

Abstract
Biliary tract cancers (BTC) is a group of malignancies that arise from the epithelial cells of the biliary tree. These cancers are typically classified by anatomic site of origin: intrahepatic cholangiocarcinoma (IHCC) and extrahepatic cholangiocarcinoma (EHCC), and gallbladder cancer (GBC). To date, complete surgical resection remains the mainstay of treatment especially for earlier stage disease. Unfortunately, most patients present with advanced or metastatic disease, when systemic chemotherapy is the only treatment option. Due to the paucity of effective treatments, BTCs have a dismal prognosis. There is a tremendous need to better understand the disease biology, discover new therapies, and improve clinical outcomes for this challenging disease. Next-generation sequencing has produced a more accurate and detailed picture of the molecular signatures in BTCs. The three BTC histologic subtypes are, in fact, quite molecularly distinct. IHCC commonly contain FGFR2 fusions and IDH 1 and 2 mutations, whereas EHCC and GBC tend to carry mutations in EGFR, HER2, and MAPK pathway. In light of this emerging knowledge, clinical trials have become more biomarker-driven, which allows capturing of subsets of patients that are most likely to respond to certain therapies. Many new and promising targeted therapeutics are currently in the pipeline. Here we review the genetic landscape of BTCs while focusing on new molecular targets and targeted therapeutics currently being investigated in biomarker-driven clinical trials.
Keywords: Biliary tract cancer (BTC), cholangiocarcinoma, gallbladder cancer (GBC), biologics
Introduction
Biliary tract cancers (BTC) is a group of rare and aggressive malignancies arising from the epithelium of the biliary duct system. They are classified based on their anatomical site in the biliary tree [intrahepatic cholangiocarcinoma (IHCC) or extrahepatic cholangiocarcinoma (EHCC)] or gallbladder cancer (GBC) (Figure 1). IHCC is the most common BTC and the second most common hepatic malignancy, accounting for 10–20% of all primary hepatic malignancies (1,2). Owing to the insidious nature of this group of cancers, they are usually diagnosed at an advanced stage and carry a dismal prognosis. Surgery is potentially curative in early stage disease. In cases of advanced and metastatic disease, the current standard of care is systemic chemotherapy with gemcitabine and cisplatin. Clinical response rates to these cytotoxic chemotherapies are low, with a 5-year survival of less than 10% for all three BTC subtypes. In recent years, we have made strides in our understanding of the disease biology, as well as advancements in diagnostic techniques and novel therapeutic strategies. Notably, the genomic revolution has ushered in an era of high-throughput and deep molecular profiling, which has provided invaluable insight into actionable molecular alterations, as well as their prognostic significance. We have also developed a greater appreciation for the molecular heterogeneity across the BTC subtypes, realizing that these anatomically classified subgroups exhibit distinct molecular architectures. Considering this emerging knowledge, clinical trial design has steered away from the “one-size-fits-all” mentality and has become more biomarker-driven. Currently there are several ongoing clinical studies investigating the efficacy of targeted therapies aimed at populations that underwent biomarker selection. In this review, we will highlight actionable molecular targets and their novel targeted therapeutics in current clinical trials.
Figure 1.
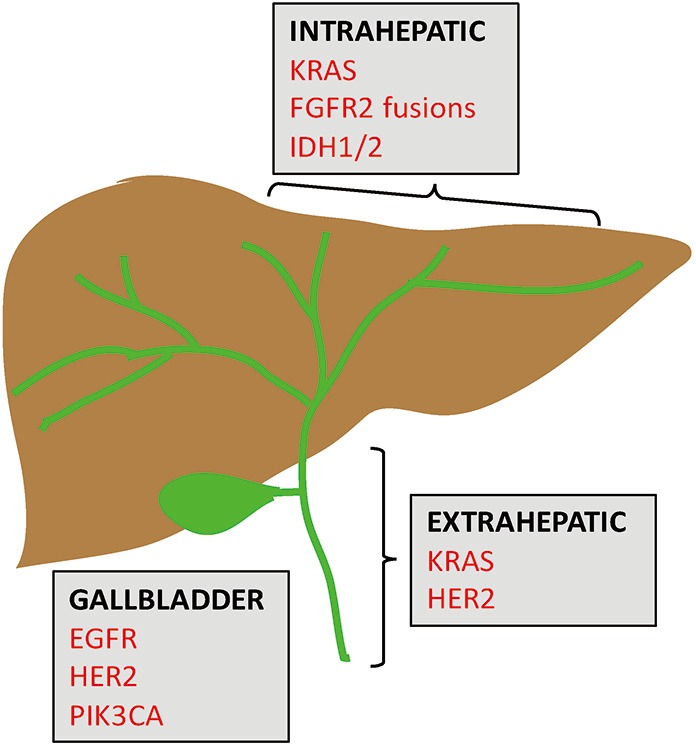
Distinct molecular signatures of BTCs. Shown is a schematic of the biliary tract and gallbladder, along with common genetic aberrations seen in intrahepatic and extrahepatic cholangiocarcinoma, and GBC. Red font indicates targetable genes.
Epidemiology
The incidence of BTC varies by geography and demographics, likely due to distinct environmental risk factors and genetic predisposition. Though BTCs are traditionally more common in Asian countries, their incidence has been rising in Western countries in recent decades (3). Though it is a rare disease, the global incidence is rising. Chronic inflammation and bile stasis in the biliary tract are thought to be major risk factors underlying the pathogenesis of these cancers. Specific risk factors include primary sclerosing cholangitis, liver fluke (Clonorchis, Opisthorchis) infection, hepatitis B and C infections, cholelithiasis or choledocholithiasis, cirrhosis, alcohol, smoking, and fatty liver disease (3,4).
Current management
Overall, the 5-year survival rate of BTCs is extremely low (10% for CCAs and less than 5% for GBC) (5,6). Surgery is the only potentially curative modality but most patients are asymptomatic until late in the disease course and present with locally advanced or metastatic disease. Thus, only 10–15% of BTCs are amenable to surgery at initial presentation (7). Even though improved surgical techniques and better patient selection based on more advanced radiologic techniques have resulted in better tumor resection rates, the recurrence rates of these aggressive cancers remain high at 50–60% (7,8). The role of adjuvant therapy is poorly-defined and standard regimen is unclear due to the relative rarity of this disease which hinders large scale prospective studies (9,10). Therefore, the benefit of adjuvant treatment is commonly appraised from meta-analyses of multiple small retrospective studies that usually include more than one, if not all, subtypes of BTC. To more clearly determine the role of adjuvant treatment, two phase III randomized controlled trials are currently ongoing in the Europe to determine the role of adjuvant gemcitabine plus cisplatin (ACTICCA-1 trial, NCT02170090) or oxaliplatin (NCT01313377) versus observation for patients with resected BTC. Before results from these trials are available, current NCCN guidelines recommend adjuvant fluoropyrimidine or gemcitabine-based chemotherapy with consideration of radiation for patients with node-positive disease or R1/R2 resections.
For patients presenting with unresectable BTCs (locally advanced, recurrent, or metastatic), the current standard first-line therapy is a combination of gemcitabine and cisplatin. This regimen was established by the ABC-02 trial, the largest randomized phase III study to date, which showed a survival benefit of the combination as opposed to gemcitabine alone (11.7 vs. 9 months) (11). Other chemotherapy combinations (e.g., oxaliplatin, 5-FU, capecitabine, irinotecan) have demonstrated only marginal improvements in survival (12). Targeted therapies such as anti-EGFR or anti-VEGF antibodies have so far struggled to succeed in phase I or II clinical trials. Performing randomized control trials (RCT) for advanced BTCs has proven challenging due to the rarity of these malignancies, lack of effective agents, potential high heterogeneity within this diagnostic entity, and possibly fundamental differences among the three BTC subtypes (IHCC, EHCC, and GBC). In fact, next generation sequencing (NGS) and transcriptomic analyses have revealed that these BTC subtypes are molecularly distinct from one another, and therefore may respond differently to the same treatment strategy and should not be approached as a single entity for clinical trial design (13,14). To improve patient outcome, future clinical trial design must better stratify patients based on considerations of histologic and molecular subtypes, and allocate patients to the appropriate targeted agents driven by biomarkers that could predict treatment response.
Genetic landscape
Before the advent of NGS, our knowledge of genetic aberrations in BTCs was limited because older methodologies restricted mutational profiling to a few select oncogenes or hotspots (15). That technology previously allowed us to identify key signaling pathways altered in BTCs, such as the EGFR and vascular endothelial growth factor receptor (VEGFR) pathways. Thus, many of the first generation BTC trials targeted EGFR and VEGFR, but these targeted agents ultimately proved ineffective at improving clinical outcome (12). NGS, which allows for characterization of an entire genetic landscape through gene panels, whole exome, or transcriptome sequencing, has led to the discovery of many novel actionable mutations in BTCs (15). Thus, pre-clinical and clinical studies have expanded from targeting well-established pathways like EGFR and VEGFR to promising, novel alterations.
Recent studies employing NGS have shed light on distinctive molecular spectra across the BTC subtypes (13,14). FGFR2 gene fusions and mutations in IDH1/2 are predominantly observed in IHCC. KRAS and HER2 mutations are preferentially found in EHCC. Lastly, GBCs are enriched for mutations in EGFR, HER2, and PIK3CA. Figure 1 and Table 1 highlight these key genomic alterations along the biliary tract and gallbladder. Next, we will discuss key actionable aberrations in BTCs and the novel agents that target them in biomarker-driven clinical trials.
Table 1. Prevalence of key genetic alterations in biliary tract cancers.
| Variables | IHCC (%) | EHCC (%) | GBC (%) | References |
|---|---|---|---|---|
| Tyrosine kinase signaling | ||||
| EGFR | 4 | 3 | 4–18 | (16,17) |
| HER2 | 1.5–3 | 11–18 | 10–16 | (16,18-20) |
| KRAS | 17–30 | 12–40 | 0–13 | (16,17,19-21) |
| BRAF | 4–7 | 3 | 1–6 | (16,19,22,23) |
| PIK3CA | 5–6 | 7–9 | 8–14 | (19,21,24,25) |
| FGFR2 fusions | 6–50 | 0–5 | 0–3 | (17,19,26-29) |
| IDH pathway | 10–28 | 0–7 | 0 | (19,21,27,30-32) |
| Chromatin-remodeling genes | ||||
| ARID1A | 17 | 12 | 13 | (19,27) |
| BAP1 | 11 | 8 | 0 | (17,27) |
| PBRM1 | 8 | 5 | 7 | (17,27) |
Tyrosine kinase signaling
EGFR
The EGFR family comprises four tyrosine kinase receptors (ERBB1–4) that regulate cell proliferation, survival, angiogenesis, and invasion through ligand binding and subsequent activation of signal transduction cascades involving the MAPK pathway (Ras-Raf-MEK-ERK) and the PI3K/AKT pathway (33) (Figure 2). Aberrant activation of the EGFR pathway is a common oncogenic event in BTCs and is associated with tumor recurrence and worsened outcome (16,18,26,34). Of the EGFR family members, EGFR (ERBB1) and HER2 (ERBB2) are most commonly altered in BTCs. Overexpression of EGFR occurs in 11–27% of IHCC, 5–19% of EHCC (26), and 12% in GBCs (35), whereas activating EGFR mutations are preferentially seen in GBC (4–18%), but rarely in CCAs (Table 1) (16,17).
Figure 2.

Key signaling pathways and current targeted therapies. Molecular targeted therapies including drugs currently assessed in phase II/III trials are highlighted.
Given that EGFR activation regulates several cellular functions important for carcinogenesis and is one of the most altered pathways in BTCs, there was strong rationale to evaluate it as a therapeutic target. However, extensive clinical testing with EGFR inhibitors has failed to show a survival benefit in advanced BTCs. Although earlier single arm phase II trials suggested possible benefits of EGFR antagonists cetuximab and panitumumab either as single agents or in combination with chemotherapy (36-39), larger RCTs of erlotinib, cetuximab or panitumumab in combination with gemcitabine plus oxaliplatin failed to show a progression-free survival (PFS) or overall survival (OS) benefit over chemotherapy alone in advanced BTCs (40,41).
Of note, almost all of these trials have been performed without stratifying patients by molecular signatures that could predict response to anti-EGFR agents. In fact, none has used EGFR genomic alterations as a biomarker. Additionally, lessons from the colorectal cancer world have informed us that KRAS mutations negate response to anti-EGFR therapy (42-44). However, only a few of the BTC trials have used KRAS status to stratify patients. A recent phase II trial stratified BTC patients based on KRAS status, but failed to demonstrate that KRAS status predicted the population most likely to benefit from anti-EGFR therapy (45). Furthermore, two biomarker-driven trials that was restricted to KRAS wild-type patients failed to show a clinically significant improvement in PFS or OS using panitumumab combined with chemotherapy (46,47). These studies call into question the utility of KRAS status as a clinically relevant biomarker predictive of EGFR therapy response in BTC, as opposed to colon cancer. The relative importance of mutations in other EGFR pathway genes, such as BRAF, are being investigated as mechanisms of resistance to anti-EGFR agents (47,48).
HER2
HER2 overexpression and amplification are predominantly seen in EHCC and GBCs (10–18% for both) and rarely in IHCC (Table 1) (16,19,20,26,34,35). Like EGFR-directed agents, similarly disappointing results came out of trials with HER2 antagonists (including trastuzumab, lapatinib, afatinib) combined with chemotherapy in advanced BTC (49-51). Currently, there is an ongoing phase II trial with trastuzumab aimed at a selected group of HER2-positive BTC patients (Table 2).
Table 2. Biomarker-driven clinical trials of biliary tract cancers.
| Drug(s) | Target | Biomarker selection | Phase | NCT number |
|---|---|---|---|---|
| Trastuzumab | HER2 | HER2 | II | NCT02999672 |
| Dabrafenib + trametinib | BRAF, MEK | BRAF V600E | II | NCT02034110 |
| BGJ398 | FGFR2 | FGFR alterations | II | NCT02150967 |
| BGJ398 | FGFR2 | FGFR alterations | II | NCT02160041 |
| Ponatinib | FGFR2 | FGFR2 fusion | II | NCT02265341 |
| Ponatinib | FGFR2 | FGFR alteration | II | NCT02272998 |
| INCB054828 | FGFR2 | FGFR2 translocation | II | NCT02924376 |
| Erdafitinib | FGFR2 | FGFR alteration | II | NCT02699606 |
| ARQ087 | FGFR2 | FGFR alteration | I/II | NCT01752920 |
| INCB054828 | FGFR2 | FGFR alteration | I | NCT02393248 |
| AG-120 | IDH1 | IDH1 mutation | III | NCT02989857 |
| AG-221 | IDH2 | IDH2 mutation | I/II | NCT02273739 |
| Dasatinib | Multiple kinases | IDH1/2 mutation | II | NCT02428855 |
| AG-120 | IDH1 | IDH1 mutation | I | NCT02073994 |
VEGF
VEGF is the ligand that binds VEGFR, which initiates signals to promote cancer growth and metastasis through stimulating angiogenesis. VEGF is overexpressed in BTCs and associated with enhanced metastasis, increased tumor recurrence, and worsened prognosis (34). Studies with antagonists of the VEGF pathway, including bevacizumab, cediranib, sorafenib have not yielded encouraging results (52-56).
MAPK pathway
Aberrations in cell-surface receptors and their ligands (e.g., EGFR, VEGF) can lead to constitutive activation of downstream cascades, including the MAPK arm (RAS-RAF-MEK-ERK, Figure 2). KRAS is a member of the RAS family and gain of function mutations in KRAS are one of the most common events in BTCs, with highest rates seen in EHCC, followed by IHCC, and lowest in GBC (16,17,19,20,57). KRAS is associated with lower median survival and perineural invasion (58). Its frequency also increases with disease stage (22). BRAF belongs to the RAF family of kinases that lie directly downstream of RAS (Figure 2). BRAF mutations are less frequent in BTCs (less than 10% across all subtypes) and are considered mutually exclusive with KRAS mutations (16,19,22,59). The most common BRAF mutation is V600E, but the mutational frequency is highly varied in BTCs ranging from 0–33% (60). The clinical significance of BRAF mutations is less well-established, with one study demonstrated an association with advanced tumor stage, higher likelihood of lymph node involvement, and worsened survival (22).
Targeting the MAPK pathway has remained a challenge. Recently, the phase I ABC-04 study of selumetinib, a MEK inhibitor, in combination with gemcitabine and cisplatin failed to show clinical benefit in in advanced or metastatic BTC (61). Even attempts to block multiple components of the MAPK pathway using multikinase inhibitors like sorafenib have not proved fruitful (62-65). These disappointing results are in stark contrast to melanomas, which frequently harbor the BRAF V600 mutations, where use of the BRAF inhibitors vemurafenib or dabrafenib has achieved a striking survival benefit (66-68). Recently, dual inhibition of BRAF with vemurafenib or dabrafenib and MEK with trametinib in BRAF V600-mutated melanoma patients has led to further survival improvements (69-71). Currently, there is an ongoing phase II trial with dabrafenib combined with trametinib for BRAF V600-mutated rare cancers including BTCs (Table 2).
Multiple signaling pathways seem to be involved in the pathogenesis of BTCs, rendering the decision of which pathways to target challenging. Moreover, no oncogene addiction pathway has been pinpointed. Targeting single pathways either as monotherapy or in combination with chemotherapy has shown varying degrees of improvements in response rates, but these have not translated to clinically significant increases in PFS or OS. Currently, some clinical trials are using a multi-target approach by using multikinase inhibitors or a combinatorial approach with multiple agents aimed at different pathways (12,72). Results from studies using multikinase inhibitors regorafenib and pazopanib are anxiously awaited.
Novel targets
Over the recent years, genomic profiling using NGS has revealed the presence of novel alterations in BTCs such as recurrent fusion events (FGFR2 and ROS1 fusions), somatic mutations in metabolic enzymes (IDH1 and 2) (17-19,21,23,26-31,57,73,74), and chromatin-remodeling genes (ARID1A, BAP1, PBRM1) (17,19,27).
FGFR2 fusions
FGFR2 is a member of the fibroblast growth factor family of receptor tyrosine kinases that regulate cell proliferation, differentiation, apoptosis (75). Alterations in this pathway through activating mutations, amplifications, or chromosomal translocation have been implicated in malignant transformation (76). Chromosomal fusions occur between exons 1–19 of FGFR2 and various genomic partners (e.g., AHCYL1, BICC1, PARK2, KCTD1, MGEA5, TACC3, TXLNA) in BTCs (17,19,26-29). The resulting fusion protein undergoes ligand-independent dimerization and subsequent autophosphorylation, which leads to constitutive activation of downstream signaling pathways, such as MAPK (76) (Figure 2). The oncogenic potential of FGFR2 fusions has been demonstrated in vitro (23,28,77,78) and in vivo (28). Screening for fusions by massive parallel sequencing or FISH-based assays has revealed a wide range of IHCC (6–50%) containing FGFR2 fusions, whereas EHCC and GBC rarely do (Table 1).
In preclinical studies, the presence of FGFR2 fusions seems to predict high sensitivity to FGFR2 inhibitors (23,28,73,77,78). This provided the catalyst to target the FGFR pathway specifically in tumors harboring these fusions. FGF pathway antagonists include small molecule tyrosine kinase inhibitors that act at the receptor level to suppress oncogenic signaling (28). Clinical efficacy of FGFR2 inhibitors is being investigated in biomarker-driven clinical trials aimed at patients harboring FGFR2 pathway alterations (Table 2). The pan-FGFR inhibitor BGJ398 has potent activity against FGFR1–3 and is under evaluation in advanced CCAs with FGFR genetic alterations in two phase II studies (Table 2). Preliminary results from one of the studies (NCT02150967) was recently reported. Amongst the 26 patients with advanced or metastatic CCA harboring FGFR2 fusions or other alterations, the disease control rate was 82% (79). The drug was well tolerated except for hyperphosphatemia.
Ponatinib is an example of a non-selective pan-FGFR inhibitor that is far along in clinical development. In a preclinical study, treatment with ponatinib resulted in biochemical CA 19-9 response with tumor shrinkage in a patient with the FGFR-MGEA5 fusion (73). Another patient in the study with FGFR-TACC3I fusion whose disease had progressed on pazopanib (another non-selective FGFR inhibitor) was treated with ponatinib therapy, resulting in stabilization of disease (73). This preliminary evidence supported assessing the anti-tumor activity of ponatinib in clinical trials. Ponatinib is being investigated in a phase II trial of advanced BTCs harboring FGFR2 gene fusions detected by either NGS or FISH (NCT02265341, Table 2). Another ongoing phase II trial is assessing the efficacy of ponatinib in advanced malignancies including CCA with any FGFR aberrations (mutations, fusions, amplifications) (NCT02272998, Table 2).
Other ongoing phase II studies include oral pan-FGFR selective small molecular inhibitors INCB054828, erdafitinib (JNJ-42756493), ARQ087 (Table 2). Preclinical and phase I studies have suggested that these compounds have potent and selective anti-tumor activity against FGFR-mutated cancers (80-83). A recently developed monoclonal antibody against FGFR2 (BAY1179470) showed tumor suppressive potential in tumors with high FGFR2 expression (84). Phase I testing of this antibody just recently completed (NCT01881217). Another phase I trial with oral pan-FGFR inhibitor AZD4547 also just completed (NCT00979134).
IDH1/2
IDH1 and IDH2 encode metabolic enzymes that participate in the Krebs cycle. Mutations in IDH1 and IDH2 result in the accumulation of the oncometabolite 2-hydroxyglutarate, which affects cell differentiation, survival, as well as DNA methylation. The epigenetic alterations caused by mutations in IDH1/2 lead to a blockade of hepatocyte differentiation, causing an increase in hepatic progenitor cells, which eventually results in tumorigenesis (85). IDH mutations have been seen in solid tumors, including gliomas, and recently identified in BTCs. They occur primarily in IHCC (10–28%) and rarely in EHCC and GBCs (19,21,27,30,31) (Table 1). The most common IDH1 and IDH2 mutations cluster at the hotspot codons 132 and 172, respectively (86). The prognostic significance of these mutations remains to be fully elucidated, as there is some conflicting data. Two studies have correlated IDH mutations with decreased survival in IHCC compared to wild-type cases (27,31). Another study failed to demonstrate an association between IDH mutation status and survival (32). In contrast, a large cohort of IHCC samples (n=326) showed IDH mutations were associated with longer time to recurrence (30).
The efficacy of pharmacologically targeting the mutant IDH enzymes has been demonstrated in other types of tumors. IDH1 inhibitor AGI-5198 slowed the growth of IDH-mutant glioma cells (87) and IDH2 inhibitor AGI-6780 selectively inhibited the growth of leukemic cells carrying mutant IDH2/R140Q (30). The role of IDH inhibitors in IHCC is currently being investigated. AG-120, an IDH1 inhibitor, has been shown to transiently stabilize disease progression in patients with IDH1-mutant IHCC. The expansion phase is currently underway (NCT02073994, Table 2). AG-120 is also being tested in the ongoing phase III RCT “ClarIDHy” in patients with advanced or metastatic CCA carrying an IDH1 mutation (Table 2). A phase I/II trial with AG-221 (IDH2 inhibitor) has just completed. A recent study showed that a subset of IHCC tumors with IDH mutations are exquisitely sensitive to the multikinase inhibitor dasatinib (88). This evidence paved the way for designing a phase II trial using dasatinib in IHCC cases harboring mutations in IDH1 or 2 (Table 2). Other agents that have demonstrated preclinical efficacy and are now in phase I testing include BAY1436032 (IDH1 inhibitor), IDH305 (IDH1 inhibitor), and AG-881 (IDH1/2 inhibitor) (89).
ROS1
ROS1 kinase fusions between kinase domain of ROS and FIG have been found in 8.7% of CCAs (74). The resultant FIG-ROS1 fusion protein has oncogenic potential in vitro and in vivo and can be inhibited by pharmacological targeting (74,90). A phase II trial of crizotinib (ALK/ROS1 inhibitor) in patients with ALK, MET, or ROS1 alterations is underway (Table 2). LDK378, a ALK/ROS1 inhibitor, is being investigated in ROS1-overexpressing advanced CCAs (Table 2). Entrectinib, another ALK/ROS1 inhibitor, is being tested in CCAs carrying ROS1 gene fusions.
Conclusions
BTCs are highly aggressive tumors that carry a dismal prognosis. Historically, the BTC subtypes have been studied as a single entity. Application of NGS technologies has allowed for enhanced characterization of the distinct genetic landscapes in the various BTC subtypes. FGF and IDH pathway alterations are commonly seen in IHCC, whereas alterations in the EGFR-MAPK-PI3K pathway occur more frequently in EHCC and GBC. The molecular heterogeneity across these subtypes likely confers differential responses to various treatments. Thus, therapy should be customized based on mutational spectra. To optimize clinical trial design, targeted therapies should be matched to specific molecular alterations through patient biomarker selection. Past investigations into agents targeting receptor tyrosine kinase and MAPK pathways have not shown significant benefit over standard chemotherapy regimen. However, improvements in genetic profiling have unveiled novel actionable mutations, such as FGFR2 fusion proteins and mutated IDH1/2. Agents targeted against these newly discovered aberrations are being actively investigated in clinical trials and hold the promise of improving clinical outcomes in this devastating orphan disease.
Acknowledgements
Funding: This work is supported by 5T32 GM07200 National Research Science Award-Medical Scientist (DY Zhao).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology 2009;136:1134-44. 10.1053/j.gastro.2009.02.038 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7-30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- 3.Tyson GL, Ilyas JA, Duan Z, et al. Secular trends in the incidence of cholangiocarcinoma in the USA and the impact of misclassification. Dig Dis Sci 2014;59:3103-10. 10.1007/s10620-014-3276-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan SA, Toledano MB, Taylor-Robinson SD. Epidemiology, risk factors, and pathogenesis of cholangiocarcinoma. HPB (Oxford) 2008;10:77-82. 10.1080/13651820801992641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet 2014;383:2168-79. 10.1016/S0140-6736(13)61903-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misra S, Chaturvedi A, Misra NC, et al. Carcinoma of the gallbladder. Lancet Oncol 2003;4:167-76. 10.1016/S1470-2045(03)01021-0 [DOI] [PubMed] [Google Scholar]
- 7.Nathan H, Pawlik TM, Wolfgang CL, et al. Trends in survival after surgery for cholangiocarcinoma: a 30-year population-based SEER database analysis. J Gastrointest Surg 2007;11:1488-96; discussion 96-7. 10.1007/s11605-007-0282-0 [DOI] [PubMed] [Google Scholar]
- 8.Mavros MN, Economopoulos KP, Alexiou VG, et al. Treatment and Prognosis for Patients With Intrahepatic Cholangiocarcinoma: Systematic Review and Meta-analysis. JAMA Surg 2014;149:565-74. 10.1001/jamasurg.2013.5137 [DOI] [PubMed] [Google Scholar]
- 9.Horgan AM, Amir E, Walter T, et al. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934-40. 10.1200/JCO.2011.40.5381 [DOI] [PubMed] [Google Scholar]
- 10.McNamara MG, Walter T, Horgan AM, et al. Outcome of adjuvant therapy in biliary tract cancers. Am J Clin Oncol 2015;38:382-7. 10.1097/COC.0b013e31829e19fb [DOI] [PubMed] [Google Scholar]
- 11.Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273-81. 10.1056/NEJMoa0908721 [DOI] [PubMed] [Google Scholar]
- 12.Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget 2016;7:46750-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain A, Javle M. Molecular profiling of biliary tract cancer: a target rich disease. J Gastrointest Oncol 2016;7:797-803. 10.21037/jgo.2016.09.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016;122:3838-47. 10.1002/cncr.30254 [DOI] [PubMed] [Google Scholar]
- 15.Jain A, Kwong LN, Javle M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr Treat Options Oncol 2016;17:58. 10.1007/s11864-016-0432-2 [DOI] [PubMed] [Google Scholar]
- 16.Li M, Zhang Z, Li X, et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat Genet 2014;46:872-6. 10.1038/ng.3030 [DOI] [PubMed] [Google Scholar]
- 17.Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10. 10.1038/ng.3375 [DOI] [PubMed] [Google Scholar]
- 18.Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014;19:235-42. 10.1634/theoncologist.2013-0352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross JS, Wang K, Thomas Catenacci DV, et al. Comprehensive genomic profiling of biliary tract cancers to reveal tumor-specific differences and genomic alterations. J Clin Oncol 2015;33:abstr 231.
- 20.Holcombe RF, Xiu J, Pishvaian MJ, et al. Tumor profiling of biliary tract carcinomas to reveal distinct molecular alterations and potential therapeutic targets. J Clin Oncol 2015;33:abstr 285. [DOI] [PMC free article] [PubMed]
- 21.Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012;17:72-9. 10.1634/theoncologist.2011-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson S, Hyder O, Dodson R, et al. The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum Pathol 2013;44:2768-73. 10.1016/j.humpath.2013.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087. 10.1038/ncomms7087 [DOI] [PubMed] [Google Scholar]
- 24.Deshpande V, Nduaguba A, Zimmerman SM, et al. Mutational profiling reveals PIK3CA mutations in gallbladder carcinoma. BMC Cancer 2011;11:60. 10.1186/1471-2407-11-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simbolo M, Fassan M, Ruzzenente A, et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014;5:2839-52. 10.18632/oncotarget.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One 2014;9:e115383. 10.1371/journal.pone.0115383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3. 10.1038/ng.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427-34. 10.1002/hep.26890 [DOI] [PubMed] [Google Scholar]
- 29.Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630-8. 10.1016/j.humpath.2014.03.014 [DOI] [PubMed] [Google Scholar]
- 30.Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091-100. 10.1038/onc.2012.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Javle M, Rashid A, Churi C, et al. Molecular characterization of gallbladder cancer using somatic mutation profiling. Hum Pathol 2014;45:701-8. 10.1016/j.humpath.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kipp BR, Voss JS, Kerr SE, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol 2012;43:1552-8. 10.1016/j.humpath.2011.12.007 [DOI] [PubMed] [Google Scholar]
- 33.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 2006;12:5268-72. 10.1158/1078-0432.CCR-05-1554 [DOI] [PubMed] [Google Scholar]
- 34.Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer 2008;98:418-25. 10.1038/sj.bjc.6604129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakazawa K, Dobashi Y, Suzuki S, et al. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol 2005;206:356-65. 10.1002/path.1779 [DOI] [PubMed] [Google Scholar]
- 36.Gruenberger B, Schueller J, Heubrandtner U, et al. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: a phase 2 study. Lancet Oncol 2010;11:1142-8. 10.1016/S1470-2045(10)70247-3 [DOI] [PubMed] [Google Scholar]
- 37.Rubovszky G, Lang I, Ganofszky E, et al. Cetuximab, gemcitabine and capecitabine in patients with inoperable biliary tract cancer: a phase 2 study. Eur J Cancer 2013;49:3806-12. 10.1016/j.ejca.2013.07.143 [DOI] [PubMed] [Google Scholar]
- 38.Hezel AF, Noel MS, Allen JN, et al. Phase II study of gemcitabine, oxaliplatin in combination with panitumumab in KRAS wild-type unresectable or metastatic biliary tract and gallbladder cancer. Br J Cancer 2014;111:430-6. 10.1038/bjc.2014.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sohal DP, Mykulowycz K, Uehara T, et al. A phase II trial of gemcitabine, irinotecan and panitumumab in advanced cholangiocarcinoma. Ann Oncol 2013;24:3061-5. 10.1093/annonc/mdt416 [DOI] [PubMed] [Google Scholar]
- 40.Lee J, Park SH, Chang HM, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2012;13:181-8. 10.1016/S1470-2045(11)70301-1 [DOI] [PubMed] [Google Scholar]
- 41.Malka D, Cervera P, Foulon S, et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): a randomised, open-label, non-comparative phase 2 trial. Lancet Oncol 2014;15:819-28. 10.1016/S1470-2045(14)70212-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757-65. 10.1056/NEJMoa0804385 [DOI] [PubMed] [Google Scholar]
- 43.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626-34. 10.1200/JCO.2007.14.7116 [DOI] [PubMed] [Google Scholar]
- 44.Hecht JR, Douillard JY, Schwartzberg L, et al. Extended RAS analysis for anti-epidermal growth factor therapy in patients with metastatic colorectal cancer. Cancer Treat Rev 2015;41:653-9. 10.1016/j.ctrv.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 45.Chen JS, Hsu C, Chiang NJ, et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann Oncol 2015;26:943-9. 10.1093/annonc/mdv035 [DOI] [PubMed] [Google Scholar]
- 46.Jensen LH, Lindebjerg J, Ploen J, et al. Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Ann Oncol 2012;23:2341-6. 10.1093/annonc/mds008 [DOI] [PubMed] [Google Scholar]
- 47.Ferraro D, Goldstein D, O'Connell RL, et al. TACTIC: a multicentre, open-label, single-arm phase II trial of panitumumab, cisplatin, and gemcitabine in biliary tract cancer. Cancer Chemother Pharmacol 2016;78:361-7. 10.1007/s00280-016-3089-4 [DOI] [PubMed] [Google Scholar]
- 48.Berlin J. Beyond exon 2--the developing story of RAS mutations in colorectal cancer. N Engl J Med 2013;369:1059-60. 10.1056/NEJMe1307992 [DOI] [PubMed] [Google Scholar]
- 49.Ramanathan RK, Belani CP, Singh DA, et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol 2009;64:777-83. 10.1007/s00280-009-0927-7 [DOI] [PubMed] [Google Scholar]
- 50.Peck J, Wei L, Zalupski M, et al. HER2/neu may not be an interesting target in biliary cancers: results of an early phase II study with lapatinib. Oncology 2012;82:175-9. 10.1159/000336488 [DOI] [PubMed] [Google Scholar]
- 51.Kwak EL, Shapiro GI, Cohen SM, et al. Phase 2 trial of afatinib, an ErbB family blocker, in solid tumors genetically screened for target activation. Cancer 2013;119:3043-51. 10.1002/cncr.28120 [DOI] [PubMed] [Google Scholar]
- 52.Zhu AX, Meyerhardt JA, Blaszkowsky LS, et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol 2010;11:48-54. 10.1016/S1470-2045(09)70333-X [DOI] [PubMed] [Google Scholar]
- 53.Lubner SJ, Mahoney MR, Kolesar JL, et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: a phase II Consortium study. J Clin Oncol 2010;28:3491-7. 10.1200/JCO.2010.28.4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iyer RV, Pokuri VK, Groman A, et al. A Multicenter Phase II Study of Gemcitabine, Capecitabine, and Bevacizumab for Locally Advanced or Metastatic Biliary Tract Cancer. Am J Clin Oncol 2016. [Epub ahead of print]. 10.1097/COC.0000000000000347 [DOI] [PubMed] [Google Scholar]
- 55.Valle JW, Wasan H, Lopes A, et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): a randomised phase 2 trial. Lancet Oncol 2015;16:967-78. 10.1016/S1470-2045(15)00139-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knox JJ, Qin R, Strosberg JR, et al. A phase II trial of bevacizumab plus temsirolimus in patients with advanced hepatocellular carcinoma. Invest New Drugs 2015;33:241-6. 10.1007/s10637-014-0169-3 [DOI] [PubMed] [Google Scholar]
- 57.Borger DR, Zhu AX. IDH mutations: new genetic signatures in cholangiocarcinoma and therapeutic implications. Expert Rev Anticancer Ther 2012;12:543-6. 10.1586/era.12.32 [DOI] [PubMed] [Google Scholar]
- 58.Chen TC, Jan YY, Yeh TS. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann Surg Oncol 2012;19 Suppl 3:S675-81. 10.1245/s10434-012-2224-7 [DOI] [PubMed] [Google Scholar]
- 59.Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829-40. 10.1053/j.gastro.2013.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goeppert B, Frauenschuh L, Renner M, et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod Pathol 2014;27:1028-34. 10.1038/modpathol.2013.206 [DOI] [PubMed] [Google Scholar]
- 61.Bridgewater J, Lopes A, Beare S, et al. A phase 1b study of Selumetinib in combination with Cisplatin and Gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer 2016;16:153. 10.1186/s12885-016-2174-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Khoueiry AB, Rankin CJ, Ben-Josef E, et al. SWOG 0514: a phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest New Drugs 2012;30:1646-51. 10.1007/s10637-011-9719-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JK, Capanu M, O'Reilly EM, et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br J Cancer 2013;109:915-9. 10.1038/bjc.2013.432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moehler M, Maderer A, Schimanski C, et al. Gemcitabine plus sorafenib versus gemcitabine alone in advanced biliary tract cancer: a double-blind placebo-controlled multicentre phase II AIO study with biomarker and serum programme. Eur J Cancer 2014;50:3125-35. 10.1016/j.ejca.2014.09.013 [DOI] [PubMed] [Google Scholar]
- 65.Bengala C, Bertolini F, Malavasi N, et al. Sorafenib in patients with advanced biliary tract carcinoma: a phase II trial. Br J Cancer 2010;102:68-72. 10.1038/sj.bjc.6605458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012;366:707-14. 10.1056/NEJMoa1112302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 2012;13:1087-95. 10.1016/S1470-2045(12)70431-X [DOI] [PubMed] [Google Scholar]
- 68.Ascierto PA, Minor D, Ribas A, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 2013;31:3205-11. 10.1200/JCO.2013.49.8691 [DOI] [PubMed] [Google Scholar]
- 69.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30-9. 10.1056/NEJMoa1412690 [DOI] [PubMed] [Google Scholar]
- 70.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015;386:444-51. 10.1016/S0140-6736(15)60898-4 [DOI] [PubMed] [Google Scholar]
- 71.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877-88. 10.1056/NEJMoa1406037 [DOI] [PubMed] [Google Scholar]
- 72.Moeini A, Sia D, Bardeesy N, et al. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin Cancer Res 2016;22:291-300. 10.1158/1078-0432.CCR-14-3296 [DOI] [PubMed] [Google Scholar]
- 73.Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 2014;10:e1004135. 10.1371/journal.pgen.1004135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu TL, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 2011;6:e15640. 10.1371/journal.pone.0015640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rizvi S, Borad MJ. The rise of the FGFR inhibitor in advanced biliary cancer: the next cover of time magazine? J Gastrointest Oncol 2016;7:789-96. 10.21037/jgo.2016.08.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116-29. 10.1038/nrc2780 [DOI] [PubMed] [Google Scholar]
- 77.Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013;3:636-47. 10.1158/2159-8290.CD-13-0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanizaki J, Ercan D, Capelletti M, et al. Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res 2015;75:3139-46. 10.1158/0008-5472.CAN-14-3771 [DOI] [PubMed] [Google Scholar]
- 79.Javle MM, Shroff RT, Zhu A, et al. A phase 2 study of BGJ398 in patients (pts) with advanced or metastatic FGFR-altered cholangiocarcinoma (CCA) who failed or are intolerant to platinum-based chemotherapy. J Clin Oncol 2016;34:abstr 335.
- 80.Liu PC, Wu L, Koblish H, et al. Preclinical characterization of the selective FGFR inhibitor INCB054828. Cancer Res 2015;75:Abstract nr 771.
- 81.Tabernero J, Bahleda R, Dienstmann R, et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol 2015;33:3401-8. 10.1200/JCO.2014.60.7341 [DOI] [PubMed] [Google Scholar]
- 82.Hall TG, Yu Y, Eathiraj S, et al. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS One 2016;11:e0162594. 10.1371/journal.pone.0162594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Papadopoulos KP, Tolcher AW, Patnaik A, et al. Phase 1, first-in-human study of ARQ 087, an oral pan-Fibroblast Growth Factor Receptor (FGFR) inhibitor, in patients (pts) with advanced solid tumors. J Clin Oncol 2015;33:abstr 2545.
- 84.Schatz CA, Kopitz C, Wittemer-Rump S, et al. Pharmacodynamic and stratification biomarker for the anti-FGFR2 antibody (BAY1179470) and the FGFR2-ADC. Cancer Res 2014;74:Abstract nr 4766.
- 85.Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110-4. 10.1038/nature13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grassian AR, Pagliarini R, Chiang DY. Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol 2014;30:295-302. 10.1097/MOG.0000000000000050 [DOI] [PubMed] [Google Scholar]
- 87.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340:626-30. 10.1126/science.1236062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saha SK, Gordan JD, Kleinstiver BP, et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov 2016;6:727-39. 10.1158/2159-8290.CD-15-1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Panknin O, Pusch S, Herbst L, et al. BAY 1436032: A highly selective, potent and orally available inhibitor of mutant forms of IDH1. Cancer Res 2016;76:Abstract nr 2645.
- 90.Saborowski A, Saborowski M, Davare MA, et al. Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc Natl Acad Sci U S A 2013;110:19513-8. 10.1073/pnas.1311707110 [DOI] [PMC free article] [PubMed] [Google Scholar]