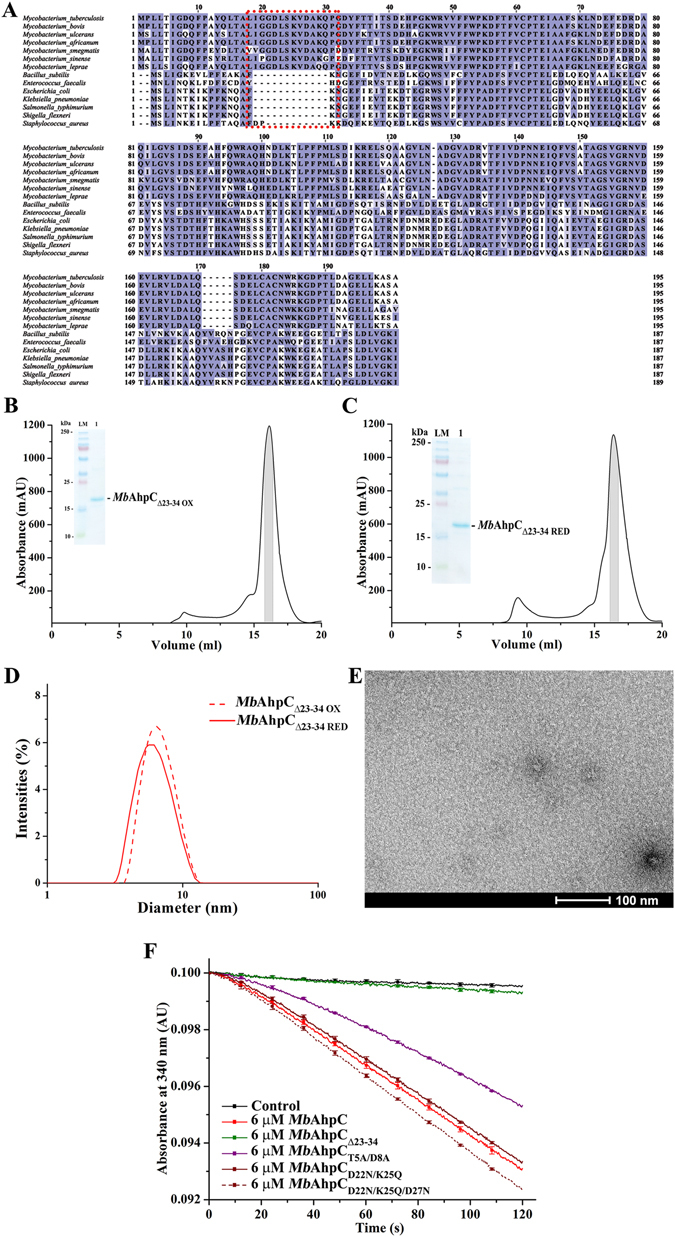
(A) Multiple sequence alignments of AhpC from different bacteria using ClustalW17. As shown, various Mycobacterium sp. presented extra 15 residues located at the N-terminus (red dashed box), which were absent in other bacteria species. Mutations with deletion from residues 23–34 were constructed and both oxidized (B) and reduced (C) mutants eluted at approximately 16 ml on a Superdex 200 HR 10/30 column and showed high purity on a 17% SDS gel (inset). (D) DLS analysis of oxidized (red dashed line) and reduced (red solid line) of MbAhpCΔ23–34. Both redox states of the protein revealed a similar diameter unlike that of wt MbAhpC. (E) Negative stained EM-images of reduced MbAhpCΔ23–34. The lack of ring-shaped particles in the micrograph indicates the inability of MbAhpCΔ23–34 to form an oligomeric ring. (F) NADPH-oxidation of MbAhpC, -AhpCΔ23–34, -AhpCT5A/D8A, -AhpCD22N/K25Q or -AhpCD22N/K25Q/D27N measured at 50 µM H2O2 is shown as a representative to highlight the effects of the mutations generated. A control without MbAhpC is represented with a black solid line. The activity of wt MbAhpC is indicated with a red solid line. The deletion of residues 23–34 (green solid line) altered the activity of the protein, hence, showing activities close to that of which resembles the background activity. However, the mutations in T5 and D8 showed the decline of activity of MbAhpCT5A/D8A as shown with the purple solid line. On the other hand, the enzymatic activity of MbAhpCD22N/K25Q (brown solid line) or MbAhpCD22N/K25Q/D27N (brown dashed line) have similar activity to wt MbAhpC.
Substitutions of N-terminal residues of MbAhpC provide insights into their enzymatic role
To specify critical residues of the N-terminus in more depth, the polar residues T5, D8, D22, K25 and D27 have been substituted in the double and triple mutants, MbAhpCT5A/D8A, -AhpCD22N/K25Q, and -AhpCD22N/K25Q/D27N, respectively (Fig. 6A–C). All mutants were purified according to the protocol of wt MbAhpC and behaved similar to wt MbAhpC with respect to the elution profile and oligomer formation under oxidized and reduced conditions (Fig. 6B and C), with the exception of the double mutant MbAhpCT5A/D8A, which showed a close to 1:1 ratio of dimer and dodecamer formation in reduced condition (Fig. 6A). These differences are also reflected in the NADPH-oxidation assay (Fig. 5F), where mutant MbAhpCD22N/K25Q revealed a similar activity profile like the wt protein, and the MbAhpCD22N/K25Q/D27N showed only a slight increase in activity, while the activity of mutant MbAhpCT5A/D8A dropped significantly. The K m and k cat of the mutant MbAhpCT5A/D8A were determined to be 1.65 ± 0.32 µM and 0.009 ± 0.001 s−1, respectively, (Fig. 2E, Table 1) resulting in a catalytic efficiency (k cat /K m (H2O2)) of 5.44 × 103 M−1 s−1. Finally, the mutant MbAhpCT5A was generated. The DLS-profile in Supplementary Figure S1 revealed that the single substitution of T5 to an alanine alters partially the change from a dodecamer formation of the reduced form to a mix of a dimer and higher oligomer form of the mutant. In comparison to the effect of the double mutant MbAhpCT5A/D8A (Fig. 6A), this also indicates that both residue T5 and D8 are important for the dodecamer formation of reduced MbAhpC.
Figure 6.
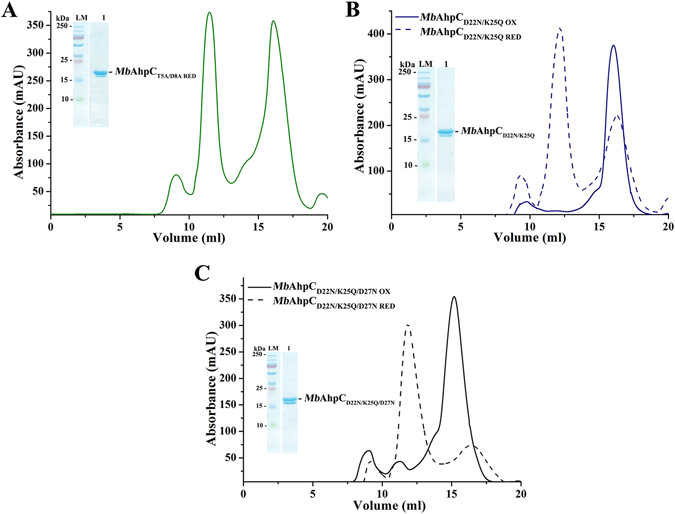
Protein purification of MbAhpC double and triple mutants. (A) The recombinant and reduced MbAhpCT5A/D8A eluted in an equal proportion of oligomer (12 ml) and dimer (16 ml) on a Superdex 200 HR 10/30 column and showed high purity on a 17% SDS gel (inset). The recombinant oxidized and reduced double mutant, (B) MbAhpCD22N/K25Q, and triple mutant, (C) MbAhpCD22N/K25Q/D27N, showed an SEC elution profile resembling wt MbAhpC. Majority of the oxidized recombinant protein (solid blue and black line) eluted at approximately 16 ml while the reduced recombinant protein eluted (dashed blue and black line) at 12 ml. Both reduced recombinant proteins revealed high purity on a 17% SDS gel (inset).
NMR titration experiment of MbTrxC with MbAhpC
Since the data above demonstrate clearly that mycobacterial TrxC provides electrons for the reduction of AhpC and thereby the reduction of H2O2 and t-bOOH, a series of NMR titration experiments were performed to examine the molecular interaction between MbTrxC and –AhpC. 1H-15N-HSQC spectra of 15N-labelled MbTrxC were collected at three different molar ratios (1:0, 1:0.5, 1:1 and 1:2) of MbTrxC and MbAhpC (Fig. 7A). During titration experiments, we observed the disappearance or the decrease of some of the cross peak intensities in the 1H-15N HSQC spectrum (Fig. 7A), slight changes of 15N- and HN-resonances for some residues and gradual line broadening with increasing molar ratios (Fig. 7B). This demonstrates the binding of MbTrxC with MbAhpC. A plot of chemical shift perturbations after the addition of MbAhpC to labeled MbTrxC at a molar ratio of 1:2 is shown in Fig. 7C. Significant changes (>0.1) in chemical shift were observed for the backbone resonances of regions including C37-V43 and E70-T71. These residues are considered as directly involved in the interaction with MbAhpC. Other residues including S4, K6, T9, W33, T52, R54, V60, L63, F75, L83, V92, V96, K99, A102 and L115 are affected indirectly by the protein-protein interaction. In addition, the NMR spectra showed also gradual line broadening and increased linewidths after the addition of MbAhpC (Fig. 7A), which supports the slow molecular tumbling and fast T2 relaxation times caused by the formation of the MbTrxC-AhpC complex. The average increased linewidths before and after binding of MbAhpC to MbTrxC is ~120% of NH and ~90% of 15N resonances, respectively. The significant changes (>150% of NH and/or 120% of 15N) in linewidths were observed for the backbone resonances of T13, S16-A18, L29, V30, D31 (disappeared), A34, W36, C37, C40, M42-A44, V46, E49-I50, D57, A61, D64-D66, T71-A72, F75, V77, S79, L83, L85 (disappeared), G97-K99, A103, L105 and R106 (Fig. 7E). Interestingly, residues in TrxC identified as the direct binding site (C34-V43 and T71) as well as additional residues including D64-D66 also showed significant changes in linewidths, suggesting that these residues are crucial in the formation of the MbTrxC-AhpC ensemble. As shown in Fig. 7D, the identified residues of MbTrxC involved in AhpC binding form an epitope based on the CSP and NMR linewidth analysis data, including the catalytic cysteine residues, which provide an ideal close proximity for electron transfer during the reduction of the catalytic cysteine-cysteine-center of mycobacterial AhpC. Moreover, the electrostatic charge distribution of MbTrxC (Fig. 7F) showed that the region of amino acids C37-V43 is mostly positively charged and the region E70-T71 is negatively charged, thereby allowing several charged interactions between mycobacterial TrxC and AhpC. Furthermore, NMR titration experiments using MbAhpCT5A/D8A or -AhpCD22N/K25Q/D27N also showed disappearance of resonances or slight changes in chemical shifts for residues located around the binding epitopes (Supplementary Figure S2), which demonstrate that the mutations on these residues do not affect the interaction between MbTrxC and MbAhpC.
Figure 7.
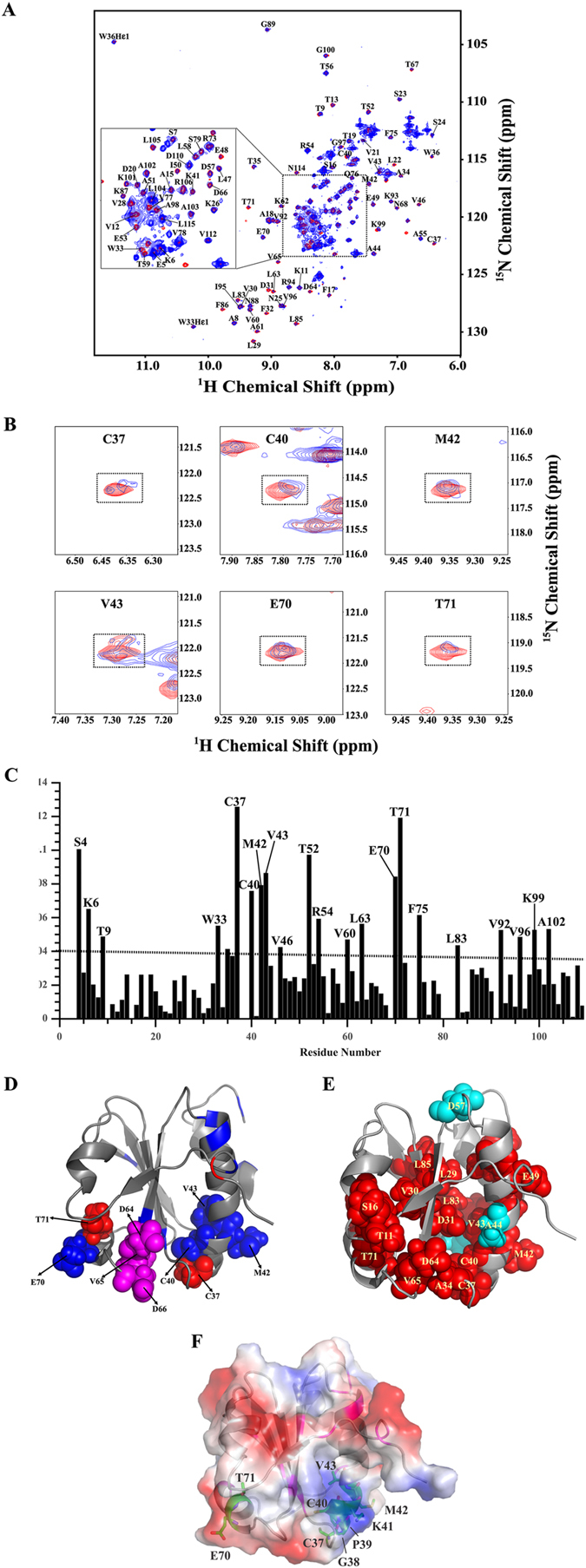
Overlayed 1H-15N HSQC-spectra of MbTrxC upon titration with MbAhpC at a molar ratio 1:2. (A) Superimposition of the 1H-15N HSQC spectrum of MbTrxC alone (red) and in the presence of MbAhpC (blue). 1H-15N HSQC-spectra were obtained on a Bruker avance 700 MHz NMR spectrometer at 298 K. Backbone resonance assignments (based on the BMRB, accession code: 17242)23 are indicated in a one-letter amino acid code and sequence number. (B) Selected sections for some residues show cross-peaks undergoing significant chemical shift perturbation. (C) Molecular interaction between MbTrxC and MbAhpC. A plot of chemical shift perturbations after addition of MbAhpC into MbTrxC at a molar ratio 1:2. Differences of chemical shifts were calculated using the following formula, Δδ = [(1Hfree − 1Hbound)2 + (15Nfree − 15Nbound)2)]1/2. (D) Ribbon representation of MbTrxC mapped by CSP results; the residues having CSP more than 0.1 are represented in red, while those showing CSP between 0.05 and 0.1 are shown in blue. Candidates for interacting residues with MbAhpC are represented as sphere model and are labeled. Additional candidates (D64-D66) based on the linewidth analysis are represented as magenta sphere. (E) Ribbon and sphere representation of MbTrxC mapped by NMR linewidths analysis; the residues showing increased in linewidth of more than 150% for NH are represented in red, while those showing increased in linewidth of more than 120% of 15N only are shown in cyan. (F) Electrostatic potential at the surface of mycobacterial TrxC (PDB ID: 2L4Q)23 with the epitopes C37-V43 and E70-T71 shown as stick representation in green and the residues involved in indirect interaction as magenta.
Model of the mycobacterial TrxC-AhpC complex
In order to model the TrxC-AhpC complex, the obtained NMR restraints for TrxC were used to drive a HADDOCK-docking simulation as described under Materials and Methods. For AhpC, the binding region is chosen to be the redox-center that contains the peroxidatic cysteine C61 of one subunit and the resolving cysteine C174 of the other subunit in the dimer interface10. The top scoring HADDOCK model from the highest scoring cluster was selected (HADDOCK score: −35 ± −5.9; restraint violation energy: 27.7 ± 14.55 Kcal/mol; buried surface area: 1124.8 ± 41.4 Å; Z-score: −0.1). The resulting TrxC-AhpC complex model (Fig. 8) indicated the spatial proximity of the two catalytic sites, with C37 of TrxC at a distance of 4 Å from C174 of AhpC. In this complex model, TrxC has several distributed interactions with one of the subunits of the dimer MtAhpCC176S and with the other subunit concentrating near the very C-terminal residues of the catalytic site. Several hydrogen bonding interactions occur in this concentrated region, such as between D172 of AhpC and the TrxC residues C37 and G38, and N177 of AhpC with C40 of TrxC. The interactions of residues C37, G38 and C40 of the C37-V43 region are nicely confirmed by the NMR titration experiment above. Furthermore, in the other interfacial region, a strong salt-bridge interaction occurs between R73 and D102 of TrxC and AhpC, respectively, which is closer to the E70-T71 epitope, wherein E70 interacts with N101 of AhpC through a hydrogen bond. In addition, K70 and T63 of AhpC also have hydrogen bonding interaction with Q76 and V78 of TrxC. Inclusion of additional residues that showed increased linewidths in the NMR experiments above for the MbTrxC-MbAhpC interactions in the docking, did not produce a better complex model.
Figure 8.
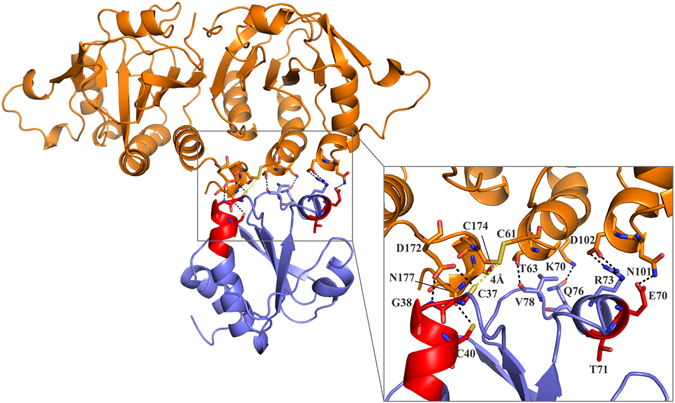
A model of mycobacterial TrxC-AhpC complex based on the NMR titration experiments. TrxC (PDB ID: 2L4Q)23 is shown in blue and MtAhpCC176S (PDB ID: 2BMX)13 in orange color. (Inset) A closer view of the various interactions between mycobacterial TrxC and AhpC in the model complex.
Discussion
The mycobacterial peroxidase system appears to play an important role in M. tuberculosis resistance against the oxidative and nitrosative stress exerted by the host immune response18. These enzymes thus represent suitable targets for novel anti-tuberculosis strategies, in particular for INH-resistant M. tuberculosis strains, where AhpC is thought to compensate for the decreased catalase-peroxidase KatG activity18, 19. We demonstrate that MbAhpC, which is identical in amino acid composition to MtAhpC, catalyses NADPH-driven hyperoxide reduction together with the mycobacterial TrxC, resulting in efficient H2O2 reduction. The K m values determined for MbTrx-AhpC with H2O2 (2.57 ± 0.36 μM) or t-bOOH (2.23 ± 0.49 µM) as a substrate are similar (Table 1), with an increased catalytic efficiency of MbAhpC with t-bOOH (Table 1). In comparison, the K m of MbTrx-AhpC with t-bOOH (2.23 ± 0.49 µM) is slightly lower compared to the reported MtTrx-AhpC complex with t-bOOH as a substrate (5.6 μM)15. The difference may in part be caused by the fact that NADH has been used as an electron donor in the studies of MtTrx-AhpC15 instead of the natural substrate NADPH (Fig. 1A) in the studies presented with MbAhpC. Together with the results of Jaeger et al.15, the data presented confirm that mycobacterial TrxC reduces efficiently AhpC and refute the proposal that only AhpD and not the thioredoxin reductase would be a redox partner of mycobacterial AhpC1. Because of its efficiency, the mycobacterial Trx-AhpC system becomes essential to protect the pathogenic bacterium against oxidative stress and therefore, the interaction of both TrxC-AhpC are important to shed light into their electron transfer and the interacting residues enabling the TrxC-AhpC complex formation.
Comparison of the MtAhpCC176S mutant structure with other peroxiredoxins showed that the helix α1 is flexible and is involved in a rigid-body movement to position the peroxidatic cysteine (C61) either in contact with the resolving cysteine (C174), or in the active center (Fig. 8)13. A small rearrangement of three phenylalanine side chains (F51, F68, F108) has been proposed to cause such displacement, and an internal cavity would be created due to the movement of the helix α1, which was proposed as a potential drug binding site13. The presented NMR-titration as well as the molecular docking studies extend the molecular model of electron transfer inside mycobacterial AhpC and the electron donor TrxC. In the derived MtTrxC-AhpC complex model, TrxC makes two crucial interactions with helix α1 of AhpC, whereby residues Q76 and V78 of TrxC have hydrogen bonding interaction with K70 and T63 of AhpC, respectively (Fig. 4). As F68 is present on helix α1, we speculate that during complex formation, the interactions mentioned above might induce the rearrangement of the phenylalanine side chains, which will aid in the helical displacement. Moreover, in the TrxC-AhpC complex model, C37 of TrxC is closer to C174 (4 Å) than C61 (5.5 Å) of AhpC. This favors an alternative mechanism, which was proposed for the interaction of mycobacterial AhpC with AhpD by Guimarães et al.13 for three cysteine preoxiredoxins. It was proposed that after the condensation reaction of peroxidatic cysteine (CP61) in helix α1 with the resolving cysteine (CR174), the disulfide bond between C61-C174 is reduced by C176 and a second intramolecular disulphide bond between C174 and C176 occurs and subsequently, the external thiol (coming from AhpD or TrxC) attacks either one of the resolving cysteines.
We also visualized the dodecameric ring formation of the reduced MbAhpC (Fig. 3D). So far, the crystallographic structure of the oxidized MtAhpCC176S mutant revealed a dimer in the asymmetric unit, and the dodecameric ring formation of the oxidized MtAhpCC176S mutant structure was proposed to be forced by crystal packing forces13. The electron microscopy data of MbAhpC in solution demonstrate that the mycobacterial protein undergoes a transfer from a dimer to a dodecameric oligomer under oxidized and reduced conditions, respectively. Deletion of and substitution inside the unique N-terminus of mycobacterial AhpC highlight the importance of this region in maintaining protein stability, redox-modulated oligomer formation and enzyme activity. Interestingly, the N-terminal deletion mutants MbAhpC16–195 and MbAhpC41–195 are unstable and formed undefined oligomers, indicating the importance of the unique N-terminal residues for the stability of mycobacterial AhpC redox- oligomarization. Moreover, the deletion mutant MbAhpCΔ23–34 demonstrated that the mutant exists as a dimer in the oxidized and the reduced state and has no activity, highlighting that the N-terminal residues 23–34 are important for maintaining not only the oligomerization but also the enzymatic activity. In the MtAhpCC176S mutant structure, the dimer-dimer interface of the dodecamer is mainly stabilized by the hydrophobic interactions of the residues F57, T58, F91, I114 and V130 of the interacting monomers13. The hydrophobic environment of F91 that faces the solvent region is occluded by the extra loop of the N-terminal amino acids 23–34, which is at a distance of 5.5 Å (Fig. 9A). The hydrophobic residues of the extra loop V26, P31, Y34 and F35 are positioned in such a way to create an ample hydrophobic environment for helix α2, that holds F91, F94, A98 and W96 (a highly-conserved residue directly associated with the redox-induced dimer-decamer switch in peroxiredoxins)13. We speculate, that by removing this extra loop, the hydrophobic environment might become destabilized for F91 and for helix α2, which in-turn might destabilize the dimer-dimer interface. Therefore, the oligomer formation would be hindered and finally the enzymatic activity would be affected. Interestingly, the double and triple mutants MbAhpCD22N/K25Q and MbAhpCD22N/K25Q/D27N did not affect the enzymatic activity, since the substitutions are made for the polar residues of the extra loop that faces the solvent region and which does not significantly contribute to the hydrophobic environment of the dimer-dimer interface (Fig. 9A).
Figure 9.
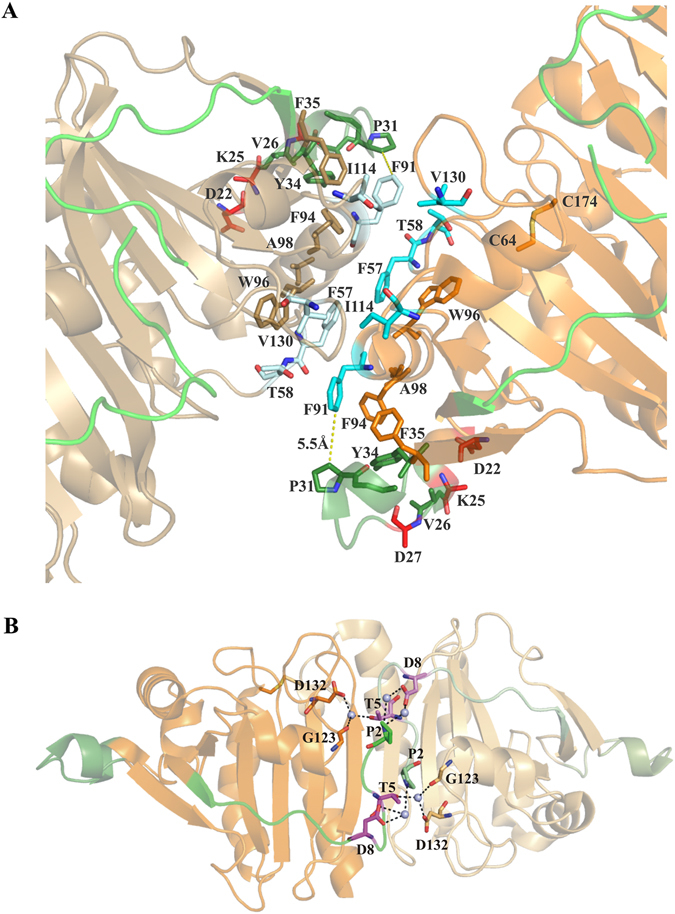
(A) Dimer-dimer interface of the MtAhpCC176S structure (PDB ID: 2BMX)13. The first monomer is shown in orange and the second monomer is shown in sand color. The N-terminal 1–15 residues are shown in green and the extra loop (23–34) in the N-terminal is shown in forest green. The hydrophobic residues F57, T58, F91, I114 and V130 that stabilize the interface are shown in cyan. The mutants D22, K25 and D27 are highlighted in red. (B) Critical residues in the dimer interface of mycobacterial AhpC (PDB ID: 2BMX)13. The mutant residues T5 and D8 are shown in magenta. The subunits are shown in orange with the second in lighter shade. The N-terminal 1–15 residues are shown in green and the extra loop (23–34) in the N-terminal is shown in forest green. The water molecules involved in the water mediated interaction are shown as light blue spheres.
Furthermore, the double mutant MbAhpCT5A/D8A at the very N-terminus reduced enzymatic activity by 2.73 × 103 M−1s−1 (33%) when compared to wt MbAhpC, indicating the critical role of these residues. The unique N-terminal stretch (amino acids 1–15) spans the complete subunit AhpC, and the very N-terminal residues bridge the functional dimer interface in the MtAhpCC176S structure13. The residue T5 of one subunit makes water-mediated hydrogen bonding interactions with D132 and G123 of another subunit (Fig. 9B), confirming its importance in dodecamer formation as shown in the MbAhpCT5A mutant (Supplementary Figure S1). In addition, amino acid D8 of one subunit has water-bridge interaction with P2 of another subunit in the functional dimer interface. Due to the substitution of these residues to a non-polar alanine in the MbAhpCT5A/D8A mutant, these interactions might be abolished, and we speculate, that this might weaken the dimer interface, thus resulting in the decreased activity.
In summary, we show that MbTrxC-AhpC forms an NADPH-dependent peroxidase ensemble for efficient reduction of H2O2 inside the mycobacterial antioxidant defense system. Identification of the amino acids involved in TrxC and AhpC interaction provide not only a new epitope for structure-guided drug design but also shed light into the electron transfer from TrxC to the catalytic cysteines of AhpC and possible rearrangements of side chains of residues F51, F68, and F108 of helix α1. AhpC undergoes a redox-modulated dimer to dodecamer formation, in which the unique mycobacterial N-terminal stretch of AhpC place a fundamental role as revealed by a variety of N-terminal mutants of MbAhpC. Since the dodecamer ring formation is essential for the proper enzyme catalysis, the important roles of this unique N-terminal stretch in dimer and oligomer formation as well as enzyme activity makes this N-terminus a novel epitope for drug design.
Materials and Methods
Cloning, production and purification of MbAhpC, and its mutants as well as MbTrxB2 and -TrxC
The coding region for the residues 1–195 of MbAhpC was amplified from the genomic DNA of M. bovis (BCG strain), which is identical in its amino acids sequence to MtAhpC (Fig. 1C), by polymerase chain reaction (PCR) using the forward primer 5′-ATG ACC ATG GTC ATG CCA CTG CTA AC-3′ and the reverse primer 5′-AAC CAG AGG ATC CTT AGG CCG AAG-3′. The coding region for the mutant MbAhpC16–195 was amplified using the forward primer 5′-CCT CCA TGG TCA CCG CTC TCA TCG GCG GTG AC-3′ and the reverse primer 5′-AGC CGG ATC CTT AGG CCG AAG CCT TGA GGA CTT C-3′. The coding region for the mutant MbAhpC41–195 was amplified using the forward primer - 5′ CCT CCA TGG TCA CCG CTC TCA TC 3′ and the reverse primer - 5′ AGC CGG ATC CTT AGG CCG AAG CCT T3′. The amplified PCR-products were gel-extracted and ligated into the pET9-d1-His6 vector20. The mutant MbAhpCΔ23–34, -AhpCT5A, -AhpCT5A/D8A, -AhpCD22N/K25Q and -AhpCD22N/K25Q/D27N were created by In-Fusion-based mutagenesis21. The whole pET9-d1-His6 20 plasmid carrying MbAhpC gene was used as the template. The deletion of residues 23 to 34 (MbAhpCΔ23–34) was introduced by amplifying the whole plasmid using the primers 5′-GGT GAT AGT GGT GAA GTC ACC GCC GAT GAG AGC GGT GAG CTG-3′ and 5′-TTC ACC ACT ATC ACC AGT GAC GAA CAC CCA GGC AAG TGG CGG-3′. For MbAhpCT5A, -AhpCT5A/D8A, -AhpCD22N/K25Q and -AhpCD22N/K25Q/D27N, point mutations were made and the whole plasmid was amplified using the primer sets 5′-TGC TAG CTA TTG GCG ATC AAT TCC CCG-3′ and 5′-CCA ATA GCT AGC AGT GGC ATG ACC ATG-3′, 5′-TGC TAG CTA TTG GCG CTC AAT TCC CCG CCT ACC AG-3′ and 5′-ATT GAG CGC CAA TAG CTA GCA GTG GCA TGA CCA TG-3′, 5′-GGT AAT CTG TCC CAA GTC GAC GCC AAG CAG-3′ and 5′-GAC TTG GGA CAG ATT ACC GCC GAT GAG AGC G-3′, and 5′-GTC AAT GCC AAG CAG CCC GGC-3′ and 5′-GGC ATT GAC TTG GGA CAG ATT ACC GCC-3′, respectively.
Genomic DNA of M. bovis was used to amplify M. bovis thioredoxin (MbTrxC) and thioredoxin reductase (MbTrxB2). MbTrxC was constructed using the forward primer 5′-GCA CAA CGC CAT GGC AAT GAC CGA TTC CGA GAA GT-3′ and the reverse primer 5′-CCG GGA TCC CTA GTT GAG GTT GGG AAC CAC GTC T-3′, and MbTrxB2 was constructed using forward primer 5′-TTA ACC ATG GAT ATG ACC GCC CCG CCT GTC-3′ and reverse primer 5′-TTA TGG ATC CTC ATC GTT GTG CTC CTA TCA ATG CGT CGG-3′. In MbAhpC, -AhpC16–195, -AhpC40–195, -TrxC and -TrxB2 constructs, the restriction sites NcoI and BamHI (underlined) were incorporated into the forward and reverse primers, respectively. The amplified PCR-products were purified and ligated into the pET9-d1-His6 vector20.
The coding sequences of all constructs were verified by DNA sequencing. The final plasmids were subsequently transformed into E. coli BL21 (DE3) cells (Stratagene). To express the recombinant proteins, the cells were grown in a shaker incubator in LB medium, containing kanamycin (30 μg/ml), for about 4 h at 37 °C until an optical density OD600 of 0.6–0.7 was reached. For MbTrxC, cells were cultured in LB medium containing kanamycin (30 µg/ml) and chloroamphenicol (34 µg/ml). To induce the production of proteins, cultures were supplemented with isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, and incubated for 16–18 hours at 18 °C.
Cells producing recombinant MbAhpC, -AhpCΔ23–34, -AhpC16–195, -AhpC41–195, -AhpCT5A, -AhpCT5A/D8A, -AhpCD22N/K25Q, -AhpCD22N/K25Q/D27N, -TrxC and -TrxB2 were lysed on ice by sonication with an ultrasonic homogenizer (Bandelin, KE76 tip) for 3 × 1 min in buffer A containing 50 mM Tris/HCl, pH 7.5, 200 mM NaCl, 0.8 mM DTT and 2 mM PefablocSC. For oxidized conditions, buffer A without 0.8 mM DTT was utilized instead. After sonication, the cell lysate was centrifuged at 12 000 × g for 25 min at 4 °C (Eppendorf, Germany). The resulting supernatant was passed through a filter (0.45 µm; Millipore) and incubated with Ni2+-NTA resin pre-equilibrated in buffer A. His-tagged proteins were allowed to bind to the matrix for 1 h at 4 °C by mixing on a sample rotator (Neolab). Subsequently, the protein was eluted with an imidazole gradient (20–600 mM). The fractions containing recombinant proteins were identified by SDS–PAGE22 and further purified by size exclusion chromatography (SEC) using a Superdex 200 HR 10/30 column or Superdex 75 HR 10/30 column (GE Healthcare, Sweden), with buffer containing 50 mM Tris/HCl, pH 7.5 and 200 mM NaCl.
Enzymatic characterization of mycobacterial AhpC
Peroxide-dependent activity of the various forms of purified recombinant MbAhpC proteins were measured by coupling its activity with NADPH-oxidation (ɛ280 = 6220 M−1 s−1) catalyzed by MbTrxC and MbTrxB2. The peroxidase activity was carried out at 25 °C by monitoring the decrease in NADPH-absorbance at 340 nm for 120 sec using a stopped-flow spectrophotometer SX20 (Applied Photophysics, UK) equipped with a sample handling unit that comprises two drive syringes. The reaction mixture containing 0.1 mM NADPH, 50 mM HEPES buffer pH 7.0, 100 mM ammonium sulfate, 1 mM EDTA, 0.5 µM of MbTrxB2, 4 µM of MbTrxC, and 6 μM of MbAhpC, -AhpCΔ23–34, -AhpCT5A/D8A, -AhpCD22N/K25Q or -AhpCD22N/K25Q/D27N were loaded into drive syringe 1, and mixed with 50 µM of hydrogen peroxide, which was loaded into drive syringe 2, to initiate NADPH-oxidation. NADPH consumption measured in the absence of the respective proteins was taken as a control, and this background rate was subtracted from the experimental rate to determine the activity due to AhpC. All data reported here is the average of three independent measurements. The characterization of MbAhpC and MbAhpCT5A/D8A peroxidase activity were performed following the above mentioned, except with various increasing concentration of hydrogen peroxide (250 nm–50 μM) or t-bOOH (250 nm–30 μM), to initiate NADPH-oxidation. All the experimental curves were created using the program Origin Pro 9.0 (OriginLab Corporation).
Dynamic light scattering
Dynamic light scattering (DLS) of the oxidized and reduced (presence of 2 mM DTT) MbAhpC, -AhpCΔ23–34 and -AhpCT5A were carried out using a Malvern Zetasizer Nano ZS spectrophotometer. DLS experiments were performed in a low-volume quartz batch cuvette (ZEN2112, Malvern Instruments) using 12 μl of the respective protein solution. After 60 s equilibration time, the backscattering at 173° was collected for all proteins. Scattering intensities were analyzed using the in-built software, Zetasizer to calculate the hydrodynamic diameter (DH), size, and volume distribution.
NMR titration experiments of MbTrxC with MbAhpC
Samples for NMR experiments were prepared in 20 mM sodium phosphate, pH 6.5, 100 mM NaCl, 0.001% NaN3 and 10% D2O. All NMR measurements were performed on a Bruker Avance 700 MHz spectrometer, equipped with a 5 mm z-axis-gradient cryogenic probe at 298 K. The resonance assignments of 15N and NH resonances were achieved based on the previously reported NMR chemical shift data of mycobacterial TrxC from the BioMagnetic Resonance Bank (BMRB, accession code: 17242)23. To study the interaction between MbTrxC and MbAhpC, unlabeled MbAhpC or -AhpCT5A/D8A or -AhpCD22N/K25Q/D27N was titrated stepwise into a solution with 0.1 mM reduced 15N labeled MbTrxC. 1H-15N HSQC spectra were recorded with 2048 complex data points in in t 2 and 240 t 1 increments at molar ratios of 1:0, 1:0.5, 1:1 and 1:2 (MbTrxC:MbAhpC), respectively. Spectral width of 2270 and 11160 Hz were employed in F1 (15N) and F2 (1H), respectively. All data were processed with NMRPipe24 and NMRDraw24, and spectrums were analyzed using SPARKY25. In order to determine the interacting residues of MbTrxC with MbAhpC, the previously reported structure of reduced form of mycobacterial TrxC (PDB code: 2L4Q)23 was used, and the respective residues were visualized by PyMOL26.
Molecular docking of mycobacterial AhpC with TrxC
The interaction identified from the NMR titration experiments were used to perform docking studies to model the complex of mycobacterial AhpC and TrxC using the HADDOCK webserver27. The coordinates of mycobacterial AhpC (PDB ID: 2BMX)13 and TrxC (PDB ID: 2L4Q)23 were obtained from the Protein Data Bank. Residues experiencing significant perturbation during titration were used as restrains. In the case of TrxC, the directly interacting residues are specified as “active” and the neighbouring residues as “passive”, whereby for AhpC, the “active” region is the redox-center and the “passive” region is the surrounding region. The submitted subunits are placed in space with an approximate distance of 25 Å and a complex is formed through rigid body energy minimization, which resulted initial in 1000 structures. The top 200 lowest energy models from the above minimization are subjected to a simulated annealing in torsion-angle space procedure and subsequent flexible refinement in explicit solvents. Models which fulfilled the interfaced ligand RMSD cutoff of 5 Å were clustered into ten groups. The best model in the highest scoring cluster was then selected for analysis. The model was visualized using the PyMOL software package26.
Electron microscopy and 2D image analysis of MbAhpC and its mutants
MbAhpC and MbAhpCΔ23–34 were diluted to a final concentration of 80 μg/ml in buffer containing 50 mM Tris/HCl, pH 7.5 and 200 mM NaCl. In the case of the reduced protein, DTT was added to a final concentration of 2 mM. A volume of 4 μl of protein sample was applied to a 30 second glow discharged carbon coated copper TEM grid and negatively stained with 2% (v/v) uranyl acetate. Electron micrographs were recorded on a FEI Tecnai T12 transmission electron microscope (FEI) equipped with a 4 K CCD camera (FEI) operated at a voltage of 120 kV at a calibrated magnification of 66,350x. 37 micrographs were recorded at 0° angle for the reduced MbAhpC. A total of 4054 particles for reduced MbAhpC were selected for 2D projection analysis. The selection criteria of particles were, (i) the clear visibility of single molecules and (ii) separation from neighboring particles. Particles were picked using EMAN228 and processed with RELION 1.429.
Electronic supplementary material
Acknowledgements
This study was supported by the Academic Research Fund (AcRF) Tier 1 ID889 Ministery of Education, Singapore to G.G. and Nanyang Technological University Start Up Grant (S.B), and Tier I grant AcRF Tier 1, 2014-T1-001-019 (RG32/14) from the Ministry of Education (MOE) of Singapore (S.B). Dr. W. G. Saw and A. Kumar thank the authority of Nanyang Technological University for awarding research scholarships.
Author Contributions
C.F.W., J.S., and G.G. designed the experiments. C.F.W., J.S. M.S.S.M., W.G.S., Z.Y., S.B., A.K., and P.R. performed the experiments. C.F.W., J.S., M.S.S.M., W.G.S., Z.Y., S.B., and G.G. analyzed the data. C.F.W., J.S., M.S.S.M., and G.G. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05354-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hillas PJ, del Alba FS, Oyarzabal J, Wilks A, Ortiz De Montellano PR. The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis. J. Biol. Chem. 2000;275:18801–18809. doi: 10.1074/jbc.M001001200. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Global Tuberculosis Report 2016, http://www.who.int/tb/publications/global_report/en/ (2016).
- 3.Wengenack NL, Rusnak F. Evidence for isoniazid-dependent free radical generation catalyzed by Mycobacterium tuberculosis KatG and the isoniazid-resistant mutant KatG(S315T) Biochemistry. 2001;40:8990–8996. doi: 10.1021/bi002614m. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, et al. Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev. Mol. Med. 2011;13:e39. doi: 10.1017/S1462399411002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 6.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 2009;11:1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 2004;52:1291–1302. doi: 10.1111/j.1365-2958.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- 8.Chauhan R, Mande SC. Site-directed mutagenesis reveals a novel catalytic mechanism of Mycobacterium tuberculosis alkylhydroperoxidase C. Biochem. J. 2002;367:255–261. doi: 10.1042/bj20020545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherman DR, et al. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science. 1996;272:1641–1643. doi: 10.1126/science.272.5268.1641. [DOI] [PubMed] [Google Scholar]
- 10.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 11.Dip PV, et al. Structure, mechanism and ensemble formation of the alkylhydroperoxide reductase subunits AhpC and AhpF from Escherichia coli. Acta Crystallogr. D Biol. Crystallogr. 2014;70:2848–2862. doi: 10.1107/S1399004714019233. [DOI] [PubMed] [Google Scholar]
- 12.Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015;40:435–445. doi: 10.1016/j.tibs.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guimaraes BG, et al. Structure and mechanism of the alkyl hydroperoxidase AhpC, a key element of the Mycobacterium tuberculosis defense system against oxidative stress. J. Biol. Chem. 2005;280:25735–25742. doi: 10.1074/jbc.M503076200. [DOI] [PubMed] [Google Scholar]
- 14.Dip PV, et al. Key roles of the Escherichia coli AhpC C-terminus in assembly and catalysis of alkylhydroperoxide reductase, an enzyme essential for the alleviation of oxidative stress. Biochim. Biophys. Acta. 2014;1837:1932–1943. doi: 10.1016/j.bbabio.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Jaeger T, et al. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2004;423:182–191. doi: 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Akif M, Khare G, Tyagi AK, Mande SC, Sardesai AA. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J. Bacteriol. 2008;190:7087–7095. doi: 10.1128/JB.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higgins DG, Sharp PM. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene. 1988;73:237–244. doi: 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- 18.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 19.Master SS, et al. Oxidative stress response genes in Mycobacterium tuberculosis: role of ahpC in resistance to peroxynitrite and stage-specific survival in macrophages. Microbiology. 2002;148:3139–3144. doi: 10.1099/00221287-148-10-3139. [DOI] [PubMed] [Google Scholar]
- 20.Grüber G, et al. Expression, purification, and characterization of subunit E, an essential subunit of the vacuolar ATPase. Biochem. Biophys. Res. Commun. 2002;298:383–391. doi: 10.1016/S0006-291X(02)02468-3. [DOI] [PubMed] [Google Scholar]
- 21.Raman, M. & Martin, K. One solution for cloning and mutagenesis: In-Fusion[reg] HD Cloning Plus. Nat Meth11, doi:10.1038/nmeth.f.373 (2014).
- 22.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Olson AL, Neumann TS, Cai S, Sem DS. Solution structures of Mycobacterium tuberculosis thioredoxin C and models of intact thioredoxin system suggest new approaches to inhibitor and drug design. Proteins. 2013;81:675–689. doi: 10.1002/prot.24228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.SPARKY v. 3 (University of California, San Francisco, May 30, 2008).
- 26.Schrodinger, L. L. C. The PyMOL Molecular Graphics System, Version 1.8 (2015).
- 27.van Zundert GC, et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.