

Abstract
Background
Severe combined immunodeficiency (SCID) is fatal unless treated with hematopoietic stem cell transplant. Delay in diagnosis is common without newborn screening. Family history of infant death due to infection or known SCID (FH) has been associated with earlier diagnosis.
Objective
The aim of this study was to identify the clinical features that affect age at diagnosis (AD) and time to the diagnosis of SCID.
Methods
From 2005 to 2016, 147 SCID patients were referred to the Asian Primary Immunodeficiency Network. Patients with genetic diagnosis, age at presentation (AP), and AD were selected for study.
Results
A total of 88 different SCID gene mutations were identified in 94 patients, including 49 IL2RG mutations, 12 RAG1 mutations, 8 RAG2 mutations, 7 JAK3 mutations, 4 DCLRE1C mutations, 4 IL7R mutations, 2 RFXANK mutations, and 2 ADA mutations. A total of 29 mutations were previously unreported. Eighty-three of the 94 patients fulfilled the selection criteria. Their median AD was 4 months, and the time to diagnosis was 2 months. The commonest SCID was X-linked (n = 57). A total of 29 patients had a positive FH. Candidiasis (n = 27) and bacillus Calmette–Guérin (BCG) vaccine infection (n = 19) were the commonest infections. The median age for candidiasis and BCG infection documented were 3 months and 4 months, respectively. The median absolute lymphocyte count (ALC) was 1.05 × 109/L with over 88% patients below 3 × 109/L. Positive FH was associated with earlier AP by 1 month (p = 0.002) and diagnosis by 2 months (p = 0.008), but not shorter time to diagnosis (p = 0.494). Candidiasis was associated with later AD by 2 months (p = 0.008) and longer time to diagnosis by 0.55 months (p = 0.003). BCG infections were not associated with age or time to diagnosis.
Conclusion
FH was useful to aid earlier diagnosis but was overlooked by clinicians and not by parents. Similarly, typical clinical features of SCID were not recognized by clinicians to shorten the time to diagnosis. We suggest that lymphocyte subset should be performed for any infant with one or more of the following four clinical features: FH, candidiasis, BCG infections, and ALC below 3 × 109/L.
Keywords: severe combined immunodeficiency, family history, candidiasis, absolute lymphocyte count, newborn screening
Introduction
Severe combined immunodeficiency (SCID) is a group of genetic diseases causing profound developmental and functional impairment of T cells, affecting cellular and humoral immunities. Currently, at least 49 genes are identified to be responsible for SCID and its variants (1–3). Of all the SCID genes, the commonest gene involved is the IL-2 receptor gamma chain gene (IL2RG), which accounted for 45 and 19% of SCID cases before and after the T-cell receptor excision circle (TREC) newborn screening, and was introduced in USA (3–5). Patients typically present with recurrent infections from opportunistic pathogens and live-attenuated vaccines, such as bacillus Calmette–Guérin (BCG) (6), chronic diarrhea, and failure to thrive (FTT), eventually die within the first 2 years of life if left untreated (7). Patients typically have low absolute lymphocyte count (ALC). They have been classified by the number of B lymphocytes as B+ or B− and recently by the causative genetic mutation.
The definitive treatment for SCID is hematopoietic stem cell transplant (HSCT). In addition, gene therapy serves as an alternative for X-linked and adenosine deaminase (ADA)-deficient SCID if suitable HSC donors are not available (8). SCID patients have a 94% survival rate if they undergo HSCT within the first 3.5 months of life (9). To facilitate timely HSCT, an early diagnosis must be made. However, delay in diagnosis is common due to the lack of awareness of the distinctive presenting features of SCID, such as recurrent and persistent opportunistic infections (2). To date, the only feature that is associated with an earlier diagnosis is a positive family history of infant death due to infection or known SCID in USA (10). In addition, family history of SCID is associated with earlier HSCT before 3.5 months (9, 11). Our present study aimed to identify the clinical features that could help clinicians diagnose SCID earlier by comparing the age and time to the diagnosis of patients with or without certain clinical features.
Materials and Methods
Patient Source and Selection
The Asian Primary Immunodeficiency Network (APIN) is a primary immunodeficiency (PID) referral network established in 2009 by The University of Hong Kong as a platform for consultation and offering free genetic testing for suspected PID in over 70 centers in Asia and Africa. Its database stores clinical information provided by the referring doctors, laboratory results, and genetic test reports (12–14). From 2005 to 2016, 147 SCID patients were referred from 23 centers to the APIN for consultation and genetic testing, 42 of whom were reported in our previous study (13). In our study, we included patients with documented age at presentation (AP) and diagnosis. Among them, we selected patients with genetic diagnosis for identifying factors that affected age and time to diagnosis of SCID.
Data Collection
The referring doctors provided the clinical records of patients, together with the laboratory results. Demographic data, clinical presentation, and progress as well as investigation results including ALC and lymphocyte subsets of the patients were recorded. We only considered the clinical features and progression before the diagnosis of SCID was made. We defined AP as the age when the first clinical symptom was documented in the clinical record, age at diagnosis (AD) as the age when clinical diagnosis of SCID was made, and time to diagnosis as the duration between AP and AD. We only considered patients to have certain clinical feature if that feature was stated in the referral summary. We defined recurrent infections as more than one episode of infections affecting similar systems. We considered the infection to be severe if at least one of the following was present: life-threatening complications (such as acute respiratory distress syndrome and sepsis), intensive care unit (ICU) admission, and life support being used (intubation, ventilation, and resuscitation). We defined opportunistic infection as an infection with at least one of the following pathogens was involved: BCG, Candida, Pseudomonas aeruginosa, Acinetobacter baumannii, Pneumocystis jiroveci (PCP), Aspergillus, cytomegalovirus (CMV), and herpes zoster virus. We defined opportunistic infections by Candida if patients were documented to have invasive candidiasis, candidemia or persistent oral candidiasis (15). We presented the ALC recorded at the time of SCID diagnosis. Patients were said to have lymphopenia if ALC below 3 × 109/L as described previously (10). Since there is no universally agreed cutoff of B-cell number to distinguish B+ and B− SCID, we defined the cut-off as 134/μL based on the CD19+ B-cell counts of patients with B+ genotypes (IL2RG, IL7R, and JAK3) and those with B− genotypes (ADA, DCLRE1C, RAG1, and RAG2) (Figure 1).
Figure 1.

CD19+ cell counts of patients with B+ and B− genotypes. B+ genotypes group consisted of CD19+ cell counts of patients with mutations found in IL2RG (n = 43), IL7R (n = 3), and JAK3 (n = 5). B− genotypes group consisted of CD19+ cell counts of patients with mutations found in ADA (n = 1), DCLRE1C (n = 3), RAG1 (n = 3), and RAG2 (n = 2). The cutoff for distinguishing B+ and B− patients was 134 CD19+ cells/μL. Three patients with IL2RG mutations were classified as having B− SCID.
Genetic analysis was performed in the Department of Pediatrics and Adolescent Medicine of the University of Hong Kong using PCR and direct sequencing (Table SE1 in Supplementary Material) (13). Genetic and functional studies on PID, data archival in the APIN database, and DNA storage were approved by the Clinical Research Ethics Review Board of the University of Hong Kong and Queen Mary Hospital (Ref. no. UW 08-301) in accordance with the Declaration of Helsinki, with written informed consent obtained from parents of subjects. HGMD Pro version 2016.4 (16) and Immunodeficiency mutation databases (IDbases) (17) were used to identify unreported mutations. The nomenclatures of cDNA mutations were based on coding region. For each unreported mutation, the population frequency was analyzed by Exome Aggregation Consortium Browser (18). Effects of missense mutations on protein functions were predicted by PANTHER (19), PHD-SNP (20), SIFT (21), SNAP (22), Meta-SNP (23), and PolyPhen2 (24). The protein structure predicted to be involved was identified using NCBI Protein database (25) and UniProt Knowledgebase database (26).
Statistical Analysis
For descriptive statistics, all data were expressed in median and range (month). Univariate analysis was performed using Mann–Whitney U test; multivariate linear regression was performed for all factors that were significant (p < 0.05) in univariate analysis. We defined statistical significance as p < 0.05, and 95% confidence interval did not contain 0 in multivariate analysis. We did not include opportunistic infection group in the multivariate linear regression to avoid multicollinearity.
Patients with missing categorical data such as clinical features were considered to be without the features. Patients with missing numerical data such as ALC were not analyzed when analyzing median and range.
Results
Patients Selection
From 147 SCID patients referred to the APIN, 131 of them had documented AP and diagnosis. Among these patients, 83 of them had genetic diagnosis (Figure 2). Sixteen patients were excluded from the study due to the lack of AP (n = 4), the lack of AD (n = 9), and being diagnosed by screening (n = 3). Among the 16 patients excluded from the study, 11 of them had genetic diagnosis. Altogether, molecular diagnosis of SCID was identified in 94 patients in our cohort.
Figure 2.

Patients selection algorithm in this study. From 147 SCID entries in the Asian Primary Immunodeficiency Network (APIN) database, 131 patients were included in our study and 16 patients were excluded from our study. Three patients were excluded as they were diagnosed by screening either antenatally or at birth. Thirteen patients were excluded due to the lack of age at presentation (n = 4) or the lack of age at diagnosis (n = 9). 1Cordocentesis was performed due to positive family history of SCID, revealed low CD4+ count. 2Complete blood count, lymphocyte subsets, and immunoglobulins measurement were performed in one patient due to positive family history, revealed severe T- and B-cell lymphopenia and low serum IgA and IgM; newborn T-cell receptor excision circle (TREC) screening revealed 0 TREC copy in another patient.
Genetic Mutations in Patients
The genetic mutations of the 83 SCID patients included in our study and 11 SCID patients excluded from our study are shown in Table 1 and Table SE1 in Supplementary Material, respectively. The commonest gene identified was IL2RG (n = 65), followed by RAG1 (n = 7), RAG2 (n = 7), JAK3 (n = 5), DCLRE1C (n = 4), IL7R (n = 3), RFXANK (n = 2), and ADA (n = 1). Eighty-eight different mutations were identified in this study (49 IL2RG mutations, 12 RAG1 mutations, 8 RAG2 mutations, 7 JAK3 mutations, 4 DCLRE1C mutations, 4 IL7R mutations, 2 RFXANK mutations, and 2 ADA mutations). There was no difference in clinical features between X-linked and autosomal recessive SCID patients (Table SE2 in Supplementary Material).
Table 1.
Genetic mutations of SCID patients (n = 83).
| No | Gene | Intron (I)/exon (E) | Nucleotide change | Predicted change |
|---|---|---|---|---|
| P001 | IL2RG | E1 | c.3G>T | M1I |
| P002 | IL2RG | E2 | c.127delA | T43fsX70 |
| P003 | IL2RG | E2 | c.202G>T | E68X |
| P004a | IL2RG | E2 | c.202G>A | E68K |
| P004b | IL2RG | E2 | c.202G>A | E68K |
| P005 | IL2RG | E2 | c.202G>A | E68K |
| P006 | IL2RG | E2 | c.252C>A | N84K |
| P007 | IL2RG | I2 | g.IVS2−15A>G | Predicted aberrant splicing |
| P008 | IL2RG | I2 | g.IVS2−15A>G | Predicted aberrant splicing |
| P011e | IL2RG | E3 | c.310_311delinsG | H104fsX146 |
| P012 | IL2RG | E3 | c.340G>T | G114C |
| P013e | IL2RG | E3 | c.359dupA | K120fsX167 |
| P014 | IL2RG | E3 | c.362delA | E121fsX146 |
| P015 | IL2RG | E3 | c.365T>C | I122T |
| P016 | IL2RG | E3 | c.365T>C | I122T |
| P017e | IL2RG | E3 | c.371T>C | L124P |
| P018 | IL2RG | E3 | c.376C>T | Q126X |
| P019e | IL2RG | E3 | c.376C>T | Q126X |
| P020 | IL2RG | E3 | c.383T>C | F128S |
| P021 | IL2RG | E3 | c.386T>A | V129D |
| P022 | IL2RG | E3 | c.406_415del | R136fsX143 |
| P023e | IL2RG | E3 | c.421delC | Q141fsX146 |
| P024 | IL2RG | I3/E4 junction | g.IVS3−2A>T | Predicted aberrant splicing |
| P025e | IL2RG | E4 | c.507delG | Q169fsX170 |
| P026 | IL2RG | E4 | c.507delG | Q169fsX170 |
| P027e | IL2RG | E4 | c.562C>T | Q188X |
| P028 | IL2RG | E4 | c.562C>T | Q188X |
| P030 | IL2RG | E5 | c.658_659del | T220fsX227 |
| P031 | IL2RG | E5 | c.664C>T | R222C |
| P032e | IL2RG | E5 | c.670C>T | R224W |
| P033 | IL2RG | E5 | c.670C>T | R224W |
| P034 | IL2RG | E5 | c.676C>T | R226C |
| P036e | IL2RG | E5 | c.677G>A | R226H |
| P037 | IL2RG | E5 | c.694G>C | G232R |
| P038 | IL2RG | E5 | c.709T>C | W237R |
| P039e | IL2RG | E5 | c.711G>A | W237X |
| P040e | IL2RG | E5 | c.722G>T | S241I |
| P041 | IL2RG | E5 | c.741delG | G247fsX272 |
| P042 | IL2RG | E5 | c.741_742insG | E248fsX302 |
| P044 | IL2RG | E6 | c.811G>T | G271X |
| P045e | IL2RG | E6 | c.835delG | V279fsX293 |
| P046 | IL2RG | E6/I6 junction | c.854G>T | R285L |
| P047e | IL2RG | E6/I6 junction | c.854G>Af | Predicted aberrant splicing |
| R285Q | ||||
| P048e | IL2RG | E6/I6 junction | c.854G>Af | Predicted aberrant splicing |
| R285Q | ||||
| P049e | IL2RG | E6/I6 junction | c.854G>Af | Predicted aberrant splicing |
| R285Q | ||||
| P050e | IL2RG | E6/I6 junction | c.854G>Af | Predicted aberrant splicing |
| R285Q | ||||
| P051 | IL2RG | E6/I6 junction | c.854G>Af | Predicted aberrant splicing |
| R285Q | ||||
| P052 | IL2RG | E6/I6 junction | c.854G>Af | Predicted aberrant splicing |
| R285Q | ||||
| P053e | IL2RG | I6-I7 | g.IVS6-72_IVS7-11del | Predicted exon 7 deletion |
| P055 | IL2RG | I6/E7 junction | g.IVS6−2A>C | Predicted aberrant splicing |
| P056 | IL2RG | I6 | g.IVS6+3G>T | Predicted aberrant splicing |
| P057e | IL2RG | I6 | g.IVS6+5G>A | Predicted aberrant splicing |
| P058e | IL2RG | I6 | g.IVS6+5G>A | Predicted aberrant splicing |
| P059 | IL2RG | E7 | c.865C>T | R289X |
| P060 | IL2RG | E8 | c.929G>A | W310X |
| P062 | IL2RG | E8 | c.979_980delinsTT | E327L |
| P063a | IL2RG | E8 | c.982C>T | R328X |
| P064 | ADA | E7 | c.646G>A | G216R |
| E11 | c.1018_1019del | K340fsX348 | ||
| P065b | DCLRE1C | E1-E3 | Gross deletion | Gross deletion |
| P066b | DCLRE1C | E1-E4 | Gross deletion | Gross deletion |
| P068e | DCLRE1C | I3/E4 junction | c.IVS3−1G>T | I83-G102del |
| Exon 4 skippeda | ||||
| E8 | c.632G>T | G211V | ||
| P069e | IL7R | E1 | c.65G>T | S22I |
| E2/I2 junction | g.IVS2+2T>A | Predicted aberrant splicing | ||
| P070b,e | IL7R | E5 | c.562delC | L188X |
| P071b | IL7R | E5 | c.616C>T | R206X |
| P072 | JAK3 | E2 | c.115dupC | Q39fsX51 |
| E13 | c.1744C>T | R582W | ||
| P073 | JAK3 | E3 | c.307C>T | R103C |
| E10 | c.1333C>T | R445X | ||
| P074e | JAK3 | E13 | c.1763A>C | H588P |
| P075b,e | JAK3 | I14 | g.IVS14−11G>A | 638_639insPPX |
| c.1914_1915insCCCCCTTAGa | ||||
| P076c | JAK3 | E16 | c.2062A>T | I688F |
| P077 | RAG1 | E2 | c.994C>T | R332X |
| E2 | c.3074dupT | L1025fsX1064 | ||
| P078e | RAG1 | E2 | c.1178delG | G393fsX402 |
| E2 | c.2095C>T | R699W | ||
| P079 | RAG1 | E2 | c.1328G>A | R443K |
| E2 | c.2486_2490del | R829fsX869 | ||
| P080 | RAG1 | E2 | c.1681C>T | R561C |
| E2 | c.2561G>A | G854D | ||
| P081d | RAG1 | E2 | c.2005G>A | E669K |
| P083 | RAG1 | E2 | c.2324T>A | L775Q |
| E2 | c.2918G>A | R973H | ||
| P084e | RAG2 | E1/I1 junction | c.-28G>C | Predicted aberrant splicing |
| E2 | c.358delG | V120fsX130 | ||
| P085b | RAG2 | E2 | c.104G>T | G35V |
| P086b | RAG2 | E2 | c.104G>T | G35V |
| P087 | RAG2 | E2 | c.104G>T | G35V |
| E2 | c.475C>T | R159C | ||
| P088b | RAG2 | E2 | c.218G>A | R73H |
| P089 | RAG2 | E2 | c.442C>T | R148X |
| E2 | c.685C>T | R229W | ||
| P091 | RFXANK | E3/I3 junction | g.IVS3+1delG | Predicted aberrant splicing |
| E5 | c.299_300del | Q100fsX113 | ||
| P092b | RFXANK | E5 | c.299_300del | Q100fsX113 |
ADA, adenosine deaminase; DCLRE1C, DNA cross-link repair enzyme 1C; IL2RG, interleukin-2 receptor subunit gamma; IL7R, interleukin-7 receptor subunit alpha; JAK3, Janus kinase 3; RAG, recombinase activating genes; RFXANK, regulatory factor X-associated ankyrin-containing protein.
aFrom RT-PCR results.
bHomozygous mutations.
cP076 was a B+NK− patient with hypogammaglobulinemia (IgG 1.45 g/L, IgA 0.23 g/L, and IgM 0.26 g/L) whose mother was a heterozygous carrier.
dOnly one mutation was found.
ePatients reported in our previous study (13).
fPrevious study reported 854G>A may cause R285Q or skipping of exon 6 (27). P004a and P004b and P063a and P063b (Table SE1 in Supplementary Material) were from the same kindred.
Genetic mutations in all SCID genes were not evenly distributed, and two mutations were seen three or more times in unrelated patients. c.854G>A mutation was seen in six unrelated patients with IL2RG mutation. c.104G>T mutation was observed in three unrelated patients with RAG2 mutation.
Twenty-two C>T or G>A mutations within CpG dinucleotides were documented (8 IL2RG mutations, 5 RAG1 mutations, 4 RAG2 mutations, 3 JAK3 mutations, 1 IL7R mutation, and 1 ADA mutation). These mutations accounted for 25% of all mutations and were involved in 31 patients (18 in IL2RG, 5 in RAG1, 3 in RAG2, 2 in JAK3, 1 in IL7R, and 1 in ADA).
There were 29 unreported mutations identified in our patients, including 23 IL2RG mutations, 3 RAG1 mutations, 1 JAK3 mutation, 1 RAG2 mutation, and 1 RFXANK mutations (Table SE3 in Supplementary Material). Effects of these unreported mutations on protein functions were predicted by multiple tools and are shown in Table SE3 in Supplementary Material.
Characteristics of Patients That Fulfilled Selection Criteria (n = 83)
Characteristics of patients included in our study (n = 131) are shown in Tables 2–4. For patients that fulfilled selection criteria (n = 83), 88.0% were male (n = 73) and 75.9% were Chinese (n = 63). The median AP was 2 months (0.1–6 months), AD 4 months (0.5–18 months), and time to diagnosis 2 months (0–14 months). Twenty-nine patients (34.9%) had a family history of early infant death (FH), among them one patient had a family history of SCID and one patient had a family history of PID. Parental consanguinity was present in four kindreds. The median ALC was 1.05 × 109/L (0.134−52.2 × 109/L, n = 70) with 88.6% below 3 × 109/L (n = 62). The major immunophenotype was B+ SCID (n = 51) (Tables 2 and 3).
Table 2.
Characteristics of patients included in our study (n = 131) at SCID diagnosis.
| With genetic diagnosis | Without genetic diagnosis | |
|---|---|---|
| n = 83 | n = 48 | |
| Gender | Number (%) | Number (%) |
| Male | 73 (88.0) | 33 (68.8) |
| Female | 10 (12.0) | 15 (31.3) |
| Ethnicity | Number (%) | Number (%) |
| Chinese | 63 (75.9) | 30 (62.5) |
| Southeast Asian | 12 (14.5) | 4 (8.3) |
| Indonesian | 1 (1.2) | 0 (0) |
| Malay | 3 (3.6) | 3 (6.3) |
| Philippino | 1 (1.2) | 0 (0) |
| Thai | 5 (6.0) | 1 (2.1) |
| Vietnamese | 2 (2.4) | 0 (0) |
| Indian | 2 (2.4) | 9 (18.8) |
| Algerian | 5 (6.0) | 0 (0) |
| Arabian | 1 (1.2) | 3 (6.3) |
| Australian | 0 (0) | 1 (2.1) |
| Korean | 0 (0) | 1 (2.1) |
| Positive family history | Number (%) | Number (%) |
| Early infant death | 29 (34.9) | 13 (27.1) |
| Consanguinity | 4 (4.8) | 1 (2.1) |
| Age in months | Median (range) | Median (range) |
| Age at presentation | 2 (0.1–6) | 2 (0–19) |
| Age at diagnosis | 4 (0.5–18) | 4 (0.1–27) |
| Time to diagnosis | 2 (0–14) | 2 (0–16) |
| SCID phenotype | Number (%) | Number (%) |
| B+ | 51 (61.4) | 18 (37.5) |
| B− | 15 (18.1) | 24 (50.0) |
| Others | 17 (20.5)a | 6 (12.5)b |
| Median (range) | Median (range) | |
| Absolute lymphocyte count (109/L) | 1.05 (0.134–52.2)c | 0.77 (0.09–13.46)d |
aMaternal engraftment (n = 1), unknown (n = 16).
bUnknown (n = 6).
cIn 70 patients.
dIn 44 patients.
Table 4.
Clinical features of patients included in our study (n = 131).
| With genetic diagnosis | Without genetic diagnosis | |
|---|---|---|
| n = 83 | n = 48 | |
| Number (%) | Number (%) | |
| Classical SCID triad | ||
| Failure to thrive | 13 (15.7) | 16 (33.3)a |
| Chronic diarrhea | 42 (50.6) | 27 (56.3) |
| Recurrent infections | 50 (60.2) | 28 (58.3) |
| Infection by systems | ||
| Respiratory infection | 61 (73.5)c | 34 (70.8)d |
| Non-bacillus Calmette–Guérin (BCG) skin and soft tissue infection | 7 (8.4) | 11 (22.9) |
| Gastrointestinal infection | 42 (50.6) | 23 (48.9) |
| Urogenital infection | 2 (2.4) | 1 (2.1) |
| Musculoskeletal infection | 3 (3.6) | 1 (2.1) |
| Central nervous system infection | 1 (1.2) | 1 (2.1) |
| Sepsis | 18 (21.7) | 8 (16.7) |
| Severe infections | 39 (47.0) | 26 (54.2) |
| Intensive care unit admission | 24 (28.9) | 14 (29.2) |
| Life support | 26 (31.3) | 14 (29.2) |
| Intubation and ventilation | 21 (25.3) | 8 (16.7) |
| Resuscitation and/or inotrope support | 5 (6.0) | 9 (18.8) |
| Life-threatening complication | 40 (48.2) | 18 (37.5) |
| Sepsis | 18 (21.7) | 8 (16.7) |
| Respiratory distress/failure | 26 (31.3) | 13 (27.1) |
| Acute heart failure | 2 (2.4) | 1 (1.2) |
| Opportunistic infections | 50 (60.2) | 28 (58.3) |
| Bacterial | 8 (9.6) | 1 (2.1) |
| Pseudomonas aeruginosa | 6 (7.2) | 0 (0) |
| Acinetobacter baumanii | 4 (4.8) | 1 (2.1) |
| Viral | 9 (10.8) | 12 (25.0) |
| Cytomegalovirus (CMV) | 8 (9.6)e | 12 (25.0)b |
| Herpes zoster | 1 (1.2) | 0 (0) |
| Bacillus Calmette–Guérin (BCG) infection | 19 (22.9)f | 8 (16.7) |
| Local | 8 (9.6) | 2 (4.2) |
| Regional | 2 (2.4) | 1 (2.1) |
| Disseminated | 9 (10.8) | 5 (10.4) |
| Candidiasis | 27 (32.5)g | 16 (33.3) |
| Persistent oral thrush | 22 (26.5) | 10 (20.8) |
| Gastrointestinal tract infection | 3 (3.6) | 2 (4.2) |
| Candidemia | 2 (2.4) | 4 (8.3) |
| Fungal | 3 (3.6) | 2 (4.2) |
| Pneumocystis jiroveci | 2 (2.4) | 1 (2.1) |
| Aspergillosis | 1 (1.2) | 1 (2.1) |
| Hepatosplenomegaly | 12 (14.5) | 9 (18.8) |
ap = 0.0189.
bp = 0.0185.
cA total of 53 patients with genetic diagnosis had pneumonia (63.9%).
dA total of 32 patients without genetic diagnosis had pneumonia (66.7%).
eThe median age for CMV infection documented was 2.25 months (n = 8).
fThe median age for BCG infection documented was 4 months (n = 14).
gThe median age for candidiasis documented was 3 months (n = 25).
Table 3.
Lymphocyte subset for patients included in our study (n = 131).
| Patient | Mutation gene | ALC (×109/L) | CD3+ cells/μL (%) | CD19+ cells/μL (%) | CD16/56+ cells/μL (%) |
|---|---|---|---|---|---|
| B+ SCID | |||||
| P006 | IL2RG | 0.4 | 9.2 (2.3) | 385.6 (96.4) | 2.4 (0.6) |
| P008 | IL2RG | 0.95 | 0 (0) | 931 (98) | 9.5 (1) |
| P011 | IL2RG | 1.16 | 3.48 (0.3) | 972 (83.8) | 2.3 (0.2) |
| P013 | IL2RG | 0.31 | 0 (0) | 270 (87) | 0 (0) |
| P014 | IL2RG | 2.93 | 468 (16) | 2,344 (80) | 58.6 (2) |
| P015 | IL2RG | 0.51 | 0 (0) | 459 (90) | 10.2 (2) |
| P017 | IL2RG | 0.7 | 0.7 (0.1) | 663 (94.7) | 36.4 (5.2) |
| P018 | IL2RG | 1 | 20 (2) | 890 (89) | 0 (0) |
| P019 | IL2RG | 2.63 | 26.3 (1) | 2,525 (96) | 26.3 (1) |
| P020 | IL2RG | 0.38 | 22.8 (6) | 345.8 (91) | 0 (0) |
| P021b | IL2RG | 1.66 | 596 (35.9) | 1,061 (63.9) | 2 (0.12) |
| P022 | IL2RG | 0.33 | 7.59 (2.3) | 313.5 (95) | 6.6 (2) |
| P024b | IL2RG | 3.43 | 504 (14.7) | 2,867 (83.6) | 58.3 (1.7) |
| P025 | IL2RG | 1.4 | 0 (0) | 1,302 (93) | 42 (3) |
| P026 | IL2RG | 0.33 | 16.5 (5) | 290.4 (88) | 9.9 (3) |
| P027 | IL2RG | 1.1 | 5.5 (0.5) | 1,022 (92.9) | 14.3 (1.3) |
| P028 | IL2RG | 5 | 4,600 (92) | 300 (6) | 100 (2) |
| P030 | IL2RG | 0.94 | 16 (1.7) | 620.4 (66) | 192.7 (20.5) |
| P031 | IL2RG | 5.1 | 948 (18.6) | 3,042 (59.7) | 928.7 (18.2) |
| P032 | IL2RG | 1.1 | 11 (1) | 979 (89) | 110 (10) |
| P033 | IL2RG | 0.99 | 5 (0.5) | 585.1 (59.1) | 17.8 (1.8) |
| P034 | IL2RG | 1.34 | 0 (0) | 1,112 (83) | 160.8 (12) |
| P036 | IL2RG | 1.11 | 11.1 (1) | 455.1 (41) | 577 (52) |
| P038 | IL2RG | 0.6 | 240 (40) | 324 (54) | 33 (5.5) |
| P039 | IL2RG | 1.72 | 0 (0) | 1,170 (68) | 498.8 (29) |
| P041 | IL2RG | 0.62 | 12.4 (2) | 545.6 (88) | 37.2 (6) |
| P044 | IL2RG | 0.9 | 0 (0) | 846 (94) | 18 (2) |
| P045 | IL2RG | 1.84 | 0 (0) | 1,748 (95) | 73.6 (4) |
| P047 | IL2RG | 1.41 | 155.1 (11) | 1,197 (84.9) | 18.3 (1.3) |
| P048 | IL2RG | 4.94 | 0 (0) | 4,841 (98) | 98.8 (2) |
| P049 | IL2RG | 2.1 | 0 (0) | 2,079 (99) | 0 (0) |
| P050 | IL2RG | 0.53 | 5.3 (1) | 424 (80) | 21.2 (4) |
| P051 | IL2RG | 1.1 | 0 (0) | 1,067 (97) | 11 (1) |
| P052 | IL2RG | 1.86 | 223.2 (12) | 1,600 (86) | 0 (0) |
| P053 | IL2RG | 1 | 52 (5.2) | 892 (89.2) | N/A (N/A) |
| P055 | IL2RG | 0.9 | 9 (1) | 828 (92) | 9 (1) |
| P056 | IL2RG | 1.3 | 1 (0.08) | 1,282 (98.6) | 9.6 (0.74) |
| P058 | IL2RG | 1.3 | 0 (0) | 611 (47) | 18.2 (1.4) |
| P059 | IL2RG | 1.18 | 0 (0) | 1,133 (96) | 35.4 (3) |
| P060 | IL2RG | 1.5 | 45 (3) | 1,350 (90) | 30 (2) |
| P063a | IL2RG | 0.62 | 61.4 (9.9) | 484.2 (78.1) | 46.5 (7.5) |
| P069 | IL7R | 1.89 | 183.3 (9.7) | 1,111 (58.8) | 565.1 (29.9) |
| P070 | IL7R | 1.21 | 147.6 (12.2) | 756.3 (62.5) | 410.2 (33.9) |
| P071 | IL7R | 0.785 | 1.6 (0.2) | 148.4 (18.9) | 433.3 (55.2) |
| P072 | JAK3 | 2.52 | 1,738 (69) | 730.8 (29) | 0 (0) |
| P073 | JAK3 | 0.47 | 4.7 (1) | 437.1 (93) | N/A (N/A) |
| P074 | JAK3 | 0.35 | 91 (26) | 196 (56) | N/A (N/A) |
| P075 | JAK3 | 0.49 | 6.4 (1.3) | 266.1 (54.3) | 19.6 (4) |
| P076 | JAK3 | 1.5 | 12.6 (0.84) | 1,377 (91.77) | 54.5 (3.63) |
| P078 | RAG1 | 7.64 | 3,965 (51.9) | 267.4 (3.5) | 3,705 (48.5) |
| P091 | RFXANK | 1.59 | 624.9 (39.3) | 936.5 (58.9) | 47.7 (3) |
| P094 | N/A | 2.06 | 195.7 (9.5) | 1,788 (86.8) | 76.2 (3.7) |
| P098 | N/A | 1.23 | 764.2 (62.13) | 156.9 (12.76) | 263.1 (21.39) |
| P099 | N/A | 13.46 | 9,826 (73) | 1,346 (10) | 1,750 (13) |
| P109 | N/A | 0.88 | 295.7 (33.6) | 460.2 (52.3) | 89.5 (10.2) |
| P110 | N/A | 2.06 | 68 (3.3) | 1,593 (77.3) | 345.7 (16.8) |
| P111 | N/A | 1.46 | 18.3 (1.25) | 1,387 (95) | 6.6 (0.45) |
| P112 | N/A | 2.42 | 217.8 (9) | 1,500 (62) | 532.4 (22) |
| P115 | N/A | 1.84 | 18.4 (1) | 1,472 (80) | 294.4 (16) |
| P116 | N/A | 1.3 | 26 (2) | 1,040 (80) | 130 (10) |
| P117 | N/A | 2.1 | 396.9 (18.9) | 573.3 (27.3) | 136.5 (6.5) |
| P120 | N/A | 1.09 | 21.8 (2) | 1,030 (94.5) | 3.3 (0.3) |
| P121 | N/A | 0.5 | 20 (4) | 245 (49) | 205 (41) |
| P122 | N/A | 0.41 | 32.8 (8) | 278.8 (68) | 86.1 (21) |
| P123b | N/A | 1.28 | 65.3 (5.1) | 833.3 (65.1) | 381.4 (29.8) |
| P124 | N/A | 0.8 | 15.2 (1.9) | 724 (90.5) | 43.2 (5.4) |
| P126 | N/A | 0.84 | 342.7 (40.8) | 207.5 (24.7) | 197.4 (23.5) |
| P128 | N/A | 3.5 | 2,485 (71) | 455 (13) | 455 (13) |
| P137 | N/A | 0.9 | 9.9 (1.1) | 136.8 (15.2) | 419.4 (46.6) |
| B− SCID | |||||
| P001 | IL2RG | 0.14 | 0 (0) | 70 (50) | 1.4 (1) |
| P002 | IL2RG | 0.67 | 636.5 (95) | 13.4 (2) | 0 (0) |
| P003 | IL2RG | 3.6 | 3,456 (96) | 0 (0) | N/A (N/A) |
| P005 | IL2RG | 0.18 | 7.2 (4) | 3.6 (2) | 145.8 (81) |
| P064 | ADA | 0.21 | 4 (1.9) | 1 (0.48) | 16.8 (8) |
| P065 | DCLRE1C | 0.65 | 110.5 (17) | 26 (4) | 78 (12) |
| P066 | DCLRE1C | 1.2 | 36 (3) | 24 (2) | 1,080 (90) |
| P068 | DCLRE1C | 0.72 | 7.2 (1) | 0.72 (0.1) | 672.5 (93.4) |
| P079 | RAG1 | 0.96 | 144 (15) | 1.9 (0.2) | 796.8 (83) |
| P080 | RAG1 | 0.134 | 132 (98.6) | 0.04 (0.03) | 1.5 (1.1) |
| P083 | RAG1 | 0.34 | 80.6 (23.7) | 3.1 (0.9) | 190.1 (55.9) |
| P084 | RAG2 | 0.74 | 7.4 (1) | 7.4 (1) | 666 (90) |
| P087 | RAG2 | 2.55 | 2.6 (0.1) | 132.6 (5.2) | 2,020 (79.2) |
| P088 | RAG2 | 28.36 | 26,772 (94.4) | 0 (0) | 623.9 (2.2) |
| P092 | RFXANK | 1.019 | 276.1 (27.1) | 19.4 (1.9) | 25.5 (2.5) |
| P093 | N/A | 0.31 | N/A (N/A) | 2 (0.65) | 120.9 (39) |
| P095 | N/A | 3.38 | 3,191 (94.4) | 33.8 (1) | 33.8 (1) |
| P097 | N/A | 2.49 | 2,366 (95) | 18.9 (0.76) | 49.8 (2) |
| P100 | N/A | 0.489 | 477.8 (97.7) | 2.9 (0.6) | 4.9 (1) |
| P101 | N/A | 0.242 | 15 (6.19) | 2.9 (1.2) | 206.4 (85.3) |
| P102 | N/A | 0.09 | 41.1 (45.7) | 1.4 (1.6) | 19.5 (21.7) |
| P103 | N/A | 1.8 | 1,499 (83.3) | 3.6 (0.2) | 257.4 (14.3) |
| P104 | N/A | 0.138 | 26.2 (19) | 1.4 (1) | 93.8 (68) |
| P105 | N/A | 0.72 | 144 (20) | 0.72 (0.1) | 537.1 (74.6) |
| P106 | N/A | 0.42 | 408.2 (97.2) | 1.3 (0.3) | 4.6 (1.1) |
| P107 | N/A | 0.65 | 76.7 (11.8) | 29.3 (4.5) | 490.8 (75.5) |
| P108 | N/A | 0.8 | 40 (5) | 14.4 (1.8) | 656 (82) |
| P114 | N/A | 0.59 | 11.8 (2) | 15.9 (2.7) | 472 (80) |
| P118 | N/A | 0.19 | 0 (0) | 39.9 (21) | 0.38 (0.2) |
| P125 | N/A | 0.84 | 579.6 (69) | 100.8 (12) | 134.4 (16) |
| P127 | N/A | 0.53 | 312.2 (58.9) | 73.1 (13.8) | 19.6 (3.7) |
| P130 | N/A | 0.29 | 70.8 (24.4) | 45.8 (15.8) | 150.8 (52) |
| P131 | N/A | 0.8 | 16 (2) | 0 (0) | 768 (96) |
| P132 | N/A | 0.74 | 583.1 (78.8) | 17.8 (2.4) | 96.2 (13) |
| P134 | N/A | 0.7 | 539 (77) | 0 (0) | 1.1 (0.16) |
| P135 | N/A | 1.03 | 20.6 (2) | 30.9 (3) | 875.5 (85) |
| P136 | N/A | 0.28 | 254.8 (91) | 2.8 (1) | 19.6 (7) |
| P138 | N/A | 0.22 | 72.6 (33) | 2.2 (1) | 129.8 (59) |
| P139 | N/A | 0.1 | 42.8 (42.8) | 4.1 (4.1) | 50 (50) |
| Others | |||||
| Maternal engraftment | |||||
| P077 | RAG1 | 52.23 | 49,619 (95) | 0 (0) | 2,089 (4) |
| Unknown | |||||
| P004a | IL2RG | 0.64 | 0 (0) | N/Aa | N/Aa |
| P004b | IL2RG | N/A | N/A (2) | N/A (85) | N/A (10) |
| P007 | IL2RG | N/A | N/A (0.2) | N/A (86.7) | N/A (6.7) |
| P012 | IL2RG | N/A | N/A (2) | N/A (95) | N/A (0) |
| P016 | IL2RG | N/A | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P023 | IL2RG | 0.5 | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P037 | IL2RG | 1.6 | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P040 | IL2RG | N/A | N/A (16) | N/A (82) | N/A (0) |
| P042 | IL2RG | N/A | N/A (2) | N/A (95) | N/A (1) |
| P046 | IL2RG | N/A | N/A (0) | N/A (93) | N/A (2) |
| P057 | IL2RG | N/A | N/A (0) | N/A (89) | N/A (0) |
| P062 | IL2RG | 5.4 | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P081 | RAG1 | N/A | N/A (1.5) | N/A (0.52) | N/A (74.1) |
| P085 | RAG2 | N/A | N/A (13) | N/A (0.1) | N/A (24) |
| P086 | RAG2 | N/A | N/A (0.67) | N/A (0) | N/A (76) |
| P089 | RAG2 | N/A | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P096 | N/A | 0.26 | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P113 | N/A | N/A | N/A (1) | N/A (87) | N/A (4) |
| P119 | N/A | N/A | N/A (3) | N/A (67.5) | N/A (25.4) |
| P129 | N/A | 0.22 | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P133 | N/A | 0.6 | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| P140 | N/A | N/A | N/A (2.8) | N/A (0.6) | N/A (90) |
B+ SCID was defined as having ≥134 CD19+ cells/μL and B− SCID was defined as having <134 CD19+ cells/μL. ALC, absolute lymphocyte count; N/A, not available.
aMedical record documented as “raised”.
bALC was not provided, derived by summation of CD3+ cells, CD19+ cells, and CD16/56+ cells, in P024 the ALC was documented as 2.7 × 109/L in separate test.
50.6% of patients presented with chronic diarrhea (n = 42) and 60.2% of patients recurrent infections (n = 50). The commonest site of infection was the respiratory system (n = 61), followed by gastrointestinal system (n = 42). 47.0% of infections were severe (n = 39).
Fifty patients developed opportunistic infection (60.2%). The commonest opportunistic infection was candidiasis (n = 27), followed by BCG infection (n = 19) and viral infection (n = 9). The median age for candidiasis documented was 3 months, the median age for BCG infection was 4 months, and the median age for CMV infection was 2.25 months.
For patients included in our study, clinical features were compared between those with (n = 83) and without genetic diagnosis (n = 48). Patients without genetic diagnosis had higher frequency of FTT (33.3 versus 15.7%, p = 0.0189) and CMV infections (25.0 versus 9.6%, p = 0.0185) (Table 4).
For patients with documented ALC (n = 114), 107 of them (93.9%) had at least one of the following four clinical features: FH, candidiasis, BCG infection, and ALC below 3 × 109/L. 65 of them (57.0%) had at least two of the four clinical features mentioned.
FH and Pneumonia Were Associated with Earlier AP
Factors that were found to significantly affect AP, AD, and time to diagnosis are shown in Tables 5 and 6.
Table 5.
Univariate analysis of features that affect age at presentation (AP), age at diagnosis, and time to diagnosis in patients fulfilled selection criteria (n = 83).
| Features | Median AP (months) when |
Difference in months (Group A–Group B) | p-Value | |
|---|---|---|---|---|
| Feature present (Group A) | Feature absent (Group B) | |||
| FH | 1 | 2 | −1 | 0.002 |
| Candidiasis | 2 | 2 | 0 | 0.664 |
| Bacillus Calmette–Guérin (BCG) | 2 | 2 | 0 | 0.291 |
| CMV | 1 | 2 | −1 | 0.280 |
| FTT | 2 | 2 | 0 | 0.954 |
| Chronic diarrhea | 2 | 2 | 0 | 0.778 |
| Recurrent infections | 2 | 3 | −1 | 0.008 |
| Severe infections | 2 | 2 | 0 | 0.813 |
| Pneumonia | 2 | 3 | −1 | 0.003 |
| Hepatosplenomegaly | 2.25 | 2 | 0.25 | 0.347 |
| X-linked SCID | 2 | 2 | 0 | 0.057 |
| Low ALCa | 2 | 2.25 | −0.25 | 0.771 |
| Features |
Median age at diagnosis (months) when |
Difference in months (Group C–Group D) | p-Value | |
| Feature present (Group C) | Feature absent (Group D) | |||
| FH | 3 | 5 | −2 | 0.008 |
| Candidiasis | 6 | 4 | 2 | 0.008 |
| BCG | 6 | 4 | 2 | 0.005 |
| CMV | 3 | 4 | −1 | 0.025 |
| FTT | 7 | 4 | 3 | 0.038 |
| Chronic diarrhea | 4 | 4.5 | −0.5 | 0.949 |
| Recurrent infections | 5 | 4 | 1 | 0.241 |
| Severe infections | 4 | 5 | −1 | 0.476 |
| Pneumonia | 4 | 5 | −1 | 0.111 |
| Hepatosplenomegaly | 4 | 4 | 0 | 0.544 |
| X-linked SCID | 4 | 3.5 | 0.5 | 0.689 |
| Low ALCa | 4 | 6.5 | −2.5 | 0.086 |
| Features |
Median time to diagnosis (months) when |
Difference in months (Group E–Group F) | p-Value | |
| Feature present (Group E) | Feature absent (Group F) | |||
| FH | 2 | 2 | 0 | 0.494 |
| Candidiasis | 2.5 | 1.95 | 0.55 | 0.003 |
| BCG | 3 | 2 | 1 | 0.052 |
| CMV | 1.25 | 2 | −0.75 | 0.155 |
| FTT | 4 | 2 | 2 | 0.104 |
| Chronic diarrhea | 1.15 | 2 | −0.85 | 0.617 |
| Recurrent infections | 2.5 | 1 | 1.5 | <0.001 |
| Severe infections | 2 | 2 | 0 | 0.565 |
| Pneumonia | 2 | 1.75 | 0.25 | 0.382 |
| Hepatosplenomegaly | 1.5 | 2 | −0.5 | 0.217 |
| X-linked SCID | 2 | 2 | 0 | 0.569 |
| Low ALCa | 2 | 4.5 | −2.5 | 0.124 |
aDefined as ALC below 3 × 109/L.
FH, family history of early infant death; CMV, cytomegalovirus infection; FTT, failure to thrive, ALC, absolute lymphocyte count.
Table 6.
Multivariate linear regression of features that affect age at presentation (AP), age at diagnosis (AD), and time to diagnosis in patients fulfilled selection criteria (n = 83).
| Features | Regression coefficient (months) | p-Value | 95% CI |
|---|---|---|---|
| AP | |||
| FH | −0.884 | 0.005 | −1.499 to −0.269 |
| Recurrent infections | −0.541 | 0.086 | −1.161 to 0.078 |
| Pneumonia | −0.863 | 0.009 | −1.504 to −0.221 |
| AD | |||
| FH | −1.86 | 0.007 | −3.189 to −0.529 |
| Candidiasis | 2.21 | 0.002 | 0.858 to 3.555 |
| Bacillus Calmette–Guérin | 1.11 | 0.141 | −0.375 to 2.595 |
| CMV | −1.58 | 0.147 | −3.727 to 0.569 |
| FTT | 1.15 | 0.190 | −0.584 to 2.886 |
| Time to diagnosis | |||
| Candidiasis | 1.511 | 0.018 | 0.267 to 2.754 |
| Recurrent infections | 1.845 | 0.003 | 0.655 to 3.036 |
FH, family history of early infant death; CMV, cytomegalovirus infection; FTT, failure to thrive; 95% CI, 95% confidence interval.
In univariate analysis, FH, pneumonia, and recurrent infections were associated with earlier AP (FH by 1 month, p = 0.002; pneumonia by 1 month, p = 0.003; recurrent infections by 1 month, p = 0.008). Upon multivariate analysis, only FH and pneumonia were associated with earlier AP (FH by 0.884 month, p = 0.005; pneumonia by 0.863 month, p = 0.009).
FH Was Associated with Earlier AD
In univariate analysis, FH and CMV infections were associated with an earlier AD (FH by 2 months, p = 0.008; CMV by 1 month, p = 0.025). Upon multivariate analysis, only FH was associated with earlier AD (by 1.86 months, p = 0.007).
Candidiasis and Opportunistic Infections Were Associated with Later AD
In univariate analysis, candidiasis, FTT, opportunistic infections, and BCG infection were associated with a later AD (candidiasis by 2 months, p = 0.008; FTT by 3 months, p = 0.038; opportunistic infections by 1 month, p = 0.018; BCG by 2 months, p = 0.005). Upon multivariate analysis, only candidiasis was associated with later AD (by 2.21 months, p = 0.002).
Candidiasis, Opportunistic Infections, and Recurrent Infections Were Associated with Longer Time to Diagnosis
Candidiasis, opportunistic infections, and recurrent infections were shown to be associated with longer time to diagnosis (candidiasis by 0.55 month, p = 0.003; opportunistic infections by 1 month, p = 0.005; recurrent infections by 1.5 months, p < 0.001). Upon multivariate analysis, both candidiasis and recurrent infections were associated with longer time to diagnosis (candidiasis by 1.51 months, p = 0.018; recurrent infections by 1.85 months, p = 0.003).
Other Features Were Not Significantly Associated with AD and Time to Diagnosis
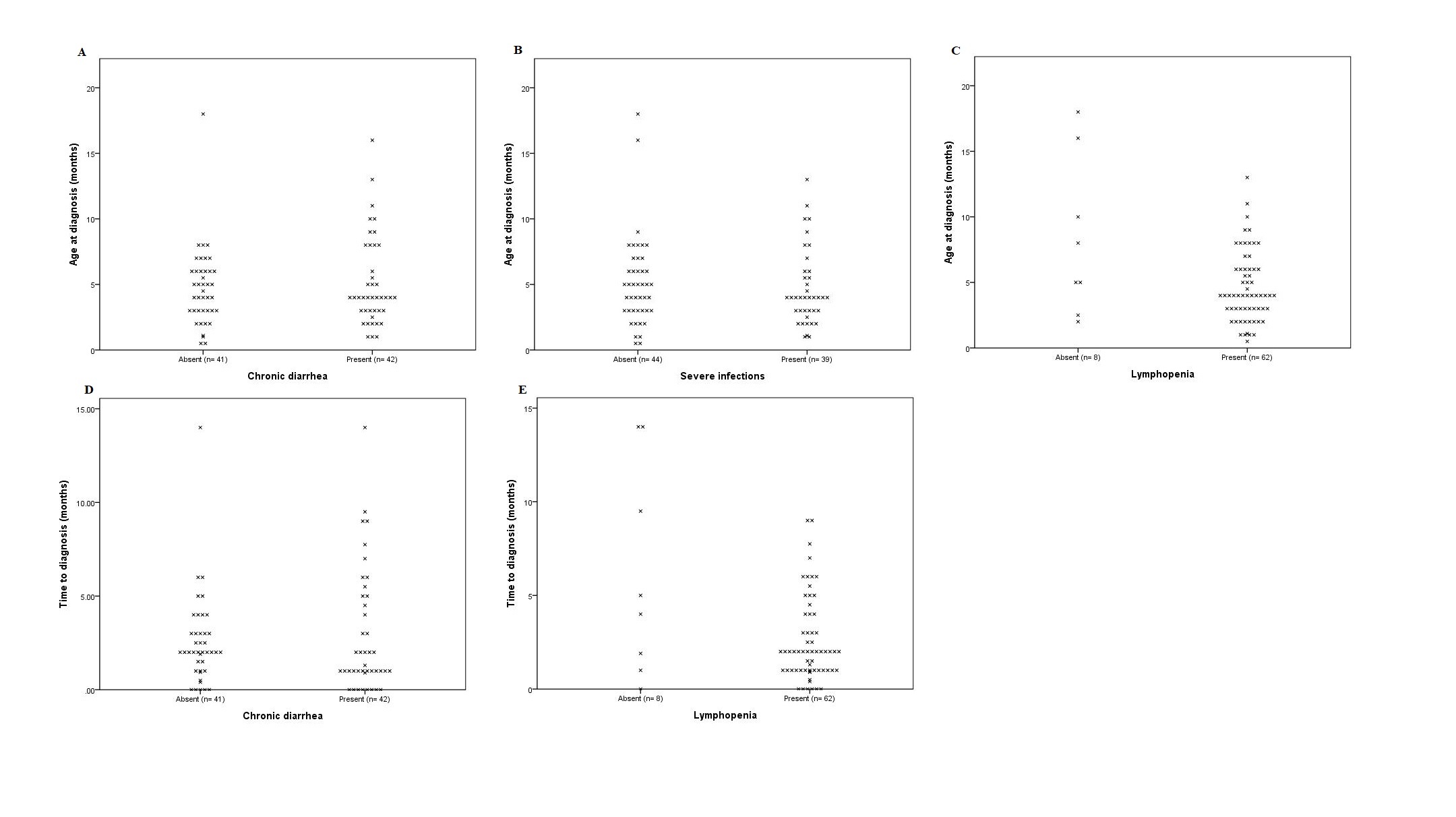
Analysis of chronic diarrhea, pneumonia, hepatosplenomegaly, severe infections, and lymphopenia revealed no association with AD and time to diagnosis. There was no difference between X-linked and autosomal recessive forms of SCID in AP, AD, and time to diagnosis.
Discussion
We found family history of early infant death was associated with earlier AP and earlier AD but not shorter time to diagnosis. Therefore, the earlier AD could be due to the heightened alertness of family with such history so that medical attention was sought earlier, rather than prompting clinicians in making quicker SCID diagnosis. The association between positive family histories and earlier AD was reported by studies in USA and France (10, 28), but they did not investigate whether the positive family history shortened the time to diagnosis. Moreover, they did not investigate whether presence of family history of early infant death alone is associated with an earlier AD. Previous studies reported 16–60% of patients with positive family histories compared to that of 32% in our study; however, the definition of family history differs between studies (Table 7). Our findings suggested that the family history of early infant death was valuable in alerting families but not clinicians who failed to recognize this clue as the time to diagnosis remained the same regardless of the presence of family history of early infant death.
Table 7.
Comparison with previous SCID studies.
| Cohort | n | Origin | Duration | AP (months) | AD (months) | Genotype (%) |
ALC (×109/L) | Present in cases (%) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL2RG | Other | Unknown | FH | Candidiasis | BCG infection | |||||||
| Our study | 131 | Asia | 2005–2016 | 2 (0–19)a | 4 (0.1–27)a | 57 (44) | 26 (20) | 48 (37) | 0.975 (0.09–52.20)a | 42/131 (32)f | 43/131 (33) | 27/131 (21)e |
| Stephan et al. (28) | 117 | France | 1970–1992 | 3 (0–19)c | 4.6 (0–27)c | 0 (0) | 0 (0) | 117 (100) | 1.608 (0–30)c | 70/117 (60) | 33/117 (28) | 10/28 (36)e |
| Mazzucchelli et al. (29) | 70 | Brazil | 1996–2011 | 2 (0–19)a | 8 (0–22)a | 0 (0) | 0 (0) | 70 (100) | N/A | 19/70 (27)g | 29/64 (45) | 39/69 (57)e |
| Yeganeh et al. (30) | 40 | Iran | 1999–2007 | 2.26 (±0.43)b | 5 (±0.67)b | 0 (0) | 0 (0) | 40 (100) | 1.26a | 20/40 (50)h | 23/40 (58) | 18/40 (45)e |
| Saleem et al. (31) | 13 | Pakistan | 2006–2011 | N/A | 5 (1.23–8.93)a | 0 (0) | 0 (0) | 13 (100) | 0.41 (0.17–2.28)a | 7/13 (54)f | N/A | N/Ae |
| McWilliams et al. (10) | 172 | USA | 1982–2013 | N/A | 4.87 (0–18)b | 77 (45) | 91 (53) | 4 (2) | 0.43 (±1.28)b | 63/172 (37)h | 74/172 (43) | 2/172 (1) |
| Dvorak et al. (32) | 50 | North America | 2010–2012 | N/A | 1.13 (0–10.13)d | 20 (40) | 27 (54) | 3 (6) | 1.22 (0.02–10.72)a | 12/50 (24)i | 8/50 (16) | 0/0 (0) |
| Rozmus et al. (33) | 40 | Canada | 2004–2010 | N/A | 4.2 (0–19.4)c | 4 (10) | 16 (40) | 20 (50) | 1.13 (0.05–14.04)a | 20/39 (51)j | 8/39 (21) | 0/0 (0) |
AP, age at presentation; AD, age at diagnosis; ALC, absolute lymphocyte count; FH, positive family history.
aMedian (range).
bMean (SD).
cMean (range).
dTypical SCID (n = 37) only.
eWith universal BCG vaccination at birth.
fFH of early infant death.
gSuggestive or confirmed FH of SCID.
hFH of early infant death due to infection or known SCID.
iFH of immunodeficiency.
jFH of SCID and/or infant death and consanguinity.
We found candidiasis was associated with later AD and longer time to diagnosis. The median age of candidiasis documented was 3 months, and the median AD of SCID for patients with candidiasis was 6 months. Therefore, clinicians required 3 months to diagnose SCID after candidiasis was first documented. This suggested that candidiasis was an overlooked feature by clinicians in Asia. Other studies reported similar percentage at candidiasis in SCID patients but no report of association between candidiasis and AD (Table 7). Although oral candidiasis is relatively common in infants under 6 months old; however, persistent, recurrent, or invasive candidiasis warrants investigation for underlying immunodeficiencies in particular SCID (15). Our finding suggested that candidiasis may be useful as a clue for earlier diagnosis since the median age of candidiasis documented was 3 months, which was earlier than the optimal time for HSCT at 3.5 months (9, 11).
We were surprised to find that BCG infection was not associated with AD and time to diagnosis. This could be due to the relatively low frequency of patients with BCG infections (21%) identified in our study, which was at a lower frequency when compared to that of 45–57% reported previously (6, 29, 30). The population coverage, immunization schedules, and virulence of BCG in countries and regions included in our study were comparable to that in Brazil and Iran (Table SE5 in Supplementary Material) (29, 34–39); therefore, the above factors of BCG policies could not account for the discrepancy in the frequency of BCG complications between our study and that from Brazil and Iran. In addition, the onset of BCG complication in our study at 4 months old was comparable to that in Brazil at 3.7 months old (29). The median AD of SCID in our study was 4 months, which was earlier than the 8 months in Brazil and 5 months in Iran, suggesting that the lower frequency of BCG infections in our study (21%) than that in Brazil (57%) and Iran (45%) (Table 7) could be due to earlier diagnosis of SCID in our study.
The median age for BCG infection documented in our patients was 4 months, which was beyond the optimal time for HSCT. Our findings were in line with a previous report in which 74% of 349 BCG-vaccinated SCID patients developed BCG infection at or after 4 months of age (6). Therefore, despite BCG infections being useful clinical features of SCID as SCID patients have approximately 400-fold increase in risk of having localized BCG complication and 33,000-fold increase in risk of having disseminated complications (6), noticing BCG infection had little value in alerting clinicians to make a timely diagnosis of SCID for optimal HSCT, which should be before 3.5 months (9, 11).
We found that opportunistic infections were associated with later AD, while recurrent infections and opportunistic infections were associated with longer time to diagnosis. Therefore, such clinical features were likely the consequences of delay in the diagnosis of SCID, reflecting that clinicians in Asia were unable to recognize these as SCID features.
We found that pneumonia was associated with an earlier AP but did not affect AD and time to diagnosis. Therefore, parents may perceive pneumonia as a severe medical condition and then brought their children to seek medical care earlier. However, pneumonia also commonly affects children without SCID in Asia (40–42), and clinicians are not alerted to the possible diagnosis of SCID.
Chronic diarrhea, severe infections, and ALC below 3 × 109/L were not associated with AD and time to diagnosis, likely due to the distributions of the AD and the time to diagnosis that were quite wide in patients with these features (Figure S1 in Supplementary Material). In addition, chronic diarrhea is common in Southeast Asia and Western Pacific region (40); thus, it may not be a useful differentiating feature for patients with SCID as compared to those without. CMV infections did not affect AD and time to diagnosis as they were documented in small number of patients in our study (n = 8). The low rate of documented CMV infections may be due to the lack of diagnostic capacity (43, 44).
This study presented the largest collection of SCID patients in China and Southeast Asia with 147 patients, including 94 SCID patients with genetic diagnosis. The median AD was 4 months, which was comparable to other cohorts in the world, given no newborn screening of TREC was performed (Table 7) (10, 28–33); however, it was later than the optimal time for HSCT.
The commonest SCID gene found to be mutated in our patients was IL2RG because of the low consanguinity rate in our population (45) as well as near absence of newborn screening in Asia. Mutations in IL2RG were unevenly distributed. Exons 3 and 5 of IL2RG were common sites for mutation, accounting for 45% of all IL2RG mutations (Table 1; Table SE1 in Supplementary Material) and 48% of all unreported IL2RG mutations (Table SE3 in Supplementary Material), which was comparable with previous study (46). Five mutation hotspots, namely cDNA 670, 676, 677, 854, and 865, were identified previously and accounted for 29% of all IL2RG mutations in one study (46). Mutations in these hotspots collectively accounted for 27% of IL2RG mutations in our study. Majority of the mutations in these hotspots involved either C>T or G>A mutations in CpG dinucleotides. The mutation frequency of the C nucleotides in CpGs is 10–50 times higher compared to any other bases (47). This is commonly thought to be due to the methylation and subsequently deamination of cytosine to form thymidine in CpG (48, 49). Apart from the mentioned hotspots, we identified 16 additional point mutations in all SCID genes involving such mechanism, suggesting that cytosine methylation and deamination to thymidine in CpG dinucleotide is a relatively common mechanism causing mutations in SCID genes.
Four patients with mutations in IL2RG were classified as having B− SCID with CD19+ B cells ranging from 0 to 70/μL (Table 3). The four patients had typical SCID presentations (Table SE4 in Supplementary Material). One patient was screened for DCLRE1C, RAG1, and RAG2 due to his B− phenotype, but no mutation found. Patients suffering from X-linked SCID but with T-B− phenotype have been described previously (28). Patients with documented IL2RG mutations but with T-B+NK+ phenotype were also described previously (50–52). Many possible mechanisms can lead to atypical SCID immunophenotypes, including concurrent mutations in other SCID genes, modifier gene(s), and mutations, that lead to sparing or disrupting developments of other lineages of lymphocytes. In addition, one patient with mutations in RAG1 was classified as having B+ SCID with 267.4 CD19+ B cell/μL (Table 3). He likely had either Omenn syndrome or maternal engraftment due to his T+B+NK+ immunophenotype as well as his clinical presentation of severe eczema and eosinophilia (Table 3; Table SE4 in Supplementary Material). The T+B+NK+ immunophenotype of this patient may be explained by his missense mutation (c.2095C>T; p.R699W). This hypomorphic mutation results in a mutant RAG1 enzyme with 19.3% residual recombinase activity (53), thus allowing the generation of B cells in this patient. These cases in our study as well as previous reports demonstrated the imperfect correlation of genotype–immunophenotype in SCID patients.
We reported two patients (P091 and P092) with RFXANK mutations, with one (P092) reported previously in Chinese literature (54). These patients were the only two with confirmed RFXANK mutations reported in Asia. RFXANK mutation causes bare lymphocyte syndrome type 2B (55), commonly observed in North Africa (56) and sometimes in other places such as France and Spain (57). MHC class II deficiency accounted for 32% of all forms of SCID and their variants in North Africa (58–62), while it accounted for 1.4% of all SCID in Asia. Such discrepancy could be explained by the higher consanguinity rate of 50% in North Africa (63) compared to that of less than 10% in Asia (45), as well as presence of founder mutations in North Africa (56, 64). These patients present with features of typical SCID and frequently with sclerosing cholangitis (56, 65). However, they have normal TREC level and cannot be identified by newborn screening (5, 66).
The care of patients with SCID in Asia is still at an early phase of development, as reflected by delay in diagnosis and suboptimal management with no easy access to HSCT (12, 14). Eighty-one of the 83 patients were the first member of their respective families to be diagnosed genetically with SCID, thus explaining the relative lack of family history of SCID in our study. For genetic counseling, we offered testing for family members of SCID patients including prenatal and newborn screening on siblings of six index patients as well as carrier screening for parents, siblings, and maternal aunts of 56 index patients (Table SE6 in Supplementary Material). Unfortunately, there is still a relative lack of clinical genetic service in Asia.
The median ALC in all studies including ours was below 3 × 109/L, and reaffirming lymphopenia is a feature commonly seen in SCID patients (Table 7). However, clinicians failed to act on this critical clue as lymphopenia did not affect the AD or time to diagnosis. Since 1994, many reports have emphasized the importance of low ALC in alerting clinicians regarding SCID (67–70), but sadly clinicians to this date still failed to appreciate the value of low ALC for the diagnosis of SCID.
In this study, we identified that FH, candidiasis, and ALC below 3 × 109/L were overlooked clinical features prompting the diagnosis of SCID. In addition, BCG infections were useful clinical features as they were the second most common opportunistic infections in our SCID patients. Ninety-four percent of patients in our study had at least one of the following four features: FH, candidiasis, BCG infections, and ALC below 3 × 109/L. Therefore, we suggest a simple guideline mandating that clinicians should order lymphocyte subset analysis for infants with any one of the following four features: FH, candidiasis, BCG infections, and ALC below 3 × 109/L.
Failure to diagnose SCID in time will lead to delay in HSCT, leading to economic losses in addition to poor outcome. Study has shown that the mean total hospital charges in patients who had HSCT after 3.5 months were four times greater than those before 3.5 months (71). Since all the clinical features we analyzed failed to help clinicians in making earlier SCID diagnosis, newborn screening is the only solution for making early enough diagnosis of SCID for timely HSCT in Asia.
Our retrospective case-series relied on reports made by referring doctors instead of analyzing original charts and results; therefore, underreport of clinical features was possible. In addition, our handling of missing data tends to underestimate the strength of association of clinical features with AD and time to diagnosis.
In conclusion, clinicians failed to recognize typical clinical features of SCID to shorten the time to diagnosis. There is an urgent unmet need to educate clinicians in Asia on SCID. Ultimately, the only solution for early diagnosis of and timely HSCT for patients with SCID is newborn screening.
Ethics Statement
Genetic and functional studies on PID, data archival in the APIN database, and DNA storage were approved by the Clinical Research Ethics Review Board of the University of Hong Kong and Queen Mary Hospital (Ref. no. UW 08-301).
Author Contributions
ADL, PL, and YL designed the study; ADL and YL wrote the manuscript with extensive appraisal from PL, K-WC, and HM; YL, K-WC, WW, and ADL analyzed the data; K-WC, WY, and WT performed the genetic and immunological studies; PL, HM, YL, XC, T-XC, JH, NK, DS, YT, YX, LJ, WL, OJ, TD, AG, SS, AR, AHL, ACL, LS, TN, TC, YC, ZL, TML, NL, BL, QL, DR, M-RB, M-KT, MA, XW, CX, HY, H-HY, TLL, FY, PC, and MH contributed clinical data and took care of patients in this study. PL and YL established the Asian Primary Immunodeficiency Network (APIN).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the Hong Kong Society for Relief of Disabled Children; the Health and Medical Research Fund (01120846); and grant from Shenzhen Development and Reform Commission ([2015]164).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00808/full#supplementary-material.
Distribution of age at diagnosis and time to diagnosis of patients with genetic diagnosis (n = 83). Distribution of age at diagnosis of patients with chronic diarrhea (A), severe infections (B), and lymphopenia (C) and distribution of time to diagnosis of patients with chronic diarrhea (D) and lymphopenia (E).
{kind=link}
Abbreviations
ADA, adenosine deaminase gene; ALC, absolute lymphocyte count; AD, age at diagnosis; AP, age at presentation; APIN, Asian Primary Immunodeficiency Network; BCG, bacillus Calmette–Guérin; CMV, cytomegalovirus; DCLRE1C, DNA cross-link repair enzyme 1C (Artemis); FH, family history of early infant death; FN3, fibronectin type-III; FTT, failure to thrive; HSCT, hematopoietic stem cell transplant; ICU, intensive care unit; IL2RG, IL-2 receptor gamma chain; IL7R, IL-7 receptor alpha chain; JAK 3, janus kinase 3; PCP, Pneumocystis jiroveci; PID, primary immunodeficiency; RAG1, recombinase activating gene 1; RAG2, recombinase activating gene 2; RFXANK, regulatory factor X associated ankyrin containing protein; SCID, severe combined immunodeficiency; TREC, T-cell receptor excision circle.
References
- 1.Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol (2015) 35(8):696–726. 10.1007/s10875-015-0201-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers (2015) 1:15061. 10.1038/nrdp.2015.61 [DOI] [PubMed] [Google Scholar]
- 3.Kwan A, Puck JM. History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol (2015) 39(3):194–205. 10.1053/j.semperi.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckley RH, Schiff RI, Schiff SE, Markert ML, Williams LW, Harville TO, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr (1997) 130(3):378–87. 10.1016/S0022-3476(97)70199-9 [DOI] [PubMed] [Google Scholar]
- 5.Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2014) 312(7):729–38. Erratum in: JAMA (2014) 312(20):2169. 10.1001/jama.2014.9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marciano BE, Huang CY, Joshi G, Rezaei N, Carvalho BC, Allwood Z, et al. BCG vaccination in patients with severe combined immunodeficiency: complications, risks, and vaccination policies. J Allergy Clin Immunol (2014) 133(4):1134–41. Erratum in: J Allergy Clin Immunol (2014) 134(1):244. 10.1016/j.jaci.2014.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sponzilli I, Notarangelo LD. Severe combined immunodeficiency (SCID): from molecular basis to clinical management. Acta Biomed (2011) 82(1):5–13. [PubMed] [Google Scholar]
- 8.Gaspar HB, Qasim W, Davies EG, Rao K, Amrolia PJ, Veys P. How I treat severe combined immunodeficiency. Blood (2013) 122(23):3749–58. 10.1182/blood-2013-02-380105 [DOI] [PubMed] [Google Scholar]
- 9.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med (2014) 371(5):434–46. 10.1056/NEJMoa1401177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McWilliams LM, Dell Railey M, Buckley RH. Positive family history, infection, low absolute lymphocyte count (ALC), and absent thymic shadow: diagnostic clues for all molecular forms of severe combined immunodeficiency (SCID). J Allergy Clin Immunol Pract (2015) 3(4):585–91. 10.1016/j.jaip.2015.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood (2011) 117(11):3243–6. 10.1182/blood-2010-08-300384 [DOI] [PubMed] [Google Scholar]
- 12.Lee PP. Primary Immunodeficiency Disorders in Southeast Asia: Needs, Priorities and Opportunities [MD Thesis]. University of Hong Kong, Pokfulam, Hong Kong SAR: (2014). [Google Scholar]
- 13.Lee PP, Chan KW, Chen TX, Jiang LP, Wang XC, Zeng HS, et al. Molecular diagnosis of severe combined immunodeficiency – identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian children. J Clin Immunol (2011) 31(2):281–96. 10.1007/s10875-010-9489-z [DOI] [PubMed] [Google Scholar]
- 14.Lee PP, Lau YL. Improving care, education, and research: the Asian primary immunodeficiency network. Ann N Y Acad Sci (2011) 1238:33–41. 10.1111/j.1749-6632.2011.06225.x [DOI] [PubMed] [Google Scholar]
- 15.Krol DM, Keels MA. Oral conditions. Pediatr Rev (2007) 28(1):15–22. 10.1542/pir.28-1-15 [DOI] [PubMed] [Google Scholar]
- 16.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat (2003) 21(6):577–81. 10.1002/humu.10212 [DOI] [PubMed] [Google Scholar]
- 17.Piirilä H, Väliaho J, Vihinen M. Immunodeficiency mutation databases (IDbases). Hum Mutat (2006) 27(12):1200–8. 10.1002/humu.20405 [DOI] [PubMed] [Google Scholar]
- 18.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature (2016) 536(7616):285–91. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, et al. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res (2017) 45(D1):D183–9. 10.1093/nar/gkw1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics (2006) 22(22):2729–34. 10.1093/bioinformatics/btl423 [DOI] [PubMed] [Google Scholar]
- 21.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc (2009) 4(7):1073–81. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 22.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics (2008) 24(24):2938–9. 10.1093/bioinformatics/btn564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capriotti E, Altman RB, Bromberg Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics (2013) 14(Suppl 3):S2. 10.1186/1471-2164-14-S3-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7(4):248–9. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.NCBI Resource Coordinators. Database resources of the national center for biotechnology information. Nucleic Acids Res (2016) 44:D7–D19. 10.1093/nar/gkv1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res (2015) 43(Database issue):D204–12. 10.1093/nar/gku989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanai N, Yanai F, Hirose S, Nibu K, Izuhara K, Tani T, et al. A G to A transition at the last nucleotide of exon 6 of the gamma c gene (868G – >A) may result in either a splice or missense mutation in patients with X-linked severe combined immunodeficiency. Hum Genet (1999) 104(1):36–42. Erratum in: Hum Genet (1999) 104(2):196. 10.1007/s004390050907 [DOI] [PubMed] [Google Scholar]
- 28.Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G, et al. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr (1993) 123(4):564–72. 10.1016/S0022-3476(05)80951-5 [DOI] [PubMed] [Google Scholar]
- 29.Mazzucchelli JT, Bonfim C, Castro GG, Condino-Neto AA, Costa NM, Cunha L, et al. Severe combined immunodeficiency in Brazil: management, prognosis, and BCG-associated complications. J Investig Allergol Clin Immunol (2014) 24(3):184–91. [PubMed] [Google Scholar]
- 30.Yeganeh M, Heidarzade M, Pourpak Z, Parvaneh N, Rezaei N, Gharagozlou M, et al. Severe combined immunodeficiency: a cohort of 40 patients. Pediatr Allergy Immunol (2008) 19(4):303–6. 10.1111/j.1399-3038.2007.00647.x [DOI] [PubMed] [Google Scholar]
- 31.Saleem AF, Ali Khawaja RD, Shaikh AS, Ali SA, Mehdi Zaidi AK. Severe combined immune deficiency syndrome. J Coll Physicians Surg Pak (2013) 23(8):570–3. [PubMed] [Google Scholar]
- 32.Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol (2013) 33(7):1156–64. 10.1007/s10875-013-9917-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozmus J, Junker A, Thibodeau ML, Grenier D, Turvey SE, Yacoub W, et al. Severe combined immunodeficiency (SCID) in Canadian children: a national surveillance study. J Clin Immunol (2013) 33(8):1310–6. 10.1007/s10875-013-9952-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zwerling A, Behr MA, Verma A, Brewer TF, Menzies D, Pai M. The BCG world atlas: a database of global BCG vaccination policies and practices. PLoS Med (2011) 8(3):e1001012. 10.1371/journal.pmed.1001012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.WHO. UNICEF: Immunization Summary, a Statistical Reference Containing Data through 2013. (2014). Available from: http://www.who.int/immunization/monitoring_surveillance/Immunization_Summary_2013.pdf
- 36.Joung SM, Ryoo S. BCG vaccine in Korea. Clin Exp Vaccine Res (2013) 2(2):83–91. 10.7774/cevr.2013.2.2.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang JF, Dai FY, Gong XL, Bao L. Commonly administered bacilli Calmette–Guerin strains induce comparable immune response. Int J Clin Exp Med (2015) 8(9):15834–9. [PMC free article] [PubMed] [Google Scholar]
- 38.Govindarajan KK, Chai FY. BCG adenitis-need for increased awareness. Malays J Med Sci (2011) 18(2):66–9. [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L, Ru HW, Chen FZ, Jin CY, Sun RF, Fan XY, et al. Variable virulence and efficacy of BCG vaccine strains in mice and correlation with genome polymorphisms. Mol Ther (2016) 24(2):398–405. 10.1038/mt.2015.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, et al. Global burden of childhood pneumonia and diarrhoea. Lancet (2013) 381(9875):1405–16. 10.1016/S0140-6736(13)60222-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudan I, Boschi-Pinto C, Biloglav Z, Mulholland K, Campbell H. Epidemiology and etiology of childhood pneumonia. Bull World Health Organ (2008) 86(5):408–16. 10.2471/BLT.07.048769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gereige RS, Laufer PM. Pneumonia. Pediatr Rev (2013) 34(10):438–56; quiz 455–6. Erratum in: Pediatr Rev (2014) 35(1):29. 10.1542/pir.34-10-438 [DOI] [PubMed] [Google Scholar]
- 43.Plosa EJ, Esbenshade JC, Fuller MP, Weitkamp JH. Cytomegalovirus infection. Pediatr Rev (2012) 33(4):156–63; quiz 163. 10.1542/pir.33-4-156 [DOI] [PubMed] [Google Scholar]
- 44.Eguchi H, Horita N, Ushio R, Kato I, Nakajima Y, Ota E, et al. Diagnostic test accuracy of antigenemia assay for polymerase chain reaction proven cytomegalovirus infection: systematic review and meta-analysis. Clin Microbiol Infect (2017). 10.1016/j.cmi.2017.05.009 [DOI] [PubMed] [Google Scholar]
- 45.Hamamy H. Consanguineous marriages: preconception consultation in primary health care settings. J Community Genet (2012) 3(3):185–92. 10.1007/s12687-011-0072-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puck JM, Pepper AE, Henthorn PS, Candotti F, Isakov J, Whitwam T, et al. Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency. Blood (1997) 89(6):1968–77. [PubMed] [Google Scholar]
- 47.Walser JC, Furano AV. The mutational spectrum of non-CpG DNA varies with CpG content. Genome Res (2010) 20(7):875–82. 10.1101/gr.103283.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RY, Kuo KC, Gehrke CW, Huang LH, Ehrlich M. Heat- and alkali-induced deamination of 5-methylcytosine and cytosine residues in DNA. Biochim Biophys Acta (1982) 697(3):371–7. 10.1016/0167-4781(82)90101-4 [DOI] [PubMed] [Google Scholar]
- 49.Ehrlich M, Zhang XY, Inamdar NM. Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat Res (1990) 238(3):277–86. 10.1016/0165-1110(90)90019-8 [DOI] [PubMed] [Google Scholar]
- 50.Ginn SL, Smyth C, Wong M, Bennetts B, Rowe PB, Alexander IE. A novel splice-site mutation in the common gamma chain (gammac) gene IL2RG results in X-linked severe combined immunodeficiency with an atypical NK+ phenotype. Hum Mutat (2004) 23(5):522–3. 10.1002/humu.9235 [DOI] [PubMed] [Google Scholar]
- 51.Mou W, He J, Chen X, Zhang H, Ren X, Wu X, et al. A novel deletion mutation in IL2RG gene results in X-linked severe combined immunodeficiency with an atypical phenotype. Immunogenetics (2017) 69(1):29–38. 10.1007/s00251-016-0949-3 [DOI] [PubMed] [Google Scholar]
- 52.Estévez OA, Ortega C, Fernández S, Aguado R, Rumbao J, Perez-Navero J, et al. A novel IL2RG mutation presenting with atypical T(−)B(+)NK+ phenotype: rapid elucidation of NK cell origin. Pediatr Blood Cancer (2014) 61(1):178–9. 10.1002/pbc.24717 [DOI] [PubMed] [Google Scholar]
- 53.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol (2014) 133(4):1099–108. 10.1016/j.jaci.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu WC, Wang W, Song HM, Ma MS, Tang XY, Jian S, et al. [A major histocompatibility complex class II deficiency case report and literature review]. Zhonghua Er Ke Za Zhi (2016) 54(8):614–8. 10.3760/cma.j.issn.0578-1310.2016.08.013 [DOI] [PubMed] [Google Scholar]
- 55.Shrestha D, Szöllosi J, Jenei A. Bare lymphocyte syndrome: an opportunity to discover our immune system. Immunol Lett (2012) 141(2):147–57. 10.1016/j.imlet.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 56.Ouederni M, Vincent QB, Frange P, Touzot F, Scerra S, Bejaoui M, et al. Major histocompatibility complex class II expression deficiency caused by a RFXANK founder mutation: a survey of 35 patients. Blood (2011) 118(19):5108–18. 10.1182/blood-2011-05-352716 [DOI] [PubMed] [Google Scholar]
- 57.Renella R, Picard C, Neven B, Ouachée-Chardin M, Casanova JL, Le Deist F, et al. Human leucocyte antigen-identical haematopoietic stem cell transplantation in major histocompatiblity complex class II immunodeficiency: reduced survival correlates with an increased incidence of acute graft-versus-host disease and pre-existing viral infections. Br J Haematol (2006) 134(5):510–6. 10.1111/j.1365-2141.2006.06213.x [DOI] [PubMed] [Google Scholar]
- 58.Mellouli F, Mustapha IB, Khaled MB, Besbes H, Ouederni M, Mekki N, et al. Report of the Tunisian registry of primary immunodeficiencies: 25-years of experience (1988–2012). J Clin Immunol (2015) 35(8):745–53. 10.1007/s10875-015-0206-9 [DOI] [PubMed] [Google Scholar]
- 59.Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, et al. First report on the Moroccan registry of primary immunodeficiencies: 15 years of experience (1998–2012). J Clin Immunol (2014) 34(4):459–68. 10.1007/s10875-014-0005-8 [DOI] [PubMed] [Google Scholar]
- 60.Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol (2009) 29(3):343–51. 10.1007/s10875-008-9260-x [DOI] [PubMed] [Google Scholar]
- 61.Kechout N, Attal N, Doudou F, Boukari R, Ardjoun M, Abbadi MC. Study of primary immunodeficiencies in Algeria. BMC Proc (2011) 5(Suppl 1):35. 10.1186/1753-6561-5-s1-p35 [DOI] [Google Scholar]
- 62.Reda SM, El-Ghoneimy DH, Afifi HM. Clinical predictors of primary immunodeficiency diseases in children. Allergy Asthma Immunol Res (2013) 5(2):88–95. 10.4168/aair.2013.5.2.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barbouche MR, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci (2011) 1238:42–52. 10.1111/j.1749-6632.2011.06260.x [DOI] [PubMed] [Google Scholar]
- 64.Naamane H, El Maataoui O, Ailal F, Barakat A, Bennani S, Najib J, et al. The 752delG26 mutation in the RFXANK gene associated with major histocompatibility complex class II deficiency: evidence for a founder effect in the Moroccan population. Eur J Pediatr (2010) 169(9):1069–74. 10.1007/s00431-010-1179-6 [DOI] [PubMed] [Google Scholar]
- 65.Elhasid R, Etzioni A. Major histocompatibility complex class II deficiency: a clinical review. Blood Rev (1996) 10(4):242–8. 10.1016/S0268-960X(96)90008-9 [DOI] [PubMed] [Google Scholar]
- 66.Kuo CY, Chase J, Garcia Lloret M, Stiehm ER, Moore T, Aguilera MJ, et al. Newborn screening for severe combined immunodeficiency does not identify bare lymphocyte syndrome. J Allergy Clin Immunol (2013) 131(6):1693–5. 10.1016/j.jaci.2013.01.019 [DOI] [PubMed] [Google Scholar]
- 67.Hague RA, Rassam S, Morgan G, Cant AJ. Early diagnosis of severe combined immunodeficiency syndrome. Arch Dis Child (1994) 70(4):260–3. 10.1136/adc.70.4.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gennery AR, Cant AJ. Diagnosis of severe combined immunodeficiency. J Clin Pathol (2001) 54(3):191–5. 10.1136/jcp.54.3.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol (2004) 22:625–55. 10.1146/annurev.immunol.22.012703.104614 [DOI] [PubMed] [Google Scholar]
- 70.Rivers L, Gaspar HB. Severe combined immunodeficiency: recent developments and guidance on clinical management. Arch Dis Child (2015) 100(7):667–72. 10.1136/archdischild-2014-306425 [DOI] [PubMed] [Google Scholar]
- 71.Kubiak C, Jyonouchi S, Kuo C, Garcia-Lloret M, Dorsey MJ, Sleasman J, et al. Fiscal implications of newborn screening in the diagnosis of severe combined immunodeficiency. J Allergy Clin Immunol Pract (2014) 2(6):697–702. 10.1016/j.jaip.2014.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of age at diagnosis and time to diagnosis of patients with genetic diagnosis (n = 83). Distribution of age at diagnosis of patients with chronic diarrhea (A), severe infections (B), and lymphopenia (C) and distribution of time to diagnosis of patients with chronic diarrhea (D) and lymphopenia (E).