

Abstract
Phosphoglycosyl transferases (PGTs) represent “gatekeeper” enzymes in complex glycan assembly pathways by catalyzing transfer of a phosphosugar from an activated nucleotide diphosphosugar to a membrane-resident polyprenol phosphate. The unique structures of selected nucleoside antibiotics, such as tunicamycin and mureidomycin A, which are known to inhibit comparable biochemical transformations, are exploited as the foundation for the development of modular synthetic inhibitors of PGTs. Herein we present the design, synthesis, and biochemical evaluation of two readily manipulatable modular scaffolds as inhibitors of monotopic bacterial PGTs. Selected compounds show IC50 values down to the 40 μm range, thereby serving as lead compounds for future development of selective and effective inhibitors of diverse PGTs of biological and medicinal interest.
Keywords: glycoconjugate biosynthesis, inhibitors, modular approach, nucleoside antibiotic, phosphoglycosyl transferase
Introduction
Phosphoglycosyl transferases (PGTs), also designated as “priming glycosyltransferases”, act at a membrane interface and catalyze the transfer of C1-phosphosugars to membrane-associated polyprenol phosphates to afford polyprenol diphosphosugar (Pren-PP-CHO) products. These pivotal intermediates then serve as acceptors for elaboration by a sequence of glycosyl transferases, producing highly diversified polyprenoldiphosphate-linked glycans for glycoprotein, proteoglycan, and glycolipid biosynthesis. Since PGTs catalyze the first membrane-committed step in many complex glycoconjugate biosynthesis pathways, they serve as “gatekeepers” of glycoconjugate production across all domains of life.[1] For example, MraY is a bacterial PGT that catalyzes transfer of phospho-MurNAc-pentapeptide to undecaprenol phosphate, mediating a key step in peptidoglycan biosynthesis,[2] and WecA is a bacterial phospho-GlcNAc transferase, which catalyzes the first step in the biosynthesis of lipopolysaccharide O-antigen.[3] Nucleoside antibiotics[4] that feature a common uridinyl motif have been demonstrated to be potent inhibitors of selected PGTs, and MraY and WecA are potently inhibited by mureidomycin A[5] and tunicamycin,[6] respectively (Figure 1). Recently, the importance of MraY in bacterial peptidoglycan synthesis has prompted major interest in the development of simplified nucleoside antibiotic analogues in the search for novel antimicrobial strategies.[7--11]
Figure 1.
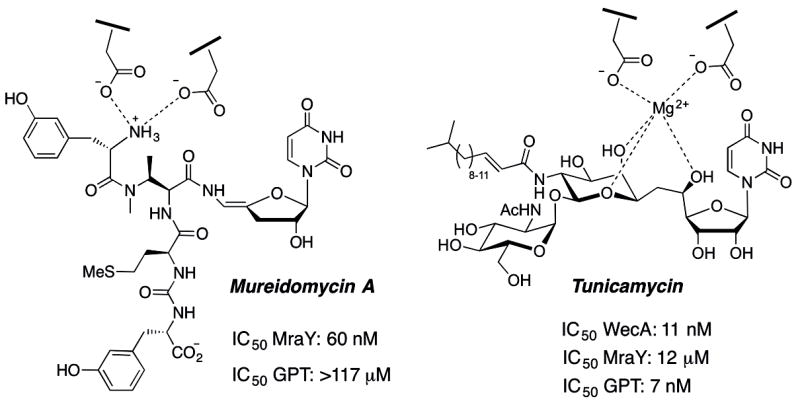
Structures of mureidomycin A and tunicamycin, and their IC50 values against selected PGTs
PGTs also feature in eukaryotic biology, most prominently at the initiation of the dolichol-dependent glycosylation pathway, where a GlcNAc-1-phosphate transferase (GPT), designated as Alg7 in Saccharomyces cerevisiae and other eukaryotes, transfers phospho-GlcNAc to dolichol phosphate.[12] Sequence analysis of WecA and Alg7 reveals that these enzymes are both integral membrane proteins with 11 predicted transmembrane helices (TMHs) and that they share key predicted active site residues, therefore it is unsurprising that Alg7 is also potently inhibited by tunicamycin[13] (Figure 1).
Unfortunately, despite the tantalizing inhibition properties of the uridinyl nucleoside antibiotics, their biological activities are hard to predict for other PGT targets, and must be empirically determined. This is exemplified by the large differences in inhibition properties of mureidomycin A and tunicamycin (Figure 1). Furthermore, the complexity of the natural product structures makes it very challenging to repurpose the structures of the natural products, by synthesis[14--16] or semisynthesis,[17--22] to target alternative PGTs with different substrate specificities. This challenge is further exacerbated when working with PGTs belonging to structural classes other than the well-studied MraY and WecA-type integral membrane proteins, which feature 10 and 11 predicted TMHs, respectively. For example, recent bioinformatics and biochemical analysis has revealed thousands of homologous small bacterial PGTs with only a single TMH and a soluble globular domain within a 20—25 kDa protein.[23] While these PGTs catalyze comparable biochemical processes and also play important roles at the initiation of diverse glycoconjugate biosynthetic pathways, there are currently no small molecule inhibitors that can be used to inform on the biology and essentiality of particular pathways and which may ultimately represent novel targets for therapeutic intervention.
A prototypic example of a small PGT is PglC from Campylobacter jejuni, a Gram-negative food-borne pathogen that is the major cause of gastroenteritis worldwide. PglC catalyzes the first membrane-committed step in the biosynthesis of virulence-associated N-linked glycoproteins in the pathogen.[24] Specifically, PglC, acting at the cytoplasmic face of the inner bacterial membrane, commences assembly of an undecaprenol diphosphate-linked heptasaccharide, which is the ultimate substrate for asparagine-linked glycosylation in the periplasm. PglC catalyzes transfer of a modified phosphosugar, di-N-acetylbacillosamine-phosphate, from the corresponding UDP-sugar donor, onto undecaprenol phosphate with release of UMP (Scheme 1).[25] The PGT reaction is isoenergetic; a diphosphate product is formed from a diphosphate starting material (UDP-sugar). However, it is likely that in nature flux through the pathway is promoted by the action of the subsequent glycosyltransferases, which elaborate the PGT product into the polyprenol diphosphate-linked glycan.
Scheme 1.
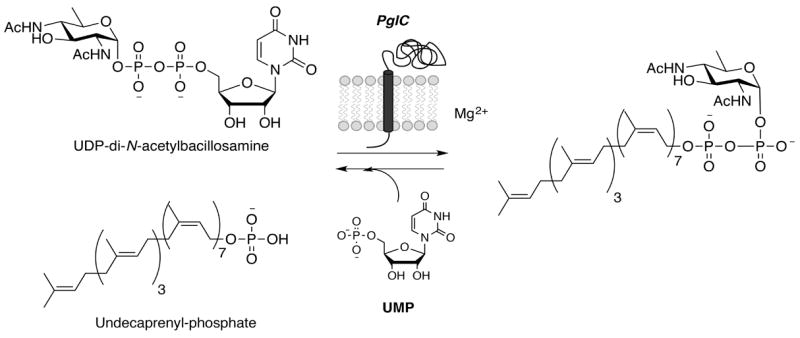
PGT reaction catalyzed by PglC.
Currently, there is no structural information available to aid in the design of inhibitors towards small PGTs such as PglC. The only PGT that has been structurally characterized is MraY,[26] and the complete absence of sequence homology to the small PGTs reinforces that very little insight can be gleaned from the MraY structure to address inhibitor development for the small PGTs. Therefore, we have turned our attention to the nucleoside antibiotics as inspiration for the blueprints of new modular scaffolds for PGT inhibitor design. Examination of the structures and reported biological activities of mureidomycin A and tunicamycin suggests important design criteria. First, the uridinyl moiety is a key “placeholder” for PGTs that act on UDP-sugar substrates and therefore this moiety should feature in projected inhibitors. Second, as illustrated in Figure 1 interception of a divalent cation-binding site, either through a strategic metal ion coordinating group(s) on the inhibitor (the tunicamycin model[1]) or by metal ion displacement with a protonated primary amine (the mureidomycin A model[22]) represents an important design feature. In this context, despite the diversity of PGT structures, Mg2+-dependence, presumably to coordinate the uridine diphosphate to prime the substrate for nucleophilic attack at the β-phosphate, is common to small PGTs, such as PglC, as well as the multi-TMH PGTs exemplified by MraY, WecA, and Alg7. Finally, the design should be compatible with extended binding determinants, including glycan, peptidic, and lipophilic components, to mimic the characteristics of the native substrates. Based on these criteria, we have devised two modular synthetic strategies, inspired by mureidomycin A and tunicamycin. Key features of both strategies include 1) a uridine moiety, 2) a functionality to either displace or coordinate to the metal ion cofactor, 3) an aromatic moiety that may intercept sugar binding, and 4) a hydrophobic acyl moiety to either bind into the undecaprenol phosphate-binding site, or potentially direct the inhibitor to the membrane interface. Strategy 1 (mureidomimetic) is based on mureidomycin A in that it includes a modification at the C5’-site to display three different lengths of alkylamines to explore placement of a positive charge adjacent to the uridine, and strategy 2 (tunicamimetic) is based on the incorporation of amino acid building blocks, once again conjugated to the uridinyl moiety, to explore an alternative, metal ion-coordinating functionality adjacent to the uridine.
The development of novel small PGT (e.g. PglC) inhibitors has also demanded attention to practical details including the establishment of reliable heterologous expression and purification procedures for the target membrane-bound enzyme[27] and the application of robust assays to provide consistent feedback on inhibitor efficiency. One of the standard approaches for assaying PGTs relies on radioactivity-based strategies that use tritium or [14C]-labeled UDP-sugars and undecaprenol phosphate together with liquid/liquid extraction of lipophilic undecaprenol dphosphosugar products. For example, PglC may be assayed in a coupled assay, in which the PglC product is further elaborated by PglA, an N-acetylgalactosamine transferase that uses UDP-[3H]GalNAc, from the pgl pathway of C. jejuni.[28] In this way, tritium-labeled GalNAc is incorporated into the undecaprenol diphospho-saccharide (Und-PP-Bac-GalNAc) and enzymatic activity is quantified by extraction of the hydrophobic product followed by scintillation counting. To test for PglC activity and inhibition directly, [3H]-labeled UDP-di-N-acetylbacillosamine was prepared by chemoenzymatic synthesis using [3H]-labeled AcCoA (see Supporting Information). Since the radioactivity-based assays require synthesis of radiolabeled UDP-Bac and are labor intensive, we also present the use of a prototype UMP/CMP-Glo assay from Promega, which is a luminescent UMP detection assay that greatly increased the reliability and throughput of activity and inhibition assays.
Results and Discussion
Strategy 1: Mureidomimetics
The first modular strategy takes its cue from mureidomycin A and includes a primary amine adjacent to the uridinyl moiety (see Figure 1). Starting from free uridine, the 2’,3’-syn diol of the ribose was protected with an isopropylidene group, and the 5’-hydroxyl was tosylated (Scheme 2). Substitution of the tosylate with mono-Boc-protected alkyldiamines (C2, C4, and C6) was accomplished under basic conditions after prolonged reaction times to yield intermediates 2--4 in good yield. Different spacer lengths between the uridine core and amino group were incorporated to investigate potential structure/function dependencies in subsequent inhibition assays (vide infra). Removal of the N-Boc and isopropylidene protecting groups was accomplished simultaneously using aqueous TFA, and products 5--7 were obtained in quantitative yield after lyophilization. Initial inhibition assays revealed that these first generation compounds 5--7 inhibited the PglC reaction completely at 5 mm, while activity was almost completely unaffected at 1 mm (data not shown). Qualitatively, small differences were observed with the three congeners, so all series were forwarded for further elaboration. In all cases, inhibition assays were carried out under conditions which minimized promiscuous binding effects resulting from inhibitor aggregation.[29]
Scheme 2.
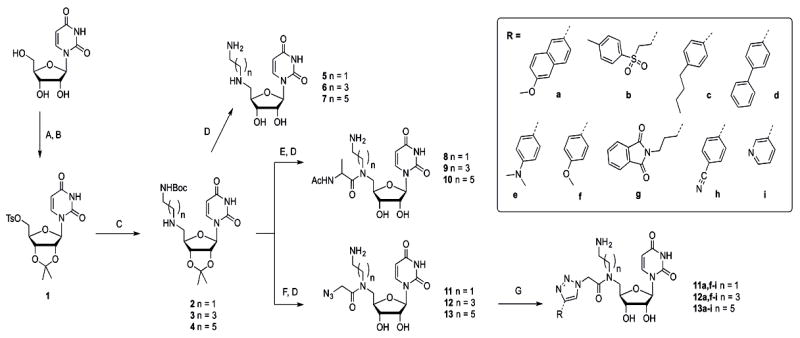
Synthesis of dyad inhibitors, varying alkyl length and alkyne. A) H2SO4, acetone (96%); B) p-TsCl, DMAP, DCM/pyridine (89 %); C) N-Boc-alkylamine, K2CO3, THF (2: 78%, 3: 91%, 4: 96%); D) TFA, H2O (quant.); E) Ac-l-Ala-OH, EDC, HOBt, DMF (towards 8: 54%, 9: 37%, 10: 74%); F) i. chloroacetic anhydride, DCM; ii. NaN3, DMF (toward 11: quant., toward 12: 94%, toward 13: quant.); G) alkyne, CuSO4, sodium ascorbate, tBuOH/H2O (quant. by LC-MS). DMAP= 4-(dimethylamino)pyridine, TFA=trifluoroacetic acid, EDC = 3-(ethyliminomethylidenamino)-N,N-dimethyl-propan-1-amine, HOBt= hydroxybenzotriazole, DCM=dichloromethane; DMF= dimethylformamide.
To reduce flexibility and potentially increase the opportunity for productive binding interactions with the enzyme, the C5’-secondary amine position was derivatized by coupling with N-acetyl-alanine using standard coupling conditions (EDC, HOBt). Although the use of the N-acetyl derivative of alanine under these conditions resulted in epimerization at the C-α center, the diasteromeric mixture was carried forward for inhibition analysis. While epimerization could have been prevented using the N-α-Fmoc-protected amino acid, deprotection under basic conditions was known to result in decomposition of the resulting tertiary amide at the 5’-N in related compounds (vide infra). Acidic treatment to remove the isopropylidine group afforded compounds 8--10, which showed improved inhibition potency, with 78±14 % inhibition at 1 mm for compound 10 (the C6 dialkylamine, Figure 2A) at substrate concentrations of 20 μm UDP-diNAcBac (KM=7.2±1.1 μm) and 20 μm Und-P (KM, app=15.6±5.1 μm) in the assay. At this point, it was possible to discriminate between the different alkyl lengths, with compound 10 being a superior inhibitor to 8 and 9 (see Figure S1, Supporting Information). The activity of compound 10 represents a significant improvement relative to uridine, which shows no inhibition of PglC at 1 mm, indicating that the uridine core alone is not sufficient for binding at this concentration (Figure 2A. In contrast, uridine monophosphate (UMP), which is also a side product of the PglC reaction, shows comparable inhibition relative to 10 at 1 mm. This is in line with our observations that the PglC reaction saturates at 30% conversion, supposedly due to UMP product inhibition. Although uridyl-phosphates are successfully elaborated towards potent glycosyltransferase inhibitors,[30--32] in the long term, the amine-substituted derivatives would be advantageous due to the positive charge, which would enhance cell permeability relative to negatively charged analogues, as has been an inspiration in several inhibitor design strategies.[33--37]
Figure 2.
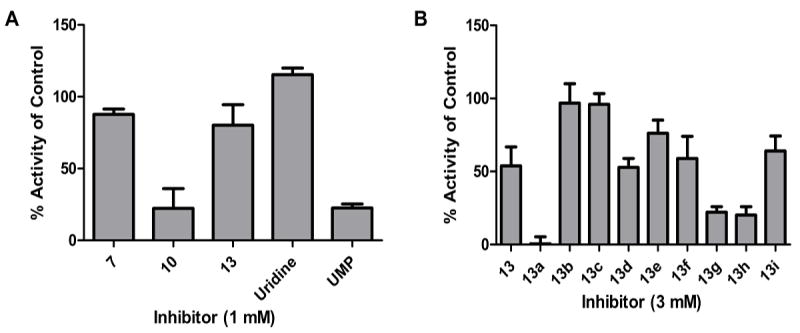
Biological evaluation of hexylamine-uridine inhibitors (A) and clicked product based on compound 13 (B), using the radioactivity-based activity assay. Error bars indicate mean ±SD.
Encouraged by these initial inhibition results, the mureidomimetic strategy was expanded to create ‘dyad inhibitors’ that would display two binding modules. Instead of the alanine that had principally been incorporated to increase rigidity, a targeted screen was performed to identify a moiety that could potentially bind in the carbohydrate-binding site of the UDP-sugar substrate, and therefore our attention was focused on mono- and bicyclic aromatic compounds that could engage in π-stacking interactions often identified in sugar-binding sites.[38] The synthesis of azide-functionalized alkylamine-uridines 11--13 involved N-acylation with chloroacetic anhydride, followed by chloride displacement with sodium azide in DMF and global deprotection under acidic conditions. Next, a selection of commercially available terminal alkynes (a--i, Scheme 2) was conjugated to 13 using standard CuII/ascorbate conditions to catalyze the [3 + 2] cycloaddition click reaction. Reaction progress was monitored using LC-MS and purification was performed using SepPak C18 cartridges. For this study, the C6 compound 13 was used as the parent compound because the alanine-conjugated equivalent, 10, had shown the best activity in inhibition assays. Indeed, a broad range of activities was observed when compounds 13 a--i were assayed, with 13 a resulting in complete inhibition of PglC activity at 3 mm (Figure 2B). Interestingly, the parent compound 13 showed only modest inhibition at 3 mm, reinforcing the importance of the naphthyl moiety on binding. These results demonstrate the added value of the dyad design, and might also suggest the importance of a stereocenter appended to 5’-N which is lacking in 13 but present in 10. With the aim of distinguishing between the different spacer lengths at this stage of inhibitor design, we conjugated the alkynes that had showed the greatest inhibition potential (a, f--i) to the shorter parent compounds 11 and 12. Interesting trends in inhibition properties were observed (see Figure S2, Supporting Information), and although alkyne g also yielded inhibitors with improved properties, the overall conclusion was that inhibitor 13 a, in which the naphthyl moiety was combined with the C6 alkylamine, showed the highest activity.
Because the dyad inhibitor strategy left little opportunity for further functionalization, the inhibitor scaffold was modified to allow further derivatization toward ‘triad inhibitors’. As illustrated in Scheme 3, N-α-Fmoc-protected azido-alanine (Aza) was coupled to protected hexylamine-uridine 4, using standard amide coupling conditions. In this way, the scaffold has the potential to be modified first by click reactions at the azide moiety with different alkynes and second by coupling different carboxylic acid derivatives to the liberated amine. Ensuing Fmoc deprotection of compound 14 using 20% piperidine in DMF gave a high amount of the β-eliminated product, as well as decomposition back to starting compound 4 (as identified using LC-MS), which was inseparable from the product. While shorter reaction times and reduced levels of base suppressed the formation of the elimination product, decomposition was still detected. Ultimately, a systematic study of potential conditions resulted in the use of piperazine immobilized on polystyrene resin, which significantly reduced the amount of compound 4 regenerated. Subsequently, myristic acid was coupled to the amine in 15, followed by acidic hydrolysis to afford inhibitor 17. Additionally, compound 16 was subjected to click chemistry conditions using 2-ethynyl-6-methoxynaphthyl to give fully functionalized triad inhibitor 18 after acidic hydrolysis in good yield.
Scheme 3.
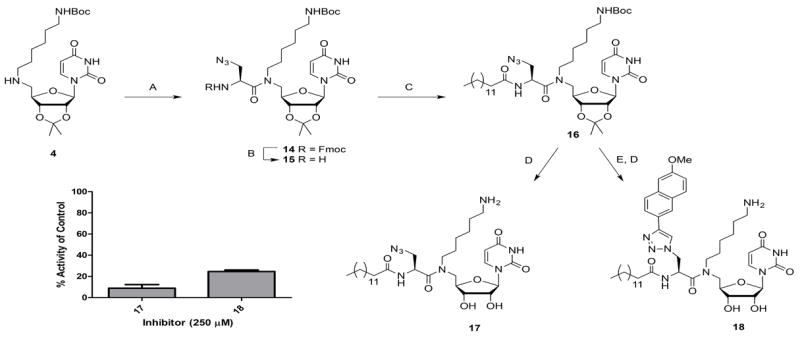
Synthesis and biological evaluation of triad inhibitors based on uridine-hexylamine, using the UMP/CMP-Glo assay. A) Fmoc-l-Aza-OH, EDC, HOBt, DMF (45%); B) immobilized piperazine, DCM; C) myristic acid, EDC, HOBt, DMF (48% 2 steps); D) TFA, H2O (17: 96%, 18: 85%); E) 2-ethynyl-6-methoxynaphthalene, CuSO4, sodium ascorbate, DMF (77%). Error bars indicate mean ±SD.
Inhibition assays using the UMP/CMP-Glo system revealed that attaching the long chain (C14) fatty acid had a positive effect on the inhibition potency, as exemplified by 90±3% inhibition of PglC at 250 μm inhibitor with substrate concentrations of 20 μm UDP-diNAcBac and 20 μm Und-P in the assay. The IC50 of compound 17 was determined to be 85±28 μm. This reinforces the importance of a hydrophobic acyl moiety in the design of PGT inhibitors, to either compete with the undecaprenol phosphate acceptor for binding, or to direct the inhibitor to the membrane interface proximal to where catalysis occurs. Interestingly, the inhibition potency decreased on going from 17 to 18, which has the added methoxynaphthyl moiety, to 75±1% inhibition at 250 μm inhibitor 18. This corresponds to an IC50 value of about 207 μm for compound 18. This decrease in inhibition potency with the added naphthyl moiety is most likely explained by the poorer solubility of inhibitor 18 in the assay conditions. Importantly, inhibition assays were carried out in the presence of 10% DMSO and 0.1% Triton X-100 in order to ensure that observed inhibition behavior was due to specific binding to the enzyme rather than promiscuous binding activity resulting from inhibitor aggregation effects.[29] In addition, the exact distance between the uridine core and the naphthyl group is slightly longer in 18 than in the precursor 13a, which could indicate a slight clash in the binding site, creating a suboptimal fit. However, it is clear from these results that increasing complexity of the modular scaffold results in increased inhibitory potency until an unfavorable interaction is introduced. Most importantly, the modular design of the mureidomycin mimetic strategy allows rapid differentiation at three positions, which will be extremely valuable in future optimization of this scaffold towards specific PGT targets.
Strategy 2 - Tunicamimetics
Having established that the combination of an alkylamine, a hydrophobic acyl group, and an aromatic moiety appended to a uridine core produced inhibitors with three points of variation and micromolar IC50 values, we then investigated an alternative modular scaffold that would display similar functionalities, but would facilitate easier differentiation at three modular positions. As depicted in Scheme 4, this strategy started with isopropylidene-protected 5’-amino-uridine (compound 19), and involved amide coupling chemistry to introduce the various modules. Instead of a primary amine that would displace the metal in the active site, this strategy exploits a strategic carboxylic acid functionality to investigate if such compounds might offer advantages in binding by directly coordinating a bound divalent cation at the active site.[39, 40] Such a strategy would mimic the proposed mechanism of tunicamycin-like nucleosides, which are thought to intercept binding as illustrated in Figure 1. To this end, Fmoc-l-Asp(tBu)-OH was coupled to compound 19, followed by Fmoc removal using 20% piperidine. To generate the first generation inhibitor (compound 21), the free amine in 20 was capped with an acetyl, and the tert-butyl and isopropylidene groups were simultaneously removed using aqueous TFA. Next, the Aza building block was conjugated to allow differentiation using click chemistry, and compound 22 was obtained in good yield. Fmoc removal under mild conditions proceeded smoothly to afford compound 23, which was either capped with an acetyl group to give compound 24, or coupled with myristic acid to generate compound 27. The acetylated precursor 24 was either directly deprotected under acidic conditions to give inhibitor 25, or subjected to click conditions with 2-ethynyl-6-methoxynaphthyl to generate a new class of tunicamimetic dyad inhibitor, 26, after acidic hydrolysis of the protecting groups. To generate the other dyad inhibitor displaying the hydrophobic acyl group, myristic acid was coupled to compound 23 using standard amide-coupling conditions. This resulted in compound 27, which was either fully deprotected using aqueous TFA to give dyad inhibitor 28, or subjected to click conditions with 2-ethynyl-6-methoxynaphthyl to produce the triad inhibitor 29. Because this second synthetic strategy was more straightforward than the first strategy, it allowed for thorough purification and isolation of intermediate compounds throughout the sequence of reactions. Advantageously, evaluating the inhibitory potential of the intermediates would provide information about the additive effects of the different modules on the overall potency. The results of the comparative inhibition assays are displayed in Scheme 4. Using the UMP/CMP-Glo assay, it was demonstrated that acid-functionalized uridine 21 inhibited PglC activity by 57±6% at 250 μm. Notably, this inhibitory potential is considerably higher than the inhibition observed with first-generation inhibitors based on the alkylamine module (Scheme 1, compound 7). The addition of the Aza building block did not influence the inhibitory potency to a great extent (25 in Scheme 4). In contrast, the addition of either a 2-ethynyl-6-methoxynaphthyl through click chemistry (compound 26), or a fatty acid moiety (compound 28) improved the inhibition of PglC, resulting in 77±10% and 88±5% inhibition at 250 μm respectively. Using the UMP/CMP-Glo assay, we were able to determine the IC50 values to be ~205 μm for 26 and 43±5 μm for 28. This implies a modest improvement over the analogous dyad inhibitor 17 from the first strategy. Interestingly, a similar trend in inhibition potential was observed with the triad inhibitor 29, which showed 81±3% inhibition at 250 μm, a slight decrease in potency compared to 28. However, this corresponded to an IC50 of 64±10 μm, which is an order of magnitude better than the corresponding alkylamine inhibitor 18.
Scheme 4.
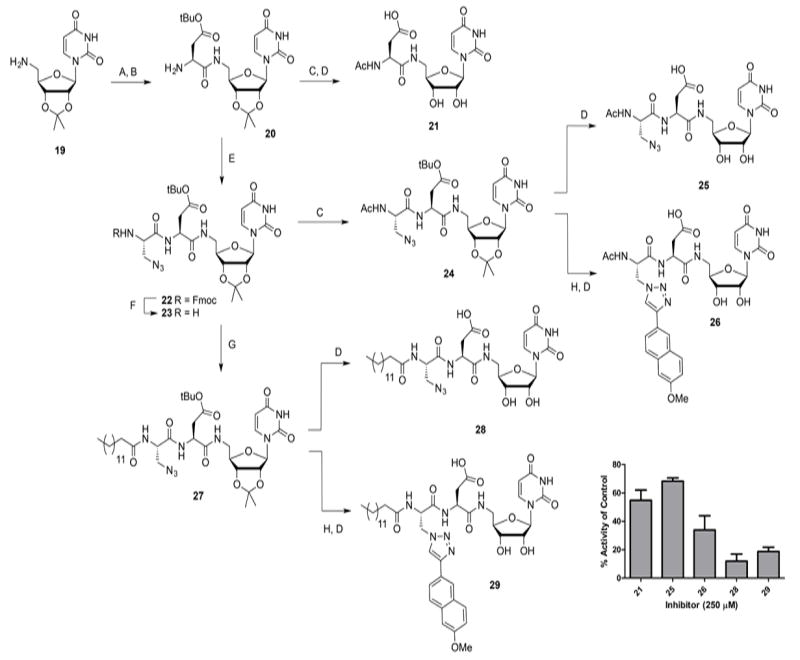
Synthesis and biological evaluation of triad inhibitors based on a peptidic backbone, using the UMP/CMP-Glo assay. A) Fmoc-l-Asp(tBu)-OH, EDC, HOBt, DMF; B) 20% piperidine, DMF (62% 2 steps); C) Ac2O, Et3N, MeOH (24: 89%); D) TFA, H2O (21: 86% 2 steps, 25: 74% 3 steps, 26: 61% 2 steps, 28: quant., 29: 66%); E) Fmoc-l-Aza-OH, EDC, HOBt, DMF (71 %); F) immobilized piperazine, DCM; G) myristic acid, EDC, HOBt, DMF (63% 2 steps); H) 2-ethynyl-6-methoxynaphthalene, CuSO4, sodium ascorbate, DMF (toward 29: 93%). Error bars indicate mean ±SD.
Conclusion
The development of inhibitors for PGTs is a relatively unexplored field, and it holds great potential to aid our understanding of this important class of enzymes, and to generate target molecules that inhibit crucial bacterial enzymes. Apart from diverse nucleosides that have been targeted to inhibit MraY, there is little known about molecular scaffolds that inhibit other PGTs specifically. Bioinformatics and structural investigations have revealed that PGTs display highly diverse topologies, hinting at diverse active site architectures to catalyze a similar reaction. Herein, we have presented the de novo design of two modular scaffolds that resulted in compounds with low micromolar inhibition of PglC, the monotopic PGT that is the gatekeeper enzyme of N-linked glycoprotein biosynthesis in C. jejuni. The modular approach allowed for evaluation of the effect of adding different modules on the inhibitory potential of PglC. Thus, it was clear that, starting from uridine, the attachment of aspartate (21, tunicamimetic strategy) was superior to the hexylamine moiety (7, mureidomimetic strategy), resulting in 57% inhibition at 250 μm with the former. Attachment of an aromatic moiety (i.e. naphthyl group in 26), and to a greater extent a hydrophobic long-chain acyl group (17 and 28) resulted in a further increase in inhibitory potential, while the combination of both modules resulted in a slight decrease (18 and 29). This detrimental effect might be attributed to a steric clash of the elaborated scaffold in the active site, or to the poor solubility of the triad inhibitors. Interestingly, the inhibition potentials of these scaffolds are similar to that of tunicamycin, which was found to have an IC50 of 100±8 μm for PglC (see Supporting Information). Together, these studies provide the proof-of-concept for our approach, and pave the way for increasing the potency and specificity for diverse monotpic PGTs and potentially the more complex polytopic PGTs by performing structure optimization at the diverse point of contact with the enzyme. Of paramount importance in these studies was the establishment of a reproducible enzyme purification protocol, and the development of a reliable activity assay, which in contrast to existing fluorescence-based assays for MraY,[41, 42] represents a significant challenge. We found that both assays described herein report accurately on PglC activity. In general, the radiochemical assay is more robust with a variety of inhibitors, but necessitates preparation of radiolabeled UDP-Bac and is also considerably more labor intensive due to the need for manual liquid--liquid extractions for each quenched reaction aliquot. In contrast, provided that appropriate controls are carried out to check for interference with the assay constituents, the UMP/CMP-Glo assay allows for rapid evaluation of inhibitory potentials in a simple, 96-well format assay.
Our efforts in both of these arenas have greatly contributed to the generation of PglC inhibitors. In conclusion, the chemical and biochemical studies lay the foundation for a diversity-oriented approach to explore the chemical space allowed for inhibitors in the active sites of PGTs. Moreover, the results will direct diversification of existing uridine libraries.[43, 44]
Experimental Section
General experimental procedures
All chemicals were used as received unless stated otherwise. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 (400/100 MHz), a Varian 500 (500/125 MHz), and a Bruker AV-600 (600/150 MHz) spectrometer. Chemical shifts (δ) are given in ppm and coupling constants are given in Hz. All given 13C spectra are proton-decoupled. Flash chromatography was performed on Silicycle Siliflash P60 silica gel (40—60 μm). TLC analysis was conducted on Agela Technologies TLC plates with detection by UV absorption (254 nm) where applicable and by spraying with 20% sulfuric acid in ethanol followed by charring at about 150°C or by spraying with a solution of (NH4)6Mo7O24·H2O (25 g/L) and (NH4)4Ce(SO4)4·2H2O (10 g/L) in 10 % sulfuric acid in water followed by charring at about 150°C. LC-MS analysis was performed on an HP 1000 series HPLC system and Finnegan LCQDeca mass spectrometer. Standard eluents used were A: 0.1% TFA in H2O, B: 0.1 % TFA in acetonitrile. The column used was a YMC-Pack ODSAQ column (3 μm, 100×3.0 mm I.D.). All analyses were carried out over 15 min, with a flow-rate of 0.1 mL/min. The UMP/CMP-Glo assay (Promega) was used as received. Luminescent readings were performed on a SynergyH1 platereader (Biotek) according to the protocol from Promega. Scintillation counting was performed on a Beckman Coulter LS6500 scintillation counting system.
General procedure A: Amino acid coupling
Fmoc-protected amino acid (2 equiv) was preactivated by mixing with EDC·HCl (2 equiv) and HOBt (2 equiv) in DMF (0.3 m) for 10 min at room temperature, and this mixture was added to the amine (1 equiv). The mixture was stirred overnight, diluted with DCM and washed with H2O (4×). The organic fraction was dried over Na2SO4, concentrated in vacuo, and re-dissolved in DMF (0.3 m). Piperidine (20 vol%) was added, and the mixture was stirred until complete consumption of starting material was observed using LC-MS. The mixture was concentrated in vacuo and purified using flash column chromatography (silica gel, DCM/MeOH) to obtain the desired product.
General procedure B: Click reaction in tBuOH/H2O
The azide (2 μmol, stock in DMSO) and alkyne (2.4 μmol, 100 mm stock in DMSO) were together dissolved in tBuOH/H2O (800 μL, 1/1, v/v). CuSO4 (2 μmol, 125 mm stock in H2O) and sodium ascorbate (4.8 μmol, 250 mm stock in H2O) were added and the resulting solution was incubated on a shaker at room temperature until complete consumption of the azide was observed using LC-MS (24—48 h). The mixture was lyophilized, redissolved in H2O/MeCN, and purified using SepPak C18 cartridges. Product fractions were lyophilized and redissolved in DMSO to give 50 mM stock solutions.
General procedure C: Click reaction in DMF
A solution of the azide (1 equiv) in DMF (0.025 m) was treated with 2-ethynyl-6-methoxynaphthalene (4 equiv), CuSO4 (1 equiv, 1 m stock in H2O) and sodium ascorbate (2 equiv, 1 m stock in H2O). Complete consumption of the azide was confirmed using LC-MS (1—3 h) and the mixture was diluted with DCM, washed with NH4Cl (4×), dried over Na2SO4, and concentrated in vacuo. The product was purified using flash column chromatography (silica gel, DCM/MeOH).
General procedure D: Acidic hydrolysis
Protected compound (10 mg) was treated with TFA/H2O (500 μL, 3/2, v/v) until complete removal of the protecting groups was observed using LC-MS (3—24 h). The mixture was lyophilized repeatedly to obtain the final compound.
Expression and purification of PglC
PglC was expressed in E. coli with a His6-SUMO-tag for expression, solubility, and purification purposes. The membrane fraction was isolated by centrifugation, and the sample was homogenized in 1% n-dodecyl β-d-maltoside (DDM) to transfer PglC to detergent micelles. Buffers used for subsequent purification steps included DDM at 0.03% (three times the critical micelle concentration) to ensure that the protein was solubilized in detergent micelles. PglC was purified using affinity chromatography with Ni-NTA resin, followed by gel filtration chromatography. The specific activity of PglC was calculated to be 3.43 μmol min-1mg-1. Purified enzyme was stored at -80°C. A detailed protocol is provided in the Supporting Information.
Radioactivity-based activity assay with PglC
Assays contained 20 μm Und-P, 10% DMSO, 0.1% Triton X-100, 50 mm HEPES pH 7.5, 100 mm NaCl, 5 mm MgCl2, 20 μm [3H]-UDP-diNAcBac (5.4 mCi/mmol), and 1 nm PglC in a final volume of 60 μL. Inhibitors were added in DMSO, in a volume such that the total concentration of DMSO in the reaction was equal to 10% (v/v). PglC was pre-incubated in the reaction mixture lacking [3H]-UDP-diNAcBac for five minutes at room temperature. After initiation of the reaction with [3H]-UDP-diNAcBac, aliquots (20 μL) were taken at twenty minute time points and quenched in 1 mL CHCl3:MeOH. The organic layer was washed three times with 400 μL PSUP (Pure Solvent Upper Phase, composed of 15 mL CHCl3, 240 mL MeOH, 1.83 g KCl in 235 mL H2O). The resulting aqueous layers were combined with 5 mL EcoLite (MP Biomedicals) liquid scintillation cocktail. Organic layers were combined with 5 mL OptiFluor (PerkinElmer). Both layers were analyzed using scintillation counting. The data was plotted as percentage remaining activity compared to the positive control (no inhibitor). A typical uninhibited reaction yielded approximately 600 dpm (quench compensated), corresponding to a turnover of roughly 12% of the total substrate in the reaction.
Luminescence assay using UMP/CMP-Glo
The quenching solution was prepared as described by Promega. Assays contained 20 μm Und-P, 10% DMSO, 0.1% Triton X-100, 50 mm HEPES pH 7.5, 100 mm NaCl, 5 mm MgCl2, 20 μm UDP-diNAcBac, and 1 nm PglC in a final volume of 25 μL. Inhibitors were added in DMSO, in a volume such that the total concentration of DMSO in the reaction was equal to 10% (v/v). PglC was pre-incubated in the reaction mixture lacking UDP-diNAcBac for five minutes at room temperature. The PglC reaction rate was dependent on the amount of enzyme in the assay and the specific quantity of PglC used was that which afforded a linear reaction rate over 20 min. After initiation of the reaction with UDP-diNAcBac, the reaction was halted after 20 min by the addition of 25 μL quenching buffer. The mixture was transferred to a 96-well plate (white, half area, Corning) and placed in the plate reader. The plate was shaken at low speed for 16 min, and incubated at room temperature for 44 min, after which time the luminescence was read. The data was plotted as percentage remaining activity compared to the positive control (no inhibitor). The IC50 values were obtained by plotting residual activity versus concentration (GraphPad Prism). To eliminate any false negatives based on luminescence quenching and/or inhibition of the luminescence assay, a control experiment was included in which the luminescence assay cocktail was incubated with and without inhibitor (in the presence of 2 μm UMP). Background inhibition of the UMP/CMP-Glo assay was established for each inhibitor, and the luminescence values were adjusted accordingly.
Comparison of radioactivity-based and UMP/CMP-Glo assays
Using the exact same assay conditions, a similar turnover is detected using both the radiochemical and UMP/CMP-Glo assays. In the radiochemical assay, approximately 600 DPM is generated in an uninhibited reaction, which corresponds to about 12% conversion of UDP-diNAcBac. Using the UMP Glo assay, RLU values between 2000--2500 are typically generated, which correspond to a conversion of about 10 % UDP-diNAcBac to UMP and Und-PP-diNAcBac (calculated using a UMP standard curve).
Supplementary Material
Acknowledgments
This project was funded by the NIH (R21 AI101807) to B.I. We thank Dr. Hicham Zegzouti (Promega) for supplying the UMP/CMP-Glo assay. We also thank Dr. Debasis Das for helpful discussions.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201503986. It includes experimental and spectroscopic data for all compounds and overview of the click library. Experimental details of the chemo-enzymatic synthesis of radiolabeled UDP-diNAcBac, the purification of PglC, the radioactivity-based activity assay with PglC, the luminescence assay using UMP/CMP-Glo. IC50 curves and Michaelis--Menten plots for UDP-diNAcBac and Und-P.
Imperiali et al @MIT have developed a modular approach to phosphoglycosyl transferase inhibitors
Inspired by the functional characteristics of mureidomycin A and tunicamycin, two novel scaffolds are synthesized that display low-micromolar IC50 against the monotopic phosphoglycosyltransferase PglC (see scheme).
References
- 1.Price NP, Momany FA. Glycobiology. 2005;15:29R–42R. doi: 10.1093/glycob/cwi065. [DOI] [PubMed] [Google Scholar]
- 2.Bouhss A, Mengin-Lecreulx D, Le Beller D, Van Heijenoort J. Mol Microbiol. 1999;34:576–585. doi: 10.1046/j.1365-2958.1999.01623.x. [DOI] [PubMed] [Google Scholar]
- 3.Lehrer J, Vigeant KA, Tatar LD, Valvano MA. J Bacteriol. 2007;189:2618–2628. doi: 10.1128/JB.01905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winn M, Goss RJ, Kimura K, Bugg TDH. Nat Prod Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 5.Inukai M, Isono F, Takatsuki A. Antimicrob Agents Chemother. 1993;37:980–983. doi: 10.1128/aac.37.5.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A. J Bacteriol. 2008;190:7141–7146. doi: 10.1128/JB.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howard NI, Bugg TDH. Bioorg Med Chem. 2003;11:3083–3099. doi: 10.1016/s0968-0896(03)00270-0. [DOI] [PubMed] [Google Scholar]
- 8.Grüschow S, Rackham EJ, Elkins B, Newill PL, Hill LM, Goss RJ. Chem Bio Chem. 2009;10:355–360. doi: 10.1002/cbic.200800575. [DOI] [PubMed] [Google Scholar]
- 9.Tanino T, Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A, Oyama H, Ichikawa S, Matsuda A. J Med Chem. 2011;54:8421–8439. doi: 10.1021/jm200906r. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto K, Sakagami M, Feng F, Togame H, Takemoto H, Ichikawa S, Matsuda A. J Org Chem. 2012;77:1367–1377. doi: 10.1021/jo202159q. [DOI] [PubMed] [Google Scholar]
- 11.Fer MJ, Olatunji S, Bouhss A, Calvet-Vitale S, Gravier-Pelletier C. J Org Chem. 2013;78:10088–10105. doi: 10.1021/jo4014035. [DOI] [PubMed] [Google Scholar]
- 12.Burda P, Aebi M. Biochim Biophys Acta. 1999;1426:239–257. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 13.Keller RK, Boon DY, Crum FC. Biochemistry. 1979;18:3946–3952. doi: 10.1021/bi00585a016. [DOI] [PubMed] [Google Scholar]
- 14.Suami T, Sasai H, Matsuno K, Suzuki N. Carbohydr Res. 1985;143:85–96. [Google Scholar]
- 15.Myers AG, Gin DY, Rogers DH. J Am Chem Soc. 1993;115:2036–2038. [Google Scholar]
- 16.Li J, Yu B. Angew Chem Int Ed. 2015;54:6618–6621. doi: 10.1002/anie.201501890. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:6718–6721. [Google Scholar]
- 17.Sarabia F, Martín-Ortiz L, López-Herrera FJ. Org Biomol Chem. 2003;1:3716–3725. doi: 10.1039/b307674a. [DOI] [PubMed] [Google Scholar]
- 18.Danishefsky SJ, Deninno SL, Chen S, Boisvert L, Barbachyn M. J Am Chem Soc. 1989;111:5810–5818. [Google Scholar]
- 19.Karpiesiuk W, Banaszek A. Carbohydr Res. 1997;299:245–252. doi: 10.1016/s0008-6215(97)00018-9. [DOI] [PubMed] [Google Scholar]
- 20.Ramza J, Zamojski A. Tetrahedron. 1992;48:6123–6134. [Google Scholar]
- 21.Bozzoli A, Kazmierski W, Kennedy G, Pasquarello A, Pecunioso A. Bioorg Med Chem Lett. 2000;10:2759–2763. doi: 10.1016/s0960-894x(00)00560-6. [DOI] [PubMed] [Google Scholar]
- 22.Gentle CA, Harrison SA, Inukai M, Bugg TDH. J Chem Soc Perkin Trans. 1999;1:1287–1294. [Google Scholar]
- 23.Lukose V, Luo L, Kozakov D, Vajfa S, Allen KN, Imperiali B. Biochemistry. 2015 doi: 10.1021/acs.biochem.5b01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly J, Jarrell H, Millar L, Tessier L, Fiori LM, Lau PC, Allan B, Szymanski CM. J Bacteriol. 2006;188:2427–2434. doi: 10.1128/JB.188.7.2427-2434.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glover KJ, Weerapana E, Chen MM, Imperiali B. Biochemistry. 2006;45:5343–5350. doi: 10.1021/bi0602056. [DOI] [PubMed] [Google Scholar]
- 26.Chung BC, Zhao J, Gillespie RA, Kwon DY, Guan Z, Hong J, Zhou P, Lee SY. Science. 2013;341:1012–1016. doi: 10.1126/science.1236501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner S, Baars L, Ytterberg AJ, Klussmeier A, Wagner CS, Nord O, Nygren PA, vanWijk KJ, de Gier JW. Mol Cell Proteomics. 2007;6:1527–1550. doi: 10.1074/mcp.M600431-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Morrison JP, Troutman JM, Imperiali B. Bioorg Med Chem. 2010;18:8167–8171. doi: 10.1016/j.bmc.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng BY, Shoichet BK. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee LV, Mitchell ML, Huang SJ, Fokin VV, Sharpless KB, Wong CH. J Am Chem Soc. 2003;125:9588–9589. doi: 10.1021/ja0302836. [DOI] [PubMed] [Google Scholar]
- 31.Auberger N, Frlan R, Al-Dabbagh B, Bouhss A, Crouvoisier M, Gravier-Pelletier C, Le Merrer Y. Org Biomol Chem. 2011;9:8301–8312. doi: 10.1039/c1ob06124k. [DOI] [PubMed] [Google Scholar]
- 32.Tedaldi LM, Pierce M, Wagner GK. Carbohydr Res. 2012;364:22–27. doi: 10.1016/j.carres.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 33.Wang R, Steensma DH, Takaoka Y, Yun JW, Kajimoto T, Wong CH. Bioorg Med Chem. 1997;5:661–672. doi: 10.1016/s0968-0896(97)00005-9. [DOI] [PubMed] [Google Scholar]
- 34.Ballell L, Young RJ, Field RA. Org Biomol Chem. 2005;3:1109–1115. doi: 10.1039/b500418g. [DOI] [PubMed] [Google Scholar]
- 35.Mitsuhashi N, Yuasa H. Eur J Org Chem. 2009:1598–1605. [Google Scholar]
- 36.Mugunthan G, Sriram D, Yogeeswari P, Kartha KPR. J Carbohydr Chem. 2012;31:553–570. [Google Scholar]
- 37.Wang S, Cuesta-Seijo JA, Lafont D, Palcic MM, Vidal S. Chem Eur J. 2013;19:15346–15357. doi: 10.1002/chem.201301871. [DOI] [PubMed] [Google Scholar]
- 38.Cecioni S, Imberty A, Vidal S. Chem Rev. 2015;115:525–561. doi: 10.1021/cr500303t. [DOI] [PubMed] [Google Scholar]
- 39.Yamashita A, Norton E, Petersen PJ, Rasmussen BA, Singh G, Yang Y, Mansour TS, Ho DM. Bioorg Med Chem Lett. 2003;13:3345–3350. doi: 10.1016/s0960-894x(03)00671-1. [DOI] [PubMed] [Google Scholar]
- 40.Spork AP, Buschleb M, Ries O, Wiegmann D, Boettcher S, Mihalyi A, Bugg TDH, Ducho C. Chem Eur J. 2014;20:15292–15297. doi: 10.1002/chem.201404775. [DOI] [PubMed] [Google Scholar]
- 41.Moberg A, Balderud LZ, Hansson E, Boyd H. Assay Drug Dev Technol. 2014;12:506–513. doi: 10.1089/adt.2014.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stachyra T, Dini C, Ferrari P, Bouhss A, van Heijenoort J, Mengin-Lecreulx D, Blanot D, Biton J, Le Beller D. Antimicrob Agents Chemother. 2004;48:897–902. doi: 10.1128/AAC.48.3.897-902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moukha-Chafiq O, Reynolds RC. ACS Comb Sci. 2014;16:232–237. doi: 10.1021/co4001452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun D, Jones V, Carson EI, Lee RE, Scherman MS, McNeil MR, Lee RE. Bioorg Med Chem Lett. 2007;17:6899–6904. doi: 10.1016/j.bmcl.2007.09.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.