

Abstract
Improvements in the understanding of cancer biology have led to therapeutic advances in the treatment of gastrointestinal cancers. Drugs which target the vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) pathways have led the way in colon cancer. Monoclonal antibodies (mAbs) such as bevacizumab, ramucirumab, cetuximab, and panitumumab, have improved progression free survival and overall survival (OS) for colorectal cancers and were quickly adopted. Human epidermal growth factor receptor 2 (HER2) has demonstrated significant benefit for gastroesophageal cancers and in the setting of HER2 amplification, trastuzumab in combination with chemotherapy has become the standard of care. However, responses have not been as durable nor as robust as once hoped. Mechanisms of resistance for each of these biologic compounds have been hypothesized and are in the process of being better elucidated. This review will approach the innate and acquired mechanisms of resistance of the above compounds. Additionally, we will explore some ongoing clinical trials to capitalize on the mechanisms of resistance in the hopes of retaining the promise of targeting these pathways.
Keywords: Targeted therapy, resistance, gastrointestinal cancers
Introduction
Since the year 2000, many new therapies have been studied and approved in the treatment of gastrointestinal malignancies. Largely, these incremental gains are due to better characterization of biologic mechanisms in cancer growth, self-sustenance, and metastatic spread, as described in the landmark Hallmarks of Cancer (1). Targeting epidermal growth factor receptor (EGFR), vascular-endothelial growth factor (VEGF), its receptor, VEGF-R, and human epidermal growth factor receptor-2 (HER2/Neu) have each yielded FDA-approved therapies in multiple tumor types because of demonstrable responses in phase III trials (Figure 1). Across studies, changes in response rate (RR), progression-free survival (PFS), and overall survival (OS) have been demonstrated with varying levels of success.
Figure 1.
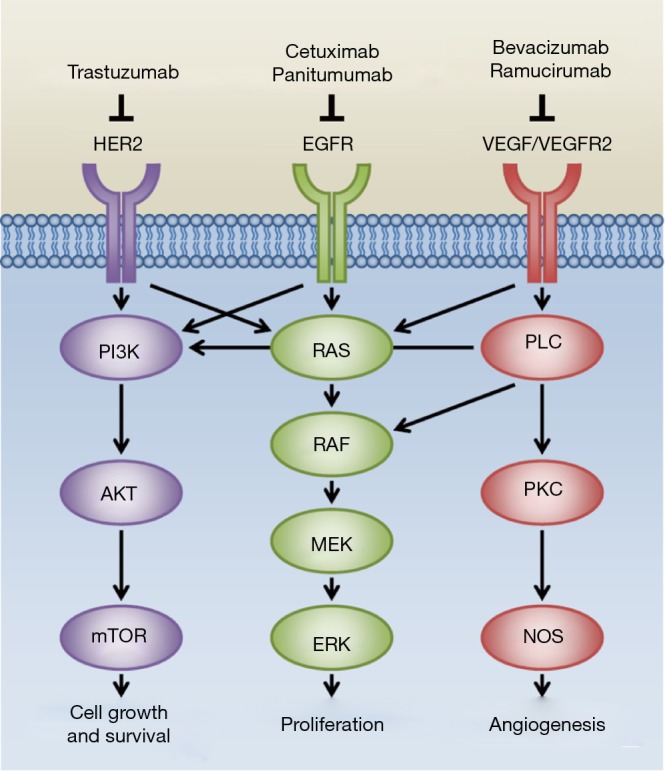
Therapies involving biologic pathways in gastrointestinal cancers and their downstream effectors.
In spite of their early promise in these areas, patients with colon and gastroesophageal cancers uniformly progress, and still die from their disease. As in other cancers, researchers are now making strides to understanding why the responses to these biologic therapies are not as durable as previously hoped.
This review will focus primarily on demonstrated mechanisms of resistance to biologic therapies. We define primary resistance as a characteristic of the tumor that exists prior to the initiation of biologic therapy that predicts for no response or a poor response. Secondary resistance we define as a tumor or host characteristic that arises during, or in response to, treatment with biologic therapies. We will review these mechanisms based on the molecular target involved, and reference the different diseases involved within each section.
Resistance to EGFR inhibitors: primary
As many tumors of the gastrointestinal tract arise from epithelial origins, the attractiveness of treating with therapies blocking EGFR is readily evident. A series of colon cancer trials have shown, improvements in RRs and OS, particularly in populations without mutations in EGFR and/or downstream effectors, which are described in the next section. In most other GI cancers, however, blocking EGFR has not demonstrated clinically relevant improvements in outcome (2-4). New strategies to investigate anti-EGFR therapy in other tumor types are focusing on selecting for patients whose tumors overexpress EGFR, or do not harbor deleterious mutations (ENRICH study, NCT 01813253) (5).
KRAS/NRAS
KRAS and NRAS belong to the family of RAS oncogenes. Mutations in these genes generate a constitutively activated tyrosine kinase which renders the tumor insensitive to upstream blockade of the EGFR/RAS/RAF pathway. The most common KRAS mutations are found in exon 2 (codon 12 or 13). Activating mutations in KRAS at exon 2 have proven to predict resistance to anti-EGFR therapies (6-11). These mutations are well-established as mediators of primary resistance to targeted biologics.
In a retrospective European consortium analysis, De Roock and colleagues analyzed tumor samples from a large cohort of patients with chemotherapy-refractory mCRC treated with cetuximab and chemotherapy (12). Forty percent of evaluable samples harbored KRAS mutations, most commonly at codon 12 or 13 (exon 2) with 2.1% at codon 61 (exon 3) and 2% at codon 146 (exon 4). Among those treated with cytotoxic chemotherapy plus cetuximab any mutation in KRAS was a negative prognostic indicator for RR, PFS, and OS. Other RAS family mutations have demonstrated importance as well; NRAS mutations were found in 2.6% of evaluable samples, mostly in codon 61, and were mutually exclusive of KRAS mutations. NRAS mutant cancers had a significantly lower RR when treated with chemotherapy and cetuximab. Numerically, lower PFS and OS were seen, but not statistically significant, perhaps owing to the low sample size of NRAS mutants (12).
The OPUS study examined the efficacy of cetuximab in combination with FOLFOX4 as first-line treatment for mCRC (7). KRAS mutations were assessed at exon 2 at codon 12 or 13 with 93% mutational status known (315/337). When treated with FOLFOX4/cetuximab versus FOLFOX alone, the KRAS exon 2 wild-type population had a better RR and median PFS. Among the KRAS exon 2 mutant populations, outcomes were reversed; adding cetuximab to FOLFOX4 resulted in worse RR, shorter PFS. OS was not significantly affected in either population. Further analysis of patients with KRAS exon 2 wild-type cancers in the cohort demonstrated that other KRAS and NRAS mutations led to resistance to anti-EGFR therapies (13).
A recent reevaluation of the CRYSTAL data assessed other RAS mutations, and found similar results (14). Alternative RAS mutations (KRAS exon 3, 4 and NRAS exons 2, 3, 4) were examined in patients with KRAS exon 2 wild-type cancers treated with FOLFIRI/cetuximab versus FOLFIRI alone. The presence of any RAS mutation showed no improvement to the addition of cetuximab in RR, PFS or OS. Similar results were found when all RAS mutations were combined. In contrast, the all-RAS wild-type population demonstrated highly significant improvements in RR, PFS and OS when treated with FOLFIRI/cetuximab compared to FOLFIRI alone (14).
Retrospective data of 579 patients treated with cetuximab in the refractory setting suggested that specific mutations in KRAS (G13D) might preserve response to cetuximab. Since this mutation is a common one in KRAS (19% of mutations in that gene, 5–8% of all mCRCs), prospective phase II studies were conducted. The ICECREAM study, published in 2016, showed in a prospective manner that anti-EGFR therapy has no role for patients with this mutation (15,16).
For treatment with panitumumab, the same concepts of any RAS mutation predicting resistance to anti-EGFR therapy hold true. Among all RAS wild-type (KRAS at exon 2, 3, 4 and NRAS 2, 3, 4) patients, benefits were observed in PFS when treated with FOLFIRI/panitumumab versus FOLFIRI alone as second-line treatment (17). Similar to results in other studies, the addition of an anti-EGFR therapy to standard chemotherapy provided no statistically significant benefit in terms of OS, PFS, or RR in the presence of any RAS mutation. As a review of the PRIME data, RAS mutations were assessed in patients treated with FOLFOX4 with and without panitumumab. Among those with a RAS mutation other than at KRAS exon 2 treated with FOLFOX4/panitumumab versus FOLFOX4 alone, there was no difference in PFS or OS. With a complete RAS mutation analysis (all KRAS at exon 2, 3, 4 and NRAS mutations at exon 2, 3, and 4), this review showed those patients with a mutation treated with chemotherapy and an anti-EGFR therapy had a significantly shorter median PFS and OS. Having no RAS mutations treated with FOLFOX4/panitumumab conferred a longer median PFS and OS compared to FOLFOX4 alone (8).
Other mutations clearly play a role, because among KRAS exon 2 wild-type patients, as many as 65% are resistant to EGFR mAbs (18). The search for which genes and which mutations cause this resistance are active areas of research.
BRAF
BRAF is an oncogene in the RAF gene family that encodes a serine-threonine protein kinase found in the RAS-RAF-MAPK cascade. Approximately 10% of colorectal cancer harbors a BRAF mutation, though this number varies depending on the population of study (19-21). The most significant and prevalent mutation occurs at the kinase domain from a substitution of valine to glutamic acid (V600E). Numerous clinical studies have suggested that the presence of this mutation predicts resistance to anti-EGFR therapies, as well as marks poor prognosis (7,12,20,22-24). In one study, BRAF mutant tumors had a significantly lower RR compared to wild-type cancers when treated with an anti-EGFR therapy, as well as shorter PFS and OS (8,12) Similarly, in a retrospective analysis of RAS and BRAF mutation status of PRIME data (8), patients with neither RAS nor BRAF mutations showed significantly better OS and PFS when treated with FOLFOX4/panitumumab compared to FOLFOX4 alone. The presence of BRAF mutations in RAS wild-type patients resulted in a worse outcome. Treatment with anti-EGFR therapy did not significant improve median PFS or OS. FOLFIRI/panitumumab versus FOLFIRI alone was examined in the second-line setting by Peeters and colleagues (17). The presence of a BRAF mutation resulted in no significant differences in PFS or OS whether patients were treated with FOLFIRI/panitumumab or FOLFIRI alone, indicating BRAF mutations may confer EGFR therapy resistance although this study was not powered for this purpose. Having a BRAF V600E mutation generated a poor prognosis regardless of treatment group.
The negative predictive influence of BRAF extends across multiple lines of therapy. FOLFIRI/panitumumab versus FOLFIRI alone was examined in the second-line setting. The presence of a BRAF mutation resulted in no significant differences in PFS or OS whether patients were treated with FOLFIRI/panitumumab or FOLFIRI alone, indicating BRAF mutations may confer EGFR therapy resistance although this study was not powered to definitively for this purpose (17).
Inhibition of BRAF has demonstrated remarkable responses in patients with V600E mutations in BRAF in melanoma (25). Colorectal cancers which harbor the same mutation do not have nearly the same successful response to this therapy (26). Preclinical studies have suggested a variety of mechanisms describing resistance to BRAF inhibition. A possible escape mechanism is activation of MAPK/ERK through an EGFR-mediated activation of RAS and CRAF. In a pre-clinical model exploring this hypothesis, BRAF mutant CRC cells displayed high basal levels of phosphorylated receptor tyrosine kinases, suggesting that activation of MAPK may take place via upstream mediators even when BRAF is inhibited. Interestingly, in this murine model, dual MAPK pathway suppression with vemurafenib and an EGFR inhibitor demonstrated less induction of GTP-bound RAS, decrease in the phosphorylation of ERK1/2 and less induction of GTP-bound RAS, corresponding with tumor inhibition and tumor regression (27).
These preclinical data led to clinical studies evaluating activity of combined EGFR and BRAF inhibition. Data presented at ASCO 2014 suggested dual inhibition with MEK1/2 inhibitors plus cetuximab may have clinical efficacy (28). Patient-derived xenograft data suggested that dabrafenib (RAF inhibitor), trametinib (MEK inhibitor), and EGFR blockade had the most antitumor efficacy in a BRAF-mutant model (29). A phase 1–2 study of dabrafenib, trametinib, and panitumumab in BRAFV600E colon cancers demonstrated safety, tolerability and efficacy with a partial response in 4/6 patients on the triplet, and the other two with stable disease (30). A similar study looking at BRAF and MEK inhibition with dabrafenib and trametinib has been published, with 12% of refractory patients demonstrating a partial response. All patients in this study who had post-treatment biopsies had reduced levels of activated ERK1/2, demonstrating the clinical plausibility of the hypothesis proposed in the preclinical model cited above (31).
PIK3CA
Phosphoinositide 3-kinases (PI3K) are a family of heterodimeric lipid kinases consisting of regulatory (p85) and catalytic (p110) subunits. These kinases are important for multiple cellular processes including cell growth, proliferation, survival and apoptosis. PI3K is downstream of EGFR signaling, so activation of this pathway might lead to resistance to anti-EGFR therapies. The PIK3CA gene encodes the catalytic subunit, p110a, which, when mutated, results in a constitutively active PI3K. PIK3CA mutations in occur in 10–20% of colorectal cancers (12,32-34). Exon 9 and 20 are responsible for more than 80% of PIK3CA mutations in colorectal cancer (35). It is mutations in these exons that generate the activated PI3K that can generate secondary resistance to anti-EGFR therapies.
Controversy exists about how critical these mutations are in generating resistance to therapy. Sartore-Bianchi and colleagues examined 110 patients with mCRC treated with either panitumumab or cetuximab (34). Of patients carried PIK3CA mutations (13.6%; 15/110), of which the majority (11/15) were located at exon 20 and 4 of 15 at exon 9, and 0/15 patients with PIK3CA mutation responded to anti-EGFR therapies compared to wild type, and PFS was lower. The authors concluded that PIK3CA mutations may be an independent predictor of resistance to anti-EGFR. Other studies have similar results (36,37). In contrast, Prenen and colleagues found no such association. PIK3CA and KRAS status were assessed in 200 chemotherapy-refractory mCRC patients subsequently treated with cetuximab as a monotherapy or in combination with irinotecan. Twenty-three (12%) of 200 carried PIK3CA mutations, of which the majority were found on exon 9. There were no differences in PIK3CA mutation status among responders and non-responders (5/39 vs. 18/160, P=0.781) (33). Furthermore, there were no differences in median PFS (24 vs. 18 weeks; P=0.760) and OS (45 vs. 39 weeks; P=0.698) when comparing mutant to wild type tumors. In the European consortium, a similar prevalence of PIK3CA mutations, 14.5%, was found (12). PIK3CA mutations at exon 20 were associated with lack of response to cetuximab whereas mutations in exon 9 did not. Most studies to date have looked at exon 9 and 20 alone, as they account for the majority of the mutations, but the remainder of the gene may have relevance, as has been shown in RAS and RAF. Clearly, further investigations are needed to clarify the role of PIK3CA mutations as predictive biomarker for the treatment of mCRC patients with anti-EGFR therapies.
Amphiregulin (AREG)/Epiregulin (EREG)
Responses are not universal for patients who do not harbor a mutation in the RAS/RAF pathway. For EGFR blockade to be effective, upregulation of EGFR may be required. High mRNA expression of EREG and AREG are found in colorectal cancers, and this overexpression is thought to portend increased response to blockade of the EGFR pathway. An analysis of the PICCOLO study assessed AREG and EREG expression; high ligand expression was associated with statistically significant improvements in PFS, but not OS for patients treated with anti-EGFR therapy, when compared to irinotecan monotherapy (38). This study in isolation is not practice-changing, but merits further study. If validated, it gives clinicians another way to stratify patients for response to anti-EGFR therapies.
Anatomy as a surrogate for biology: right- vs. left-sided colon cancers
Prognostic differences in sidedness have been identified previously, indicting a worse prognosis for those cancers on the right side of the colon, even when controlling for stage and tumor size (39). Recently a planned post-hoc analysis of the CALGB 80405 study demonstrated that right-sided colon cancers are also significantly less responsive to anti-EGFR therapies in the first-line setting (40).
Further prospective studies are warranted with retrospective analyses on-going to further characterize the key biological variables responsible for differences in response based on anatomy. Some of the potential biologic variables responsible for this treatment effect include BRAF mutations, NRAS mutations and the CpG island methylator phenotype (CIMP), though these do not completely account for the differences observed (41). Until these biological variables are completely elucidated sidedness remains and important factor in the use of anti-EGFR directed therapies. Patients whose primary cancers arise in the right side of the colon should not receive cetuximab or panitumumab in the first-line setting. Treatment with these agents could be considered in later lines of therapy though the odds of benefit may be significantly decreased.
Summary
Current best clinical practice mandates that assessment of all common mutations in KRAS, NRAS, and BRAF be undertaken at the time of diagnosis of metastatic colorectal cancer. Based on the strength of the evidence, this recommendation has been adopted by the NCCN, ASCO, and ESMO (42-44). Sidedness is also an important factor and is now standard of care to take this into account when determining the first-line treatment for patients with metastatic colon cancer. PIK3CA mutation profiling should be considered for patients as the some evidence of a potential decrease in response with these mutations. With the current level of evidence, it is difficult to recommend treatment with cetuximab or panitumumab with a known PIK3CA mutation. Alterations in AREG and EREG are interesting avenues for future study, but do not, as yet, have the strength of evidence necessary to recommend routine testing in clinical practice.
Resistance to EGFR inhibitors: acquired
Studies including anti-EGFR mAbs demonstrated improvements in RRs, but only modest improvements in PFS and OS. In CRYSTAL and PRIME, the PFS improvements were around 1.5 months when compared to cytotoxic therapy alone (45). So, even in a selected population enriched for KRAS WT patients, the acquisition of secondary resistance occurs relatively early in treatment, and prompts disease progression on these agents. For patients who have progressed on cetuximab, changing therapy to panitumumab has shown a small chance of recapitulating a modest response (46,47).
Alterations in RAS
The most common drivers of that resistance appear to be the emergence of mutations in KRAS and NRAS, particularly in exons 3 and 4 (Figure 2) (48-50). In many cases, multiple distinct genetic events arose in the same sample as assessed by ctDNA or repeat tumor biopsies. Withdrawal of treatment with EGFR mAbs appeared to decrease the prevalence of these mutations, suggesting that there is a clonal selection that takes place in response to the stress placed on the cancer cells by targeting with anti EGFR mAbs.
Figure 2.

Known genetic alterations affecting susceptibility to anti-EGFR therapies in metastatic colorectal cancer. EGFR, epidermal growth factor receptor.
Changes in the EGFR binding pocket
In vitro testing has demonstrated a point mutation in the extracellular domain of EGFR (s492R) that changes the binding pocket of EGFR, rendering it insensitive to cetuximab, but not panitumumab. For patients harboring this point mutation, there is a hypothetical benefit to a class switch from one drug to the other (51).
MET signaling
The MET signaling pathway has been implicated in the development of therapy resistance in multiple different tumor types that utilize the EGFR pathway. For example, in EGFR-mutant non-small cell lung cancer (NSCLC) MET proto-oncogene amplification and activation of PI3 kinase signaling underlies the development of resistance to TKI’s (52). Similarly, in HER2 overexpressing breast cancer, MET activation has been implicated in the development of resistance to trastuzumab (53).
There have been a number of studies that studied the role of this pathway in CRC. Liska et al. demonstrated that hepatocyte growth factor (HGF) treatment can block cell cycle arrest induced by EGFR inhibition in cultured CRC cells (54). Highly selective MET inhibition and down-regulation of MET expression by RNAi were able to abrogate HGF affect in this setting, suggesting a central role of MET pathway in anti-EGFR resistance (54). Luraghi et al. confirmed the role of MET in the development of anti-EGFR resistance by demonstrating MET amplification in patients who have become resistant to cetuximab or panitumumab. In pre-clinical models HGF/MET signaling has been implicated to underlie CRC stem cell resistance to therapy (55). Recent data suggests that MET amplification is responsible for anti-EGFR resistance in a subset of patients. In the study by Bardelli et al., tumors from three out of seven studied patients who have progressed on anti-EGFR therapy demonstrated amplification of MET gene (56). One of the three patients also had rare, but detectable, MET-amplified cells in the pretreatment tumor sample, suggesting that anti-EGFR therapy selected for this resistant clone and that primary resistance to therapy was present at the onset of treatment.
Amplification of HER2
HER2 has been seen in cell models and tumor biopsy specimens as a possible mechanism of acquired resistance to EGFR directed therapies (57,58). HER2 amplification has been seen in about 5% of KRAS wild-type cancers. In a limited sample, patients who have this amplification had significantly inferior PFS on anti EGFR therapy (2.9 vs. 8.1 mo) (59). More studies are necessary to confirm these findings, but the mechanism is biologically plausible.
Clinical trials for acquired resistance to EGFR mAbs
Other parallel pathways may be mutated or upregulated, including PI3K, MET, or other downstream effectors of the MAPK pathway (51,56). As mentioned above, changes in the secretion of AREG and EREG may alter sensitivity to anti-EGFR mAbs, but further study is needed to fully understand the role of these ligands.
There are a few different strategies currently under investigation for patients with secondary resistance to anti-EGFR mAbs. Combining mAbs with MEK inhibitors, RAF inhibitors, or anti HER2 therapies have been proposed as possibilities. In the primary BRAF mutant population, MEK and RAF inhibitors have shown promise, and are being pursued in trials of later line treatment. Newer agents that are essentially pooled anti-EGFR mAbs, Sym004 and MM151 are currently under investigation. These agents might be especially beneficial in the setting of an extracellular domain mutation in EGFR.
Resistance to VEGF inhibitors: primary and acquired
Blocking circulating VEGF and its receptors have shown evidence of clinical utility in colon cancer and gastroesophageal cancers. Bevacizumab an antibody against VEGF-A was the first biologic therapy to demonstrate an improvement in OS for patients with mCRC in addition to irinotecan or a fluoropyrimidine (60-62). Like treatment with cetuximab or panitumumab in KRAS WT patients, the responses are initially evident but eventually resistance develops, with an improvement in PFS of around 2 months (62). Mutations in KRAS, NRAS, BRAF or PIK3CA have not been shown to alter sensitivity to bevacizumab therapy (40,63)
Clinical benefit in gastric cancer with bevacizumab is mixed. In the AVAGAST study, improvements in PFS and RR were evident in patients with gastric cancer, but the primary endpoint of OS was not met (64). In the wake of bevacizumab, other agents targeting the pathway have been tested. Rather than targeting circulating VEGF, ramucirumab targets VEGFR-2, thought to be the primary driver of VEGF-A mediated angiogenesis. The RAINBOW study showed an improvement in OS by nearly two months for patients treated with ramucirumab (65). In REGARD, ramucirumab was used as a single agent with an increase in survival of 1.4 months (66). Because of these positive results, ramucirumab has received FDA approval for the late-line treatment of gastroesophageal cancers.
Alternative isoforms of VEGF
Assaying circulating biomarkers to assess for response/resistance has been challenging, largely because there are so many different/overlapping mechanisms in play regulating angiogenesis in tumor cells. Circulating VEGF-A, or short VEGF-A isoforms did not correlate with PFS/OS outcomes (67,68). Preclinical work has suggested that alternative members of the VEGF signaling family [Placental growth factor (PlGF), VEGF-C and VEGF-D] can stimulate angiogenesis in the setting of VEGF-A blockade with bevacizumab (69,70). Research out of MD Anderson using chemotherapy plus bevacizumab in colon cancer demonstrated that levels of circulating VEGF-C were increased prior to disease progression, and VEGF-D levels were increased at the time of progression. In addition, patients treated with chemotherapy plus bevacizumab had increased levels of PlGF and VEGF-D, but patients treated with chemotherapy had no such increase. These increases were transient, suggesting that increases in VEGF-D and PlGF were directly in response to treatment with bevacizumab (71). The CAIRO-2 population (capecitabine, oxaliplatin, bevacizumab ± cetuximab) was analyzed for different levels of VEGF isoforms (VEGF-A, -B, -C-, and- D, VEGF-R1, -R2), and indicated a significant effect of VEGF-D on both PFS and OS in patients treated with bevacizumab. Patients with lower levels of VEGF-D had improvements, while patients with higher levels of VEGF-D saw almost no benefit (72). In patients with gastric cancer, similar evidence of lack of benefit has been shown with elevated levels of VEGF-D (73). However, the definition of high/low levels of VEGF-D has not been validated, and there is currently no good way to apply these levels to a broader population. Conceptually, VEGF-D is an attractive biomarker, because it has been shown that VEGF-D binds to VEGFR-2 (71,74).
The MAVERICC study and CALGB 80405 may offer some insight into other biomarkers predicting response to VEGF-directed therapy, but these results have not yet been presented/published to answer the specific question.
HIF-1α
Bevacizumab is thought to promote apoptosis by decreasing tumor vascularity and by inducing hypoxia in the tumor microenvironment. Hypoxia-inducible factor 1-α (HIF-1α) promotes oxygen delivery to areas of relative hypoxia, and is integrally involved in regulation of the tumor microenvironment. Upregulation of HIF-1a promotes other proangiogenic factors by upregulating FGF2, IL-8, STAT-3, and ANGPT-2 (68). While no specific HIF-1a blockers have been tested in this population, it is an active area of research. Preclinical models suggest that IL-6 blockade could be another mechanism to overcome resistance to VEGF therapy mediated by changes in the microenvironment (75).
Resistance to HER2: primary
Blockade of HER2 has a track record of efficacy in breast cancer extending more than a decade. HER2 is overexpressed in 7–22% of esophagogastric cancers (76,77). The first randomized trial demonstrating efficacy showed an improvement in mOS of 2.7 months (13.8 vs. 11.1 mo, P=0.0046) when combined with chemotherapy (78). This benefit has been extrapolated in some studies to extend to any platinum/fluoropyrimidine containing regimen, akin to the REAL-2 data comparing cytotoxic backbones (79). In spite of these improvements, the efficacy is clearly limited to a small subset of primary GEJ cancers.
Selecting for patients with this overexpression is key, as patients whose tumors do not overexpress HER2 do not benefit from the addition of trastuzumab to chemotherapy. Much is known about HER2 overexpression in breast cancer, and similar guidelines exist for applying HER2 directed treatment against esophageal cancer as in breast cancer. Immunohistochemical expression of 0–1 is a contraindication to treatment, and expression of 3+ is an indication for treatment. IHC of 2 should prompt analysis using fluorescence in situ hybridization (80). Staining of gastric cancer is different from breast cancer, in that incomplete basolateral or lateral staining is considered positive as is complete membrane staining. This can result in inaccuracy/heterogeneity in HER2 positivity, and confirmation on multiple tumor specimens has been recommended to ensure accuracy (81). Newer technologies such as gene copy number have been used in breast cancer, and may be applied to GEJ cancers as well (82). It is this inaccuracy in molecular diagnosis that leads to at least some of the primary resistance of GEJ cancers to trastuzumab.
Even in patients whose tumors overexpress HER2, RRs was only 47% in ToGA, implying primary resistance to the target (78). Other agents in the pathway have been tried in the HER2 overexpressing population with variable results. Small molecule TKI lapatinib showed activity in the second line setting but did not improve OS in the IIT population to a significant degree (83). The recently completed GATSBY study failed to show an OS advantage for trastuzumab coupled to a taxane, T-DM1, as well (HR 1.15, P=0.86) (84). Heterogeneity in populations (Asian/non-Asian) may explain some of the variation in response across studies, but these mechanisms of resistance in a HER2 overexpressing population have not been clearly worked out. More research is needed to understand why gastroesophageal cancers have different responses to treatment with therapies that have proven efficacy in the setting of breast cancer.
Resistance to HER2: acquired
Like other targeted agents, initial responses are met with nearly inevitable progression. Much of the data about acquired resistance to HER2 directed therapies has derived from research in breast cancer. Continued research into how therapies directed against HER2 actually work highlights the different possible mechanisms of resistance.
Alteration of the HER2 receptor
As a cell surface receptor, HER2 has intra- and extracellular domains that can be affected, altering the activity of targeted therapies. Truncations in the extracellular domain, the so called p95 protein, retain its kinase activity, without showing any sensitivity to trastuzumab. Overexpression of the p95 subunit, likely through proteolytic cleavage of the extracellular domain (or a primary mutation) confers resistance to trastuzumab both pre-clinically and clinically (85). Membrane-associated glycoproteins such as MUC4 have also been shown to mask the HER2 binding site and prevent primary binding of trastuzumab to its target, reflecting primary resistance to the drug (86). Whether or not these alterations affect response to treatment with trastuzumab in esophageal cancer is as yet unclear, but is an area of active investigation.
Upregulation of HER3
Cellular proliferation via the HER2 pathway depends upon heterodimerization with other receptors. Current research suggests that HER3 is the most common and most potent dimerization partner. In models of both breast cancer and in HER2 overexpressing esophageal cancer, HER3 is upregulated in response to blockade by trastuzumab, conferring preserved cell signaling and a stimulatory growth signal. Blocking upregulated HER3 appeared to induce apoptosis in these models, suggesting that targeting HER3 and HER2 could result in improvements in response, or prevent the acquisition of resistance to trastuzumab (87).
Alternative pathways and increased intracellular signaling
Other preclinical models suggest other pathways are upregulated during treatment with trastuzumab leading to the development of resistance. Paramount among these is the PI3K-AKT pathway. Both PIK3CA mutations and inactivation of PTEN are known to promote downstream signaling even in the absence of upstream HER2 activation. Tumors with activating mutations in PI3K appear to have increased primary resistance to HER2 directed therapies in both breast cancer and gastroesophageal cancer (88). Preclinical data has been paired with pre- and post-treatment biopsies suggesting that higher expression of FGFR3 and p-AKT are seen in patients whose tumors are progressing on trastuzumab (89,90). Early phase studies are underway in gastroesophageal cancers and other solid tumors combining trastuzumab with agents targeting the PI3K pathway including direct PI3K inhibitors, mTOR inhibitors, TORC 1–2 inhibitors, and AKT inhibitors. Promising work has already been presented in breast cancer (91), but has not been completed in gastroesophageal cancer.
Conclusions
Biologic therapies have improved survival for subsets of patients with gastrointestinal malignancies. Better identification of target populations has been shown in multiple tumor types to be the single greatest factor in generating these favorable responses. Through biomarker-driven trials, we are improving understanding of these ideal patients, but also understanding the mechanisms behind the development of resistance. Additionally, the wider implementation of the routine use of molecular panels in the diagnosis and treatment of patients with gastrointestinal malignancies will aid in the search for biomarkers. Current and future clinical trials (Table 1) are dedicated to finding ways to identify resistance mechanisms and overcome them with pharmaceutical interventions or earlier transitions to subsequent lines of therapy.
Table 1. Ongoing trials targeting resistance mechanisms from clinicaltrials.gov.
| Trials registered with Clinicaltrials.gov | NCI Trial # | Agent(s) |
|---|---|---|
| EGFR resistance (colorectal) | ||
| Immune checkpoint blockade | NCT02318901 | Cetuximab + pembrolizumab |
| EGFR/ERBB2/ERBB3 blockade | NCT01862003 | AZD 8931 |
| Oligoclonal EGFR inhibition | NCT02785068 | MM-151 + 5-FU + Nal-IRI |
| Recombinant EGFR antibody | NCT02352571 | GC1118 |
| EGFR antibody mixture | NCT02568046 | Sym-004 |
| Cetuximab rechallenge after progression | NCT02296203 | Cetuximab |
| EGFR and MET inhibition | NCT02630420 | Cetuximab + savolitinib |
| EGFR and MEK inhibition | NCT02399943 | Trametinib + panitumumab |
| BRAF mutation (colorectal) | ||
| Wee1 inhibitor | NCT02906059 | AZD1775 + irinotecan |
| AKT inhibitor | NCT01902173 | Dabrafenib + trametinib + GSK2141795 |
| HER2 resistance (gastroesophageal) | ||
| Biomarkers for prediction of response | NCT02305043 | Observational |
| EGFR/HER2 blockade | NCT02378389 | Pyrotinib ± docetaxel |
| Immune checkpoint blockade | NCT02901301 | Pembrolizumab, trastuzumab |
| HER2 peptide vaccination | NCT02276300 | Vaccine targeting HER2 |
| VEGFR2 + HER2 | NCT02726399 | Ramucirumab + trastuzumab |
| AKT inhibitor | NCT02240212 | Afuresertib + paclitaxel |
| EGFR TKI | NCT01522768 | Afatinib + paclitaxel |
| Extracellular domain of HER2 | NCT02892123 | ZW25 |
| HER2 antibody drug conjugate | NCT02576548 | MEDI4276 |
| Vaccine + chemotherapy | NCT02795988 | IMU131+ chemotherapy |
| VEGF resistance (colorectal or gastroesophageal) | ||
| Wnt-Beta Catenin antagonist | NCT02413853 | Chemotherapy + Bev + PRI-724 |
| VEGFR/FGFR/PDGFR inhibition | NCT02835833 | Nintedanib + Bev |
EGFR, epidermal growth factor receptor; VEGF, vascular endothelial growth factor.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 2.Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:481-9. 10.1016/S1470-2045(13)70096-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lordick F, Rocken C. The identification of predictive factors for perioperative chemotherapy in esophago-gastric cancer. Ann Oncol 2013;24:1135-8. 10.1093/annonc/mdt111 [DOI] [PubMed] [Google Scholar]
- 4.Dutton SJ, Ferry DR, Blazeby JM, et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): a phase 3, multicentre, double-blind, placebo-controlled randomised trial. Lancet Oncol 2014;15:894-904. 10.1016/S1470-2045(14)70024-5 [DOI] [PubMed] [Google Scholar]
- 5.Wang VE, Grandis JR, Ko AH. New Strategies in Esophageal Carcinoma: Translational Insights from Signaling Pathways and Immune Checkpoints. Clin Cancer Res 2016;22:4283-90. 10.1158/1078-0432.CCR-16-0292 [DOI] [PubMed] [Google Scholar]
- 6.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643-8. 10.1158/0008-5472.CAN-06-4158 [DOI] [PubMed] [Google Scholar]
- 7.Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol 2011;22:1535-46. 10.1093/annonc/mdq632 [DOI] [PubMed] [Google Scholar]
- 8.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013;369:1023-34. 10.1056/NEJMoa1305275 [DOI] [PubMed] [Google Scholar]
- 9.Douillard JY, Rong A, Sidhu R. RAS mutations in colorectal cancer. N Engl J Med 2013;369:2159-60. 10.1056/NEJMoa1305275 [DOI] [PubMed] [Google Scholar]
- 10.Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008;26:374-9. 10.1200/JCO.2007.12.5906 [DOI] [PubMed] [Google Scholar]
- 11.Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992-5. 10.1158/0008-5472.CAN-06-0191 [DOI] [PubMed] [Google Scholar]
- 12.De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753-62. 10.1016/S1470-2045(10)70130-3 [DOI] [PubMed] [Google Scholar]
- 13.Tejpar S, Piessevaux H. Personalized medicine in metastatic colorectal cancer treated with anti-epidermal growth factor receptor agents: a future opportunity? Asia Pac J Clin Oncol 2014;10 Suppl 1:2-10. 10.1111/ajco.12176 [DOI] [PubMed] [Google Scholar]
- 14.Van Cutsem E, Lenz HJ, Kohne CH, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015;33:692-700. 10.1200/JCO.2014.59.4812 [DOI] [PubMed] [Google Scholar]
- 15.Segelov E, Thavaneswaran S, Waring PM, et al. Response to Cetuximab With or Without Irinotecan in Patients With Refractory Metastatic Colorectal Cancer Harboring the KRAS G13D Mutation: Australasian Gastro-Intestinal Trials Group ICECREAM Study. J Clin Oncol 2016;34:2258-64. 10.1200/JCO.2015.65.6843 [DOI] [PubMed] [Google Scholar]
- 16.Schirripa M, Loupakis F, Lonardi S, et al. Phase II study of single-agent cetuximab in KRAS G13D mutant metastatic colorectal cancer. Ann Oncol 2015;26:2503. [DOI] [PubMed] [Google Scholar]
- 17.Peeters M, Price TJ, Cervantes A, et al. Final results from a randomized phase 3 study of FOLFIRI {+/-} panitumumab for second-line treatment of metastatic colorectal cancer. Ann Oncol 2014;25:107-16. 10.1093/annonc/mdt523 [DOI] [PubMed] [Google Scholar]
- 18.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091-6. 10.1200/JCO.2009.21.9170 [DOI] [PubMed] [Google Scholar]
- 19.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54. 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 20.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008;26:5705-12. 10.1200/JCO.2008.18.0786 [DOI] [PubMed] [Google Scholar]
- 21.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65:6063-9. 10.1158/0008-5472.CAN-05-0404 [DOI] [PubMed] [Google Scholar]
- 22.Laurent-Puig P, Cayre A, Manceau G, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 2009;27:5924-30. 10.1200/JCO.2008.21.6796 [DOI] [PubMed] [Google Scholar]
- 23.Richman SD, Seymour MT, Chambers P, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol 2009;27:5931-7. 10.1200/JCO.2009.22.4295 [DOI] [PubMed] [Google Scholar]
- 24.Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol 2012;30:1755-62. 10.1200/JCO.2011.38.0915 [DOI] [PubMed] [Google Scholar]
- 25.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809-19. 10.1056/NEJMoa1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100-3. 10.1038/nature10868 [DOI] [PubMed] [Google Scholar]
- 27.Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol 2015;33:4023-31. 10.1200/JCO.2015.63.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahronian LG, Corcoran RB. Effective MAPK Inhibition is critical for therapeutic responses in colorectal cancer with BRAF mutations. Mol Cell Oncol 2015;3:e1048405. 10.1080/23723556.2015.1048405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Shi H, Bleam M, et al. Antitumor effects of dabrafenib, trametinib, and panitumumab as single agents and in combination in BRAF-mutant colorectal carcinoma (CRC) models. J Clin Oncol 2014;32:abstr 3513.
- 30.Bendell J, Atreya C, Andre T, et al. Efficacy and tolerability in an open-label phase I/II study of MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in combination in patients (pts) with BRAF V600E mutated colorectal cancer (CRC). J Clin Oncol 2014;32:abstr 3515.
- 31.Corcoran RB, Atreya C, Faichook G, et al. Phase 1-2 trial of the BRAF inhibitor dabrafenib (D) plus MEK inhibitor trametinib (T) in BRAF V600 mutant colorectal cancer (CRC): Updated efficacy and biomarker analysis. J Clin Oncol 2014;32:abstr 3517.
- 32.Barault L, Veyrie N, Jooste V, et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer 2008;122:2255-9. 10.1002/ijc.23388 [DOI] [PubMed] [Google Scholar]
- 33.Prenen H, De Schutter J, Jacobs B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 2009;15:3184-8. 10.1158/1078-0432.CCR-08-2961 [DOI] [PubMed] [Google Scholar]
- 34.Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009;69:1851-7. 10.1158/0008-5472.CAN-08-2466 [DOI] [PubMed] [Google Scholar]
- 35.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554. 10.1126/science.1096502 [DOI] [PubMed] [Google Scholar]
- 36.Perrone F, Lampis A, Orsenigo M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 2009;20:84-90. 10.1093/annonc/mdn541 [DOI] [PubMed] [Google Scholar]
- 37.Sood A, McClain D, Maitra R, et al. PTEN gene expression and mutations in the PIK3CA gene as predictors of clinical benefit to anti-epidermal growth factor receptor antibody therapy in patients with KRAS wild-type metastatic colorectal cancer. Clin Colorectal Cancer 2012;11:143-50. 10.1016/j.clcc.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seligmann JF, Elliott F, Richman SD, et al. Combined Epiregulin and Amphiregulin Expression Levels as a Predictive Biomarker for Panitumumab Therapy Benefit or Lack of Benefit in Patients With RAS Wild-Type Advanced Colorectal Cancer. JAMA Oncol 2016. [Epub ahead of print]. 10.1001/jamaoncol.2015.6065 [DOI] [PubMed] [Google Scholar]
- 39.Weiss JM, Pfau PR, O'Connor ES, et al. Mortality by stage for right- versus left-sided colon cancer: analysis of surveillance, epidemiology, and end results--Medicare data. J Clin Oncol 2011;29:4401-9. 10.1200/JCO.2011.36.4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venook AP, Niedzwiecki D, Innocenti F, et al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34:abstr 3504.
- 41.Brulé SY, Jonker DJ, Karapetis CS, et al. Location of colon cancer (right-sided versus left-sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur J Cancer 2015;51:1405-14. 10.1016/j.ejca.2015.03.015 [DOI] [PubMed] [Google Scholar]
- 42.Benson AB, 3rd, Bekaii-Saab T, Chan E, et al. Metastatic colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw 2013;11:141-52; quiz 52. 10.6004/jnccn.2013.0022 [DOI] [PubMed] [Google Scholar]
- 43.Allegra CJ, Rumble RB, Hamilton SR, et al. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol 2016;34:179-85. 10.1200/JCO.2015.63.9674 [DOI] [PubMed] [Google Scholar]
- 44.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386-422. 10.1093/annonc/mdw235 [DOI] [PubMed] [Google Scholar]
- 45.Haraldsdottir S, Bekaii-Saab T. Integrating anti-EGFR therapies in metastatic colorectal cancer. J Gastrointest Oncol 2013;4:285-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wadlow RC, Hezel AF, Abrams TA, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist 2012;17:14. 10.1634/theoncologist.2011-0452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saif MW, Kaley K, Chu E, et al. Safety and efficacy of panitumumab therapy after progression with cetuximab: experience at two institutions. Clin Colorectal Cancer 2010;9:315-8. 10.3816/CCC.2010.n.046 [DOI] [PubMed] [Google Scholar]
- 48.Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486:532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arena S, Bellosillo B, Siravegna G, et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res 2015;21:2157-66. 10.1158/1078-0432.CCR-14-2821 [DOI] [PubMed] [Google Scholar]
- 50.Diaz LA, Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med 2012;18:221-3. 10.1038/nm.2609 [DOI] [PubMed] [Google Scholar]
- 52.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43. 10.1126/science.1141478 [DOI] [PubMed] [Google Scholar]
- 53.Shattuck DL, Miller JK, Carraway KL, 3rd, et al. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res 2008;68:1471-7. 10.1158/0008-5472.CAN-07-5962 [DOI] [PubMed] [Google Scholar]
- 54.Liska D, Chen CT, Bachleitner-Hofmann T, et al. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res 2011;17:472-82. 10.1158/1078-0432.CCR-10-0568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luraghi P, Reato G, Cipriano E, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res 2014;74:1857-69. 10.1158/0008-5472.CAN-13-2340-T [DOI] [PubMed] [Google Scholar]
- 56.Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013;3:658-73. 10.1158/2159-8290.CD-12-0558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011;3:99ra86. 10.1126/scitranslmed.3002442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 2011;1:508-23. 10.1158/2159-8290.CD-11-0109 [DOI] [PubMed] [Google Scholar]
- 59.Raghav KPS, Overman MJ, Yu R, et al. HER2 amplification as a negative predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. J Clin Oncol 2016;34:abstr 3517. [DOI] [PubMed]
- 60.Hurwitz H. Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin Colorectal Cancer 2004;4 Suppl 2:S62-8. 10.3816/CCC.2004.s.010 [DOI] [PubMed] [Google Scholar]
- 61.Schwartzberg LS, Rivera F, Karthaus M, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol 2014;32:2240-7. 10.1200/JCO.2013.53.2473 [DOI] [PubMed] [Google Scholar]
- 62.Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013-9. 10.1200/JCO.2007.14.9930 [DOI] [PubMed] [Google Scholar]
- 63.Stintzing S, Fischer von Weikersthal L, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer-subgroup analysis of patients with KRAS: mutated tumours in the randomised German AIO study KRK-0306. Ann Oncol 2012;23:1693-9. 10.1093/annonc/mdr571 [DOI] [PubMed] [Google Scholar]
- 64.Ohtsu A, Shah MA, Van Cutsem E, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 2011;29:3968-76. 10.1200/JCO.2011.36.2236 [DOI] [PubMed] [Google Scholar]
- 65.Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 2014;15:1224-35. 10.1016/S1470-2045(14)70420-6 [DOI] [PubMed] [Google Scholar]
- 66.Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383:31-9. 10.1016/S0140-6736(13)61719-5 [DOI] [PubMed] [Google Scholar]
- 67.Goede V, Coutelle O, Neuneier J, et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer 2010;103:1407-14. 10.1038/sj.bjc.6605925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jayson GC, Kerbel R, Ellis LM, et al. Antiangiogenic therapy in oncology: current status and future directions. Lancet 2016;388:518-29. 10.1016/S0140-6736(15)01088-0 [DOI] [PubMed] [Google Scholar]
- 69.Cao R, Eriksson A, Kubo H, et al. Comparative evaluation of FGF-2-, VEGF-A-, and VEGF-C-induced angiogenesis, lymphangiogenesis, vascular fenestrations, and permeability. Circ Res 2004;94:664-70. 10.1161/01.RES.0000118600.91698.BB [DOI] [PubMed] [Google Scholar]
- 70.Hindryckx P, Waeytens A, Laukens D, et al. Absence of placental growth factor blocks dextran sodium sulfate-induced colonic mucosal angiogenesis, increases mucosal hypoxia and aggravates acute colonic injury. Lab Invest 2010;90:566-76. 10.1038/labinvest.2010.37 [DOI] [PubMed] [Google Scholar]
- 71.Lieu CH, Tran H, Jiang ZQ, et al. The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. PLoS One 2013;8:e77117. 10.1371/journal.pone.0077117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weickhardt AJ, Williams DS, Lee CK, et al. Vascular endothelial growth factor D expression is a potential biomarker of bevacizumab benefit in colorectal cancer. Br J Cancer 2015;113:37-45. 10.1038/bjc.2015.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jüttner S, Wissmann C, Jöns T, et al. Vascular endothelial growth factor-D and its receptor VEGFR-3: two novel independent prognostic markers in gastric adenocarcinoma. J Clin Oncol 2006;24:228-40. 10.1200/JCO.2004.00.3467 [DOI] [PubMed] [Google Scholar]
- 74.Rissanen TT, Markkanen JE, Gruchala M, et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res 2003;92:1098-106. 10.1161/01.RES.0000073584.46059.E3 [DOI] [PubMed] [Google Scholar]
- 75.Eichten A, Su J, Adler AP, et al. Resistance to Anti-VEGF Therapy Mediated by Autocrine IL6/STAT3 Signaling and Overcome by IL6 Blockade. Cancer Res 2016;76:2327-39. 10.1158/0008-5472.CAN-15-1443 [DOI] [PubMed] [Google Scholar]
- 76.Hofmann M, Stoss O, Shi D, et al. Assessment of a HER2 scoring system for gastric cancer: results from a validation study. Histopathology 2008;52:797-805. 10.1111/j.1365-2559.2008.03028.x [DOI] [PubMed] [Google Scholar]
- 77.Marx AH, Tharun L, Muth J, et al. HER-2 amplification is highly homogenous in gastric cancer. Hum Pathol 2009;40:769-77. 10.1016/j.humpath.2008.11.014 [DOI] [PubMed] [Google Scholar]
- 78.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687-97. 10.1016/S0140-6736(10)61121-X [DOI] [PubMed] [Google Scholar]
- 79.Gong J, Liu T, Fan Q, et al. Optimal regimen of trastuzumab in combination with oxaliplatin/ capecitabine in first-line treatment of HER2-positive advanced gastric cancer (CGOG1001): a multicenter, phase II trial. BMC Cancer 2016;16:68. BMC Cancer 2016;16:68. 10.1186/s12885-016-2092-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ajani JA, D'Amico TA, Almhanna K, et al. Esophageal and esophagogastric junction cancers, version 1.2015. J Natl Compr Canc Netw 2015;13:194-227. 10.6004/jnccn.2015.0028 [DOI] [PubMed] [Google Scholar]
- 81.Kelly CM, Janjigian YY. The genomics and therapeutics of HER2-positive gastric cancer-from trastuzumab and beyond. J Gastrointest Oncol 2016;7:750-762. 10.21037/jgo.2016.06.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gomez-Martin C, Plaza JC, Pazo-Cid R, et al. Level of HER2 gene amplification predicts response and overall survival in HER2-positive advanced gastric cancer treated with trastuzumab. J Clin Oncol 2013;31:4445-52. 10.1200/JCO.2013.48.9070 [DOI] [PubMed] [Google Scholar]
- 83.Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN--a randomized, phase III study. J Clin Oncol 2014;32:2039-49. 10.1200/JCO.2013.53.6136 [DOI] [PubMed] [Google Scholar]
- 84.Kang YK, Shah MA, Ohtsu A, et al. A randomized, open-label, multicenter, adaptive phase 2/3 study of trastuzumab emtansine (T-DM1) versus a taxane (TAX) in patients (pts) with previously treated HER2-positive locally advanced or metastatic gastric/gastroesophageal junction adenocarcinoma (LA/MGC/GEJC). J Clin Oncol 2016;34:abstr 5.
- 85.Sperinde J, Bachmeier B, Weidler J, et al. Quantitative p95HER2 protein expression is predictive of trastuzumab response in HER2-positive metastatic breast cancer. San Antonio Breast Cancer Symposium 2016;2016:P3-07-9. [Google Scholar]
- 86.Carraway KL, Perez A, Idris N, et al. Muc4/sialomucin complex, the intramembrane ErbB2 ligand, in cancer and epithelia: to protect and to survive. Prog Nucleic Acid Res Mol Biol 2002;71:149-85. 10.1016/S0079-6603(02)71043-X [DOI] [PubMed] [Google Scholar]
- 87.Ebbing EA, Medema JP, Meijer SL, et al. HER3 mediates acquired resistance to HER2-targeted therapy in esophageal adenocarcinoma. Proceedings of the American Association for Cancer Research 2015;106:abstr 350. [Google Scholar]
- 88.Sukawa Y, Yamamoto H, Nosho K, et al. HER2 expression and PI3K-Akt pathway alterations in gastric cancer. Digestion 2014;89:12-7. 10.1159/000356201 [DOI] [PubMed] [Google Scholar]
- 89.Piro G, Carbone C, Cataldo I, et al. An FGFR3 Autocrine Loop Sustains Acquired Resistance to Trastuzumab in Gastric Cancer Patients. Clin Cancer Res 2016;22:6164-75. 10.1158/1078-0432.CCR-16-0178 [DOI] [PubMed] [Google Scholar]
- 90.Niu J, Gelbspan D, Weitz D, et al. HER2-positive, trastuzumab-resistant metastatic esophageal cancer presenting with brain metastasis after durable response to dual HER2 blockade: a case report. J Gastrointest Oncol 2014;5:E103-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.André F, O'Regan R, Ozguroglu M, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol 2014;15:580-91. 10.1016/S1470-2045(14)70138-X [DOI] [PubMed] [Google Scholar]