

Abstract
The detection of rare circulating tumor cells (CTCs) in the blood of cancer patients has the potential to be a powerful and noninvasive method for examining metastasis, evaluating prognosis, assessing tumor sensitivity to drugs, and monitoring therapeutic outcomes. In this study, we have developed an efficient strategy to isolate CTCs from the blood of breast cancer patients using a microfluidic immune-affinity approach. Additionally, to gain further access to these rare cells for downstream characterization, our strategy allows for easy detachment of the captured CTCs from the substrate without compromising cell viability or the ability to employ next generation RNA sequencing for the identification of specific breast cancer genes. To achieve this, a chemical ligand-exchange reaction was engineered to release cells attached to a gold nanoparticle coating bound to the surface of a herringbone microfluidic chip (NP-HBCTC-Chip). Compared to the use of the unmodified HBCTC-Chip, our approach provides several advantages, including enhanced capture efficiency and recovery of isolated CTCs.
Graphical Abstract
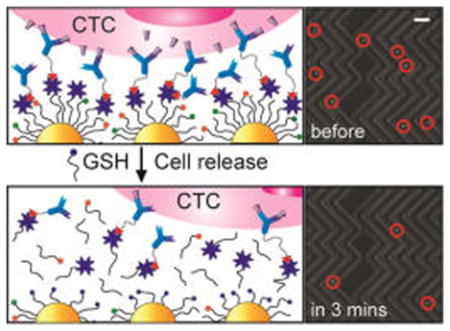
INTRODUCTION
Metastasis is responsible for the majority of cancer-related deaths and is thought to be initiated by the release of circulating tumor cells (CTCs) from the primary tumor.1 Enumeration of CTCs present in the peripheral blood of metastatic cancer patients has been shown to have prognostic utility in prostate, breast, and colorectal cancers.2 Molecular characterization of CTCs may provide a less invasive means of obtaining information from the patient’s primary tumor, helping to guide treatment and monitoring of disease progression.3 Additionally, since CTCs have been shown to contain genetic material shed from primary and metastatic tumors, they provide a unique opportunity to understand the biological mechanisms underlying metastasis.4
Although the existence of CTCs was confirmed more than 100 years ago,5 the isolation and subsequent profiling of CTCs remains a challenge due to the low number of CTCs present in the blood (as few as 1 CTC per 1 × 109 hematological cells) and their physical and biological heterogeneity within the same patient.6 The drawbacks of current CTC isolation technologies include (i) limited molecular characterization due to high residual cell background levels following CTC isolation; (ii) debulking or prelabeling steps that may cause cell stress and loss of CTC viability; (iii) the potential presence of CTC subpopulations that undergo epithelial-to-mesenchymal transitions, which are associated with different expression levels of tumor markers [e.g., epithelial cell adhesion molecule (EpCAM), cytokeratin]; and (iv) lack of access to the isolated cells due to technique or fixatives used in processing. Currently, the CellSearch system (Veridex, LLC, Raritan, NJ, USA) is the only FDA-cleared CTC diagnostic system for enumeration of CTCs in patients with breast, prostate, and metastatic colorectal cancers. Although CTC enumeration using this system provides prognostic value in cancer patients,7 CTCs are nonviable and cannot be recovered for downstream analysis or ex vivo cell culture. Therefore, there is a need to develop technologies that facilitate viable CTC recovery following the cell enrichment stage.8
Geometrically patterned microfluidic platforms with antibody-coated surfaces have been conceived as an alternative CTC isolation methodology, and high purification efficiencies have been demonstrated using this approach.9 The devices are easily fabricated at a low cost, permit viable cell isolation with a high sensitivity to low CTC concentration levels, and do not require sample preprocessing steps.10 We previously demonstrated that our microfluidic herringbone chip (HBCTC-Chip) generates microvortices within whole blood, thereby enhancing CTC capture through passive mixing and increased contact time between flowing cells and the antibody-functionalized surface.11 Clinical use of HBCTC-Chip with blood samples has enabled the determination of CTC signaling pathways by RNA sequencing,12 demonstration of dynamic changes in CTC phenotypes,13 development of an androgen receptor (AR) activity assay for prostate cancer CTCs,14 exploration of the metastatic role of CTC clusters,15 and, more recently, realization of single-point mutations in CTC deoxyribonucleic acid (DNA).16
Recently, nanostructured substrates have been incorporated into microtechnologies to enhance CTC isolation sensitivity.17 Similar to other immunoaffinity approaches, CTCs captured in this manner are irreversibly immobilized to the nanoparticles (NPs),18 nanotubes,19 and nanosheets,20 significantly limiting the ability to perform single-cell molecular analysis or long-term culture of this rare cell population. Various approaches involving polymer phase transitions (temperature-driven)21 and enzymatic degradation22 have been developed for single-cell analysis after isolation. Each one of these strategies has their advantages and limitations. For thermoresponsive substrates, they require the careful control of the surface temperature of the device to achieve uniform recovery of cells, thus, additional equipment to control the temperature is required and limits the ability to commercially scale these devices. On the other hand, the use of enzymes or chelators such as alginate lyase, EDTA, DNases, or endonucleases during recovery of the cells may compromise the viability of patient CTCs due to the over exposure to the degraded film itself and the enzymatic solution.16b,22
In this study, we utilize a thiolated ligand-exchange reaction with gold nanoparticles (AuNPs) on a herringbone chip (NP-HBCTC-Chip) to isolate and release cancer cells from whole blood. Our strategy results in a substrate that is stable during the processing of a highly complex biological fluid, yet ensures the safe release of the cancer cells for subsequent analysis and functional assays. In contrast with antibodies placed on flat silicon oxide surfaces, antibody-coated NPs were chemically assembled directly onto the HBCTC-Chip in our system. Application of this NP-mediated strategy in micro-fluidic devices such as the HBCTC-Chip provides the following additional advantages: (a) the nanoroughened structure created by the NP assemblies increases the surface area available for adhesion and binding, and this synergistically influences specific interactions between cancer cells and antibodies, ultimately enhancing tumor capture but reducing nonspecificity due to a differential adhesion profile to nanostructured substrates between cancer cells and normal blood cells;23 (b) the metal–thiol interactions can be readily disrupted in the presence of excess thiol molecules, such that the original ligands with immobilized antibodies are efficiently exchanged with biocompatible thiol molecules [i.e., glutathione (GSH)],24 resulting in the release of cancer cells from the surface; (c) the irregular surfaces of the NP assemblies provide release reagents with access to the surface area under the cells, thereby enabling successful release of captured cells for subsequent molecular analysis and ex vivo cell culture; and (d) the chemically self-assembled monolayers derived from the reversible NP bonds enable the optimization of this method for use with complex surface topographies without the need for additional process changes.
Interactions between cells and substrates play a significant role in the modulation of cell adhesion and functionality. Recently, three-dimensional nanostructures have been reported to exhibit improved cell-capturing efficiencies due to the increased frequency and duration of cell/substrate contact achieved in microfluidic channels, and this increase in sensitivity is critical for identifying cells in low concentrations, such as CTCs.25 To achieve this nanostructured surface in a HBCTC-Chip, AuNPs were used as an efficient platform for assembling tumor-specific antibodies, and a 2 nm core was chosen because the surface functionality of AuNPs with a small size can be readily manipulated with robustness and an increase in the size of NPs (e.g., 100 nm or greater) not necessarily would reflect in an increase in capture efficiency under high flow rate.26 The AuNPs used in this work were composed of a mixed monolayer of 11-mercaptoundecanoic acid (MUA) and 12-mercaptododecanoic acid N-hydroxysuccinimide ester (NHS) (Scheme 1a). The NHS ligands were used to bind an amine moiety to NeutrAvidin, thereby immobilizing the NPs on the surface. Additionally, despite the water insolubility of AuNPs, the carboxylic acid (COOH) ligands associated with MUA were used to enhance NP solubility in ethanol, thereby facilitating their use with polydimethylsiloxane (PDMS) devices. The NHS-functionalized AuNPs were immobilized within the herringbone (HB) channels through reactions with amine groups on the surface, and the remaining NHS-esters were utilized for NeutrAvidin binding. The HBCTC-Chip with bound NeutrAvidin–NP assemblies was coated with antibodies via tetravalent biotin–NeutrAvidin binding to facilitate specific tumor cell binding (Scheme 1b).
Scheme 1. Design of the NP-HBCTC-Chip for the Capture and Release of CTCsa.
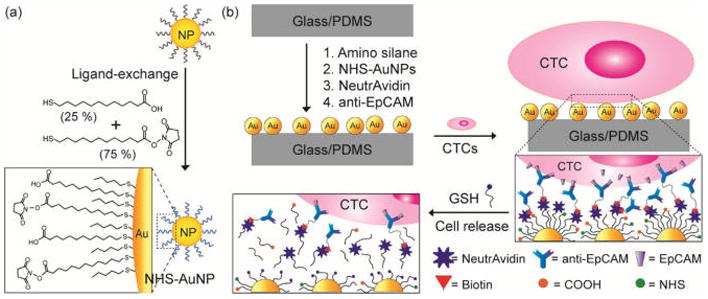
aConditions: (a) Preparation of NHS-functionalized NPs with carboxylic acid (for enhanced NP solubility in EtOH) and NHS (for the NP immobilization and avidin binding) functional groups via ligand exchange with pentanethiol-functionalized NPs. (b) Schematic illustration of each surface modification process step involved in the fabrication of the NP-HBCTC-Chip and CTC isolation on the chip and subsequent CTC release by ligand exchange with GSH.
GSH was chosen as a cell release reagent because it is a well-characterized tripeptide, and it is the most abundant thiol species in the cytoplasm. GSH performs many important physiological functions, such as controlling the redox environment in cells.27 Furthermore, the high intracellular concentration of GSH observed physiologically (up to 10 mM in liver cells) suggests that it can be used safely without causing critical damage to cells during the release process. This approach brings closer the concept “liquid biopsy”: a safe, efficient, and precise means of obtaining information about the state of blood-borne metastasis in patients.24,28
RESULTS AND DISCUSSION
As a proof-of-principle study, experiments were first performed on a flat silicon substrate instead of the herringbone chip, and surface analyses were performed using an ellipsometer and an atomic force microscope (AFM). Figure 1 shows the change in film thickness associated with each step of the assembly process for both the control and NP-mediated chip surfaces. First, the monolayer assembly of (3-mercaptopropyl)trimethoxysilane (MPTMS) and N-[3-(trimethoxysilyl)propyl]ethylenediamine (AEAPTMS) was observed on the SiO2 surface, with thicknesses of 0.4 ± 0.1 and 1.0 ± 0.1 nm in the control and NP-mediated chips, respectively. The average thickness increased to 0.3 ± 0.1 and 3.5 ± 0.4 nm with successful immobilization of N-(γ-maleimidobutyryloxy)succinimide ester (GMBS) and NHS-functionalized AuNPs, respectively, which corresponds to the relative molecular/nanoscale sizes of GMBS and the AuNPs. A significant thickness increase of 1.7 ± 0.3 nm was observed for both chips after the NeutrAvidin binding step, and this is consistent with the literature reports based on AFM analysis.29 However, with the addition of GSH, we observed a reduction in the thickness of the NP-mediated HB chip surfaces, which was equivalent to the amount gained in the NeutrAvidin binding step; this loss in thickness corresponds to the approximate size of the NeutrAvidin molecule. The topographic images of the NP-mediated chip substrates (Figure S1A,B) exhibit a corrugated surface generated by the NP assemblies and NP–NeutrAvidin binding, with surface roughness (Rq) values of 0.497 and 0.705 nm, respectively. This is compared to the GMBS and GMBS–NeutrAvidin binding on smooth substrates with Rq values of 0.249 and 0.412 nm, respectively (Figure S1C,D).
Figure 1.
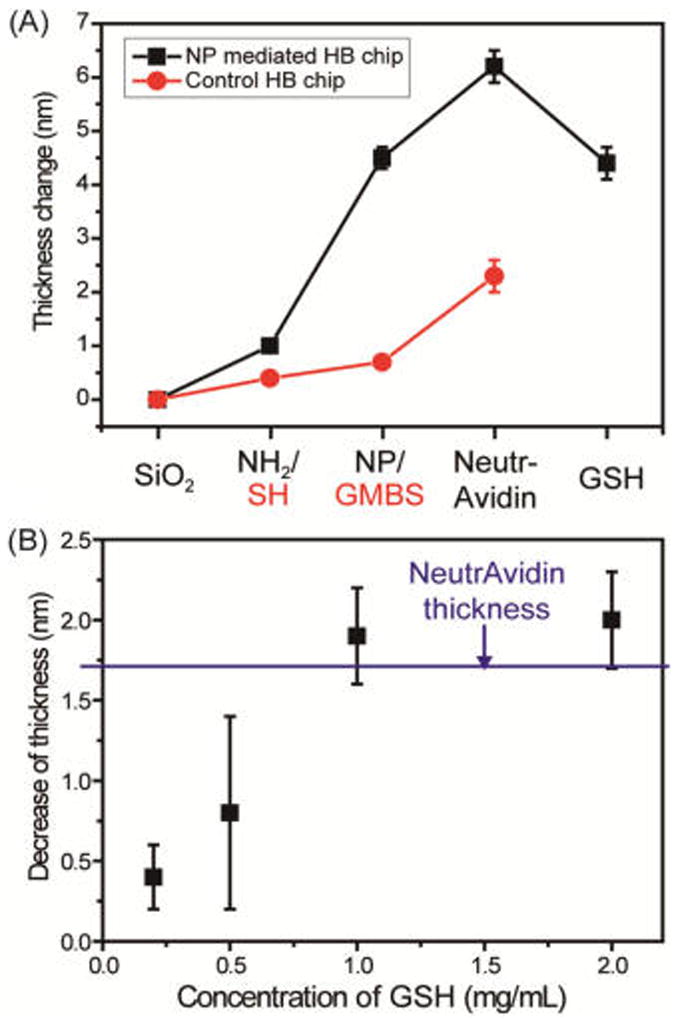
(A) Thickness changes with each surface modification step in the proof-of-principle studies comparing NeutrAvidin binding for NP-mediated and control silica substrates. (B) Thickness changes by NeutrAvidin detachment through ligand exchange as a function of GSH concentration for 30 min.
To demonstrate the successful release of cancer cells from the chip via AuNP–thiol exchange reactions, the NP–NeutrAvidin-bound substrates were dipped in different GSH solution concentrations for 30 min. A thickness decrease of 1.9 ± 0.4 nm was observed following exposure to GSH solution concentrations greater than 0.5 mg/mL, and this is indicative of successful NeutrAvidin detachment from the NP surface through ligand exchange (Figure 1B). After GSH exposure, the observed 1.9 nm reduction likely corresponds to the selective removal of NeutrAvidin and the retention of NPs on the surface due to a difficulty in accessibility of release reagent to permeate underneath the immobilized small AuNPs (2 nm).
The NP-mediated HB chip (NP-HBCTC-Chip) was fabricated using a PDMS microfluidic device with herringbone structures by the chemical modification (sequential reactions of amino silane, NHS-AuNPs, NeutrAvidin, and anti-EpCAPM onto the surface) as reported in our previous research.11 To assess coverage and uniformity of NeutrAvidin deposition onto NPs immobilized on the surface, a fluorescently labeled biotin (biotin-R-phycoerythrin) was used, and the fluorescent traces were measured and analyzed. Figure S2A,B shows the fluorescent images after the biotin treatment on the surface of the microfluidic device prepared in the presence and absence of NeutrAvidin. The strong fluorescence of the biotin able to bind NeutraAvidin clearly demonstrates the successful avidin deposition with uniform fluorescent intensity (Figure S2C,D). To further evaluate antibody coverage and effect on capture efficiency, the surface was functionalized with a fluorescently labeled secondary antibody after anti-EpCAM binding. Figure S3A shows the titration curve of the antibody coverage at the surface, showing a linear relation between antibody coverage and concentration. Importantly, we observed the correlated increase in the capture efficiency of PC3 cells with the antibody coverage up to a concentration of 10 μg/mL, but the efficiency (98.15 ± 1.1%) was saturated over than the antibody concentration (10 μg/mL) (Figure S3B). These results are in agreement with our prior publications that demonstrate that the optimal concentration of capture antibodies is 10 μg/mL.11 In addition, the steady fluorescent intensity across the surface of the device indicates the uniformly distributed antibody on the device (Figure S3C,D).
To demonstrate the impact of the corrugated NP films on cell isolation, the NP-HBCTC-Chip was compared with our standard HBCTC-Chip in a controlled experiment. Figure 2 shows the capture efficiency and nonspecific binding [NSB, an amount of white blood cells (per 3 mL) presented on the surface after running each blood sample through the microfluidic device] of both PC3 and MDA-MB-231 cancer cells with both NP-HBCTC-Chip and HBCTC-Chip substrates. PC3 cells (prostate origin and more epithelial-like) possess about 52 000 epithelial cell adhesion molecule (EpCAM) binding sites per cell, whereas MB-MDA-231 cells (breast origin and mesenchymal-like) exhibit only 1700 binding sites (approximately 30 times fewer).30 Higher capture efficiency (99% vs 82%) and lower NSB (35% decrease) were observed for PC3 cells in the NP-functionalized chip compared to that of our previous HBCTC-Chip chemistry. Moreover, a substantial increase (1–16%) in capture efficiency and an 88% decrease incredibly in NSB were achieved with the MDA-MB-231 cell line. These improvements were strongly associated with the surface nanostructure of the microfluidic chip (Figure S4). Interestingly, the reduction on nonspecificity is an important feature of our nanostructured substrates with a differential adhesion profile to nanostructured substrates between cancer cells and normal blood cells.23c To enhance the capture efficiency with MDA-MB-231 cell lines, which have a low expression level of EpCAM similar to human epidermal growth factor receptor 2 (Her2) but a high expression level of epidermal growth factor receptor (EGFR), our NP-mediated chip was further functionalized with Her2, EGFR, and a cocktail of antibodies (a combination of EpCAM, Her2, and EGFR) and tested with MDA-MB-231 cells. Figure 2B shows the high capture efficiency with the use of EGFR and cocktail that is a clear indication of the importance of antibody selection for CTCs with heterogeneous expression of various antibodies. Using a cocktail approach of three antibodies (EpCAM, HER2, and EGFR), our group has demonstrated that breast cancer cells across all stages of the EMT transition can be captured.13
Figure 2.
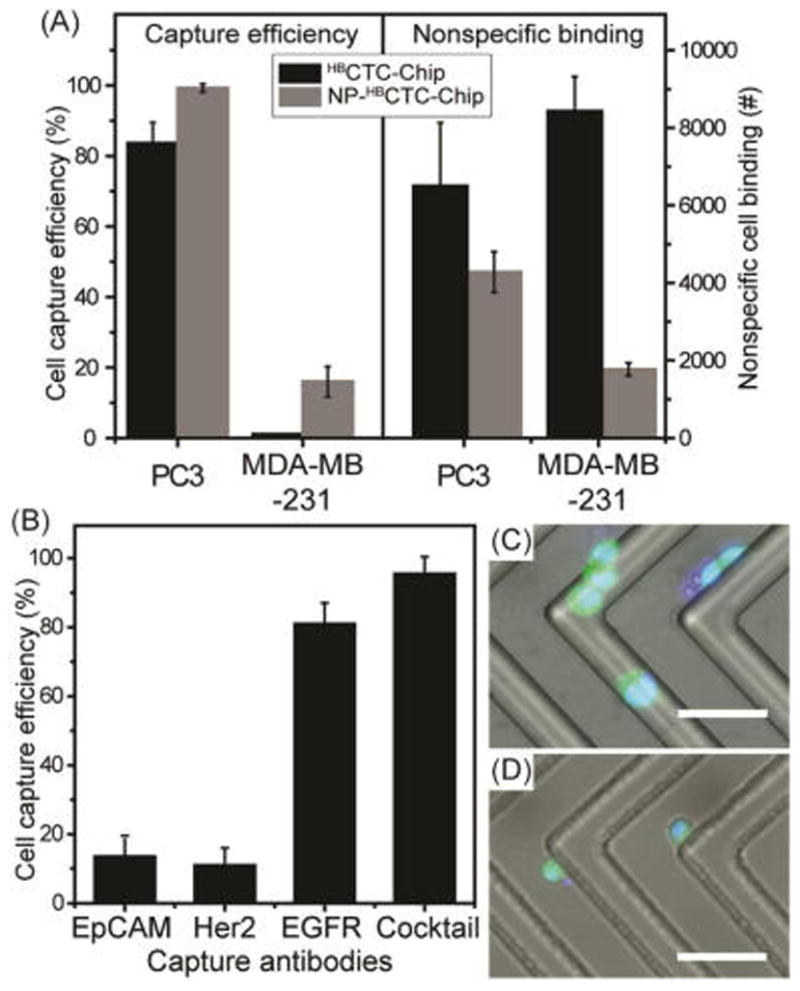
(A) Comparison of capture efficiencies and nonspecific binding of cancer cells. PC3 and MDA-MB-231 cells were spiked into whole blood and run through the original HBCTC-Chip11 and the NP-HBCTC-Chip. Cells were captured on the surface of the microfluidic device functionalized with anti-EpCAM. Capture efficiency and nonspecific cell binding were quantified using a previously developed protocol.11 (B) Capture efficiency of MDA-MB-231 cells with different capture antibodies (cocktail is a combination of EpCAM, Her2, and EGFR). Combined fluorescent and bright-field microscopic images of viable (C) PC3 and (D) MDA-MB-231 cells isolated on the NP-HBCTC-Chip (scale bar = 100 μm).
The number of CTCs present in patient blood can be highly variable, but it is almost always a rare event. Thus, to the capture sensitivity of our device at varying concentrations of cancer cells, we added cancer cell into whole blood at concentrations ranging from 5 to 1000 cells/mL of blood. A linear correlation between the number of spiked (1000 to 10 PC3 cells/mL) and captured cells (R2 = 0.9947, n = 3) was observed, with capture efficiency ranging between 96.4 ± 2.2 and 80.0 ± 1% (Figure 3A). To test ultralow concentrations of cancer cells, two different cancer cell lines (MDA-MB-231 and PC3) were investigated. For each cell line, five total cells were picked one cell at a time using a micromanipulator and spiked into 1 mL of whole blood, prior to flowing through our chip. Using this approach, for the MDA-MB-231 cells, the average capture efficiency was 68 ± 29.2%, n = 10, and for the PC3 cells, it was 72 ± 26.4%, n = 10 (Figure 3B). These results demonstrate that the NP-HBCTC-Chip can efficiently isolate cancer cells at low numbers and antibody expression levels by enhancing cell-surface contact and binding affinity between CTC antigens and substrate antibodies, regardless of phenotype.
Figure 3.
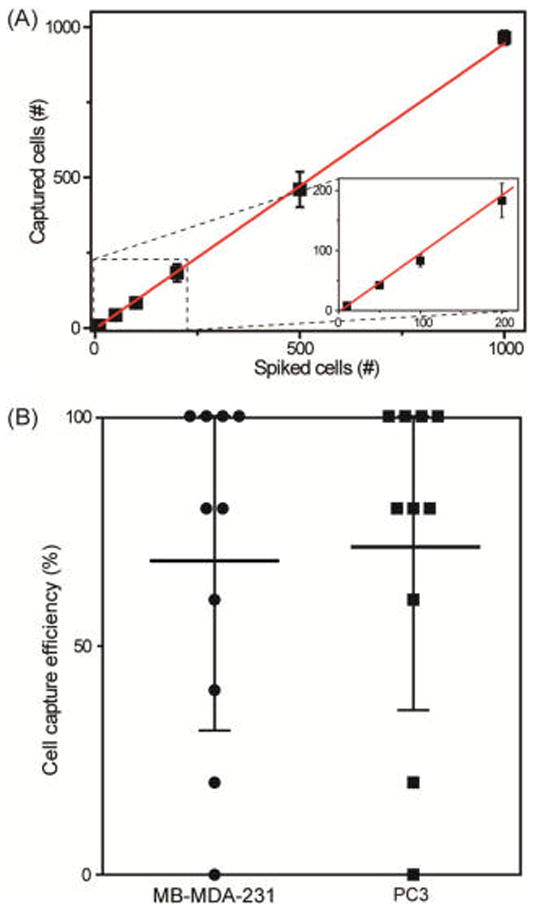
(A) Linear correlation (R2 = 0.9947) was determined between the number of spiked and captured PC3 cells with NP-HBCTC-Chip, with cancer cell lines spiked into whole blood at a concentration of 10, 50, 100, 200, 500, and 1000 (n = 3 for each condition). (B) Chip capture performance for ultralow concentrations of cancer cells (5 cells/mL of whole blood, n = 10). Each data point indicates the capture efficiency of an independent experiment for MB-MDA-231 and PC3 cells. Cell capture efficiency at such low cancer cell concentrations varied from 0 to 100%. For spiking concentrations of 1000, 500, and 200 cells per mL, a serial dilution of an initial 100 000 cells/mL was used. A micromanipulator equipped with a microneedle was used for spiking concentrations of 100, 50, 10, and 5 cancer cells/mL of whole blood.
Based on the thiol exchange reactions illustrated in Scheme 1B, the reversible thiol binding to the surface of the AuNPs facilitates detachment of isolated CTCs from the chip in the presence of excess GSH. Figure 4A shows the release efficiency achieved by flowing GSH (1 mg/mL) for 30 min and the viability of the recovered cancer cells. The NP-HBCTC-Chip showed good cell detachment performance, with release efficiencies of 92 and 91% for isolated PC3 and MDA-MB-231 cancer cells, respectively. The released cells were subsequently cultured in media for up to 5 days, with an average cell viability determined to be 78% (MDA-MB-231) and 87% (PC3) relative to our control cells (Figures 4D and S5). Optical microscopy images of isolated cancer cells on the NP-HBCTC-Chip before and 3 min after GSH treatment illustrate the efficiency of this system for cell isolation and release (Figure 4B,C). To quantify a proliferation rate of the released PC3 and MB-MDA 231 cells from our microfluidic platform, an MTT assay was used to measure the absorbance at 570 nm at different time points. During 8 days, any significant difference was not found for the control and released cells using the highest concentration (1 mg/mL) of GSH (Figure S6).
Figure 4.
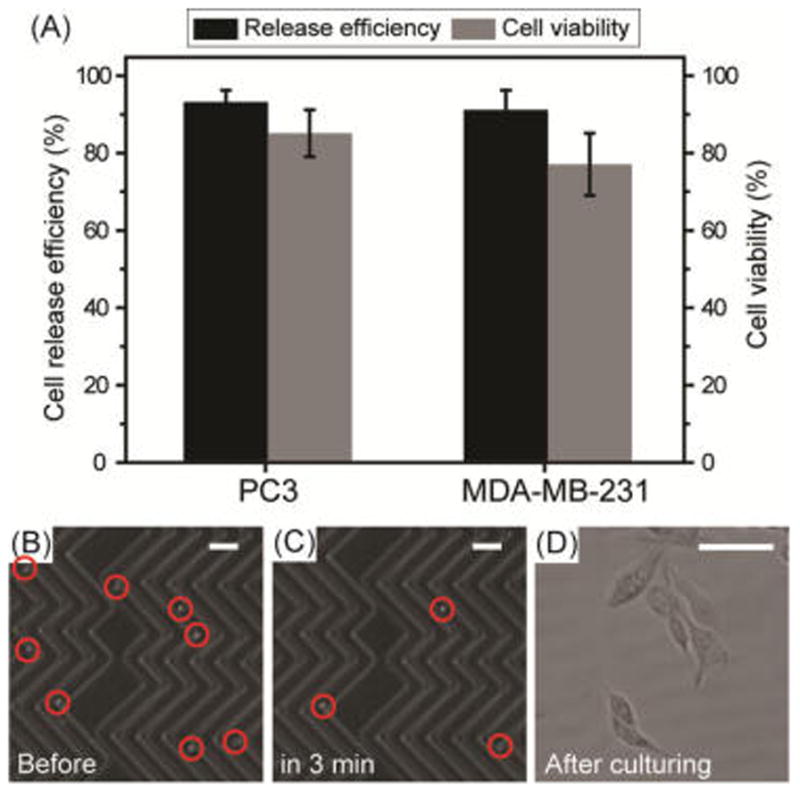
(A) Release efficiency and cell viability of recovered PC3 and MDA-MB-231 CTCs using the NP-HBCTC-Chip. After whole blood spiked with cancer cells was run, the NP-HBCTC-Chip was washed with phosphate-buffered saline, and a solution of 1% bovine serum albumin with 1 mg/mL of GSH was incubated for 30 min. Bright-field microscopy images of isolated CTCs on the NP-HBCTC-Chip (B) before and (C) 3 min after GSH treatment (scale bar = 150 μm). (D) Image of cultured individual CTCs 24 h after recovery from the chip (scale bar = 30 μm).
Blood samples from a small cohort of metastatic breast cancer patients were used to test the clinical utility of the AuNP assembly. To maximize capture efficiency from cancer patients as mentioned above, all of our patient results were obtained with the use of an antibody cocktail at 10 μg/mL.13 Blood samples from four metastatic breast cancer patients and two healthy individuals were processed through our NP-HBCTC-Chip, with an average of 3.5 mL of blood analyzed per patient. We were able to identify CTCs (Figure 5A,B) in all patients using immunofluorescence staining techniques. Cells were identified as CTCs if they stained positive for DNA with 4′,6-diamidino-2-phenylindole (DAPI), expressed the tumor markers EpCAM, and cadherin 11 (CDH11), and did not express the leukocyte marker CD45.22c A patient sample was considered positive for CTCs when at least 2 CTCs/mL were detected. This threshold was established based upon positive event levels detected in the two healthy controls (median = 0.9 CTCs/mL, mean = 0.7 ± 0.3 CTCs/mL). CTC counts ranging from 6 to 12 CTCs/mL (median = 7.4 CTCs/mL, mean = 8.2 ± 2.7 CTCs/mL) were obtained in the breast cancer patients (Figure 6C). One interesting feature of the nanocoating is that it captured not only individual CTCs (Figure 5A) but also CTC clusters present in the peripheral blood of metastatic cancer patients (Figure 5B). CTC clusters have higher metastatic potential than single CTCs owing to their biological and physical shielding from the immune system and blood flow shear stresses.31 Therefore, our NP-HBCTC-Chip approach demonstrated improved versatility and sensitivity toward CTCs.
Figure 5.
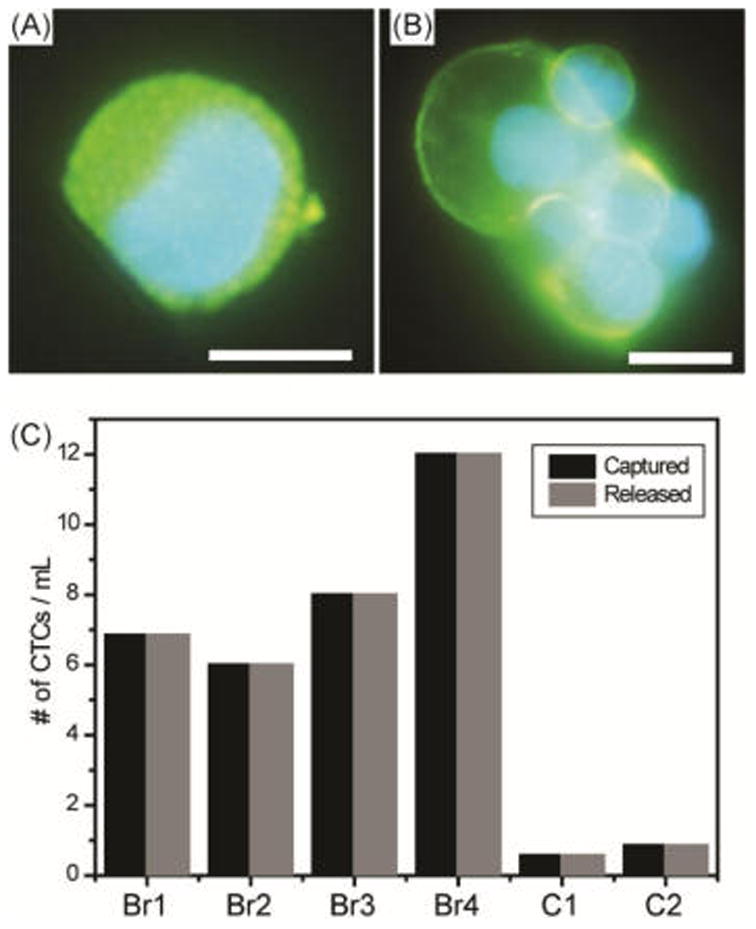
Immunofluorescence staining of cell-surface receptors of a captured (A) single CTC and (B) CTC cluster from a metastatic breast cancer patient. The images shown include EpCAM/CDH11 staining in Alexa Fluor 488 and DAPI nuclear staining in blue (scale bar = 10 μm). (C) Captured and released CTC counts from breast metastatic patients (Br1–Br4) and healthy controls (C1–C2).
Figure 6.
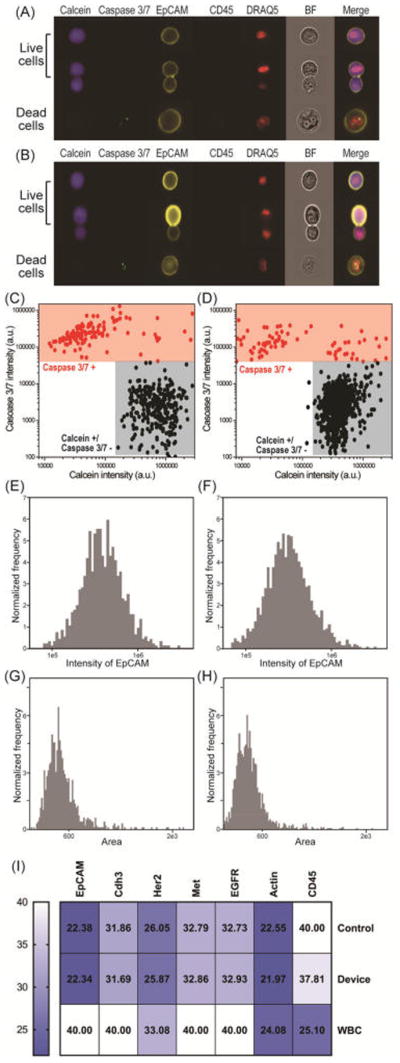
Characterization of the patient-derived breast (Brx) CTC line using imaging flow cytometry. Data compare viability, EpCAM expression, and area of control versus captured/released cells from our NP-HBCTC-Chip. Representative images of one viable, cluster, and dead Brx cell (A) obtained from culture (control) and (B) captured/released from our microfluidic device. Gate settings of (C) control and (D) captured/released Brx cells. Viable cells are defined as calcein positive and caspase 3/7 negative, whereas dead cells are caspase 3/7 positive. The intensity of EpCAM obtained from (E) control and (F) captured/released Brx cells. The area of (G) control and (H) captured/released Brx cells. (I) Heat map of the Ct values of seven genes obtained by RT-qPCR. Comparisons were across control, released Brx cells, and white blood cells (WBC).
To further demonstrate that our system is a powerful and noninvasive method for understanding CTC biology, the molecular characterization was performed using a patient-derived breast (Brx) CTC line as a biological model system.13 Our CTC cell lines have been derived from patient CTCs that are highly heterogeneous and also are more sensitive to culture and manipulation conditions than immortalized cell lines (e.g., hypoxic conditions, nonadherent culture, specialized media).32 Using imaging flow cytometry, cell viability was measured using calcein AM, and apoptotic cells were identified using a caspase 3/7 fluorescence probe. We compared control cells (obtained directly from culture) to cells captured and released using NP-HBCTC-Chip (Figure 6A,B). Also, anti-EpCAM and CD45 markers were used to distinguish cancer cells and white blood cells after release (Figure 6A,B). Figure 6C,D shows the gate used for viability quantification of control versus captured/released Brx cells. The viability oscillates between 87 and 93% with both control and released cells having almost identical scatter plots. Also, the expression levels of EpCAM for both control and released cells show nearly identical profiles with a mean EpCAM intensity of 4.5 × 105 ± 2.9 × 105 and 3.8 × 105 ± 2.7 × 105, respectively (Figure 6E,F). Also, we analyzed the size distribution to identify any potential changes as a result of microfluidic processing. Figure 6G,H shows that there are no significant changes in size distribution, with the mean area of 544 ± 210 and 480 ± 170 for control versus captured cells, respectively.
Our biocompatible and safe method was further confirmed by the identical threshold cycle (Ct) values for both control and released Brx cells using RT-qPCR, whereby lower Ct values represent higher gene expression (Figure 6I and Figures S7 and S8). Ct values for control cells versus cells released from our device were 22.38 (SD 0.23) and 22.34 (SD 0.56) for EpCAM, 31.86 (SD 0.08) and 31.69 (SD 0.12) for Cdh3, 26.05 (SD 0.05) and 25.87 (SD 0.06) for Her2, 32.79 (SD 0.19) and 32.86 (0.13) for Met, and 32.73 (SD 0.08) and 32.93 (SD 0.16) for EGFR. Taken together, these results are a clear indication that the chemical release with the use of GSH for thiol exchange does not affect viability or the molecular signature of our Brx CTC line.
To test the impact of our release mechanism on gene expression profiles of the CTCs, next generation RNA sequencing analysis was performed on CTCs isolated from metastatic breast cancer patients. For each sample, the blood sample was split between a “released” and “control” condition. After isolating RNA on-chip for both control and released conditions, the CTCs were released using our ligand-exchange approach (see Experimental Section) followed by RNA isolation. Unsupervised clustering of RNA isolated using the different processing conditions resulted in each paired patient sample (e.g., control and release) clustering together for the top 1000 of the most variant genes (Figure S9 and Table S1). When we looked at gene expression profiles for breast-cancer-specific genes, our data demonstrate remarkably negligible changes in expression levels between our control and released CTCs (Figure 7). Moreover, we were able to identify unique breast cancer gene signatures related to disease progression (e.g., KRT8, KRT18),33 patient survival (e.g., TFF3),34 risk of metastasis (e.g., CLCA2),35 and the epithelial to mesenchymal transition (e.g., S100A14, S100A16)36 of cancer cells during the dissemination of the disease (Figure 7 and Table S2).
Figure 7.
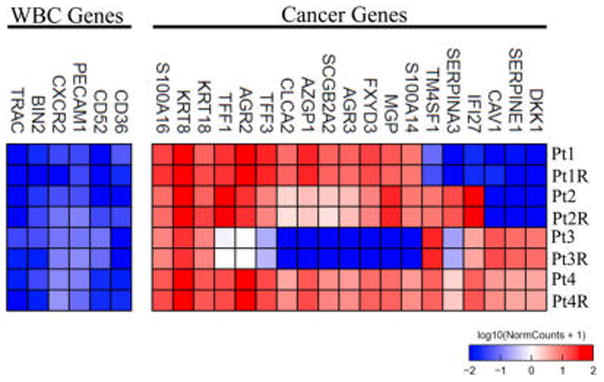
Gene expression profiles for a representative set of breast-cancer-specific genes for CTCs isolated using the NP-HBCTC-Chip. For each breast cancer patient, two processing conditions were analyzed: a control, on-chip, extraction of RNA from the captured CTCs, and a postrelease “R” extraction of RNA from the CTCs.
CONCLUSION
We have demonstrated an alternative strategy to efficiently isolate cancer cells from the peripheral blood using thiolmodified AuNPs assembled on HBCTC-Chip surfaces. Using our NP-HBCTC-Chip, the isolated cancer cells from mesenchymal and epithelial cancer cell lines as well as metastatic breast cancer patient samples can be recovered through simple thiol exchange reactions without any significant damage. The inherent advantages of the NP-HBCTC-Chip include ease of fabrication, flexibility for use with diverse ligand-exchange functional groups, and accessibility to three-dimensional surface structures, thereby facilitating cell binding and release. The approach taken for both capture and release shows limited impact on cell viability and the ability to perform sensitive downstream assays such as next generation RNA sequencing.
EXPERIMENTAL SECTION
Fabrication of the NP-Bound Microfluidic Herringbone Chip
1-Pentanethiol, 11-mercaptoundecanoic acid (MUA), 12-mercaptododecanoic acid N-hydroxysuccinimide (NHS) ester, gold-(III) chloride hydrate, sodium azide, Tween 20, bovine serum albumin (BSA), (3-mercaptopropyl) trimethoxysilane, N-(γ-maleimidobutyryloxy)succinimide ester (GMBS), and N-[3-(trimethoxysilyl)propyl]ethylenediamine (AEAPTMS) were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA) and used without further purification. The NP-bound microfluidic herringbone chip (NP-HBCTC-Chip) was fabricated using a previously described method.12 Briefly, the HBCTC-Chip consists of a 1 in. × 3 in. glass slide and a poly(dimethylsiloxane) (PDMS, Sylgard184, Dow Corning Corporation, Midland, MI, USA) layer containing eight microchannels with a herringbone pattern on the upper surface. The glass slide and PDMS layer are bonded together following oxygen plasma treatment. To chemically modify the device, the microfluidic channels were first treated with 1 wt % solution of AEAPTMS in ethanol for 1 h at room temperature, followed by repeated complete washings with ethanol. Subsequently, the chip was incubated in a 0.01 wt % ethanol solution of NHS-functionalized AuNPs for 30 min at room temperature and rinsed with ethanol to allow amide formation as a means of binding the particles to the aminated surface. Next, the channels were filled with 20 μg/mL NeutrAvidin (Pierce Biotechnology Company, Rockford, IL, USA) solution in PBS for 1 h, thereby allowing free lysines in the NeutrAvidin to react with the remaining free NHS-ester and bind the tetravalent NeutrAvidin protein to the particle surface. The NP–NeutrAvidin-bound chips were stored in a solution of NeutrAvidin at 4 °C until ready for use. Within 24 h of the experiment, a 20 μg/mL PBS solution of human EpCAM biotinylated goat antibody (R&D Systems, Inc., Minneapolis, MN, USA) containing 1 wt % BSA and 0.09 wt % sodium azide was added to the chip for 1 h, followed by rinsing with ethanol and PBS. One hour prior to running the experiments, the chip was purged with 3 wt % BSA and 0.05 wt % Tween 20.
Cell Capture and Release
Breast (MDA-MD-231) and prostate (PC3) cancer cell lines were obtained from the American Type Culture Collection (ATCC). Cancer cell lines were expanded using the appropriate protocols, and they were used at 85% cell confluence. Before the experiment, cells were stained with CellTracker Green CMFDA dye (Thermo Fisher Scientific, Waltham, MA, USA) for 15 min and then trypsinized. Stained cells were spiked in healthy blood at a concentration of 1000 cells/mL and run through the microfluidic device. A 3 mL sample of spiked blood with cancer cells was run at a flow rate of 1 mL/h. Immediately, the microfluidic chip was rinsed with PBS for 1 h. The CTC capture efficiency on the surface of the chip was evaluated with an automated protocol.11 For release of captured CTCs, each GSH solution concentration in PBS containing 1% BSA was run at a flow rate 1 mL/h through the microfluidic chip for 30 min. After being rinsed with PBS, the cells from the chip were collected in a vial containing cell media and connected to the outlet of the device. Cells remaining on the microfluidic device and in the collection vial were both imaged and counted manually using a fluorescence microscope. The viability of the cells was evaluated after the release process using propidium iodide staining to identify the membranes of compromised cells.
Blood Processing
Blood samples from healthy donors and cancer patients were collected according to an institutional review board protocol. A total of four metastatic breast cancer patients and two healthy individuals were included in this study. Blood was collected in Vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ, USA) containing the anticoagulant ethylenediaminetetraacetic acid (EDTA), and the samples without pretreatment were processed within 4 h of blood draw. Typically, for 3 mL of whole blood, the total processing time (including release) is 4 h.
Immunofluorescence Staining
CTCs were identified with mouse anti-EPCAM (3:100, Cell Signaling Technology, Inc., Danvers, MA, USA) and anti-cadherin 11 (CDH11) (1:10, R&D Systems, Inc., Minneapolis, MN, USA) conjugated to Alexa Fluor 488 dye. White blood cells were stained with mouse anti-CD45 (1:20, BD Biosciences, San Jose, CA, USA) conjugated to PE-CF594 dye. DAPI was used as a nuclear stain for the cells.
MTT Assay
Released cells were seeded at 2000 cells/mL into 96-well plates, and MTT solution was added at different time points and incubated at 37 °C for 4 h. Then, the reaction was stopped according to the manufacturer. Absorbance was measured at 570 nm and samples in triplicate.
Imaging Flow Cytometry
Imaging flow cytometry was performed using the ImageStreamX Mark II imaging flow cytometer (Amnis Corporation) equipped with a 40× objective, six imaging channels, and 405, 488, and 642 nm lasers. For analysis of cell viability and EpCAM expression, Brx cells obtained from culture or captured/released from our microfluidic device were resuspended in RPMI (supplemented with 0.3% BSA and HEPES) and stained with the following antibodies and stains where applicable: calcein blue AM (2.5 μM, ThermoFisher Scientific), CellEvent caspase 3/7 green detection reagent (1.75 μM, Life Technologies), PE-conjugated EpCAM antibody (1:260, clone VU1D9, Cell Signaling), PE-cf594-conjugated CD45 antibody (1:400, clone H130, BD Bioscience), and DRAQ5 (1 μM, Cell Signaling Technologies). Cells were acquired/gated using the nuclear marker DRAQ5. EpCAM positive cells were gated for viable cells (calcein positive and caspase 3/7 negative) versus dead cells (caspase 3/7 positive).
RT-qPCR
RNA from control and Brx cells captured/released from the microfluidic device were extracted using the RNeasy Plus micro kit (Cat# 74034, Qiagen), as per manufacturer’s direction. RNA integrity number was obtained using a 2100 Bioanalyzer instrument (Agilent Genomics), with values ranging from 8 to 9. A spectrophotometer (Biophotometer, Eppendorf) was used to ascertain purity and yields. The 260/280 ranged from 1.92 to 2.00 for all samples. The high capacity cDNA reverse transcription kit (Cat# 4368814, Applied Biosystems) was used for cDNA synthesis using 1 μg of total RNA. cDNA synthesis was performed on a C1000 Touch thermal cycler (Bio-Rad Laboratories). Quantitative PCR was performed using the TaqMan gene expression master mix (Cat# 4369016, Applied Biosystems) on the CFX96 real-time system (Bio-Rad Laboratories), with all appropriate controls (e.g., no template and no enzyme controls). TaqMan probes which span exons were all purchased from ThermoFisher as follows: EpCAM (Cat# 4331182), Cdh3 (Cat# 331182), HER2/ERBB2 (Cat# 4331182), Met (Cat# 4331182), and EGFR (Cat# 4331182). All cancer-specific probes did not amplify in samples containing purified white blood cells (WBCs), except Her2, which has been confirmed to be expressed in peripheral blood cells.37 Also, amplification of PTPRC (CD45; Cat# 4331182) confirmed Brx cells obtained directly from culture contained no contaminating WBCs (as expected), whereas cells isolated from the device contained low levels of contaminating WBCs as indicated by the high Ct value (low gene expression) of CD45. The contaminating WBCs in Brx cells isolated from our device would also contribute to the gene expression of housekeeping genes such as actin.
Library Preparation and RNA Sequencing
Captured and released CTCs were lysed with 700 μL of qiazol, and RNA was extracted using a RNAeasy mini kit from Qiagen (Hilden, Germany). RNA was amplified using a modified protocol38 and sequenced at the Broad Institute of Harvard and MIT.
Supplementary Material
Acknowledgments
Our research was supported by Stand up to Cancer, a joint program of the Entertainment Industry Foundation (EIF) and the American Association for Cancer Research (AACR) (M.T., P.T.H.), the National Institute for Biomedical Imaging and Bioengineering (NIBIB) EB008047 (M.T.), the Koch Institute Core Grant P30-CA14051 from the NCI (P.T.H.), and NIH P41 EB002503-11 (M.T.). We acknowledge all our patients and healthy blood donors. All blood specimens were obtained with informed patient consent according to an institutional review board (IRB) protocol (05-300) at the Massachusetts General Hospital. The authors also wish to express their appreciation to the Institute for Soldier Nanotechnologies at MIT, supported by the Army Research Office under contract W911NF-13-D-0001 and the Air Force under contract W911NF-07-D-0004, whose facilities and/or equipment were used to conduct the research reported in this paper. Thanks to Aditya Bardia, Lecia V. Sequist, Libby Laura, Octavio Hurtado, and Charles Vanderburg at the Advanced Tissue Resource Core (ATRC) for expert technical support. Thanks to Nicole Vincent Jordan for providing TaqMan primers, and Cindy Agpraseuth for helping with microfluidic device preparations. Thanks to Berent Aldicakti, Eric Thai, and Kevin Duc Vo for RNA extraction and RNA library preparations. S.N.T. holds a Natural Sciences and Engineering Research Council (NSERC) of Canada Postdoctoral Fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.(a) Neki K, Kawahara H, Watanabe K, Toyama Y, Akiba T, Yanaga K. Anticancer Res. 2013;33:1769–1772. [PubMed] [Google Scholar]; (b) Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen L, Meropol NJ. J Clin Oncol. 2008;26:3213–3221. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]; (c) Yoon HJ, Kozminsky M, Nagrath S. ACS Nano. 2014;8:1995–2017. doi: 10.1021/nn5004277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Pantel K, Brakenhoff RH, Brandt B. Nat Rev Cancer. 2008;8:329–40. doi: 10.1038/nrc2375. [DOI] [PubMed] [Google Scholar]; (b) Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. N Engl J Med. 2004;351:781–91. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]; (c) Lianidou ES, Strati A, Markou A. Crit Rev Clin Lab Sci. 2014;51:160–71. doi: 10.3109/10408363.2014.896316. [DOI] [PubMed] [Google Scholar]
- 3.Obermayr E, Castillo-Tong DC, Pils D, Speiser P, Braicu I, Van Gorp T, Mahner S, Sehouli J, Vergote I, Zeillinger R. Gynecol Oncol. 2013;128:15–21. doi: 10.1016/j.ygyno.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 4.Fong ZV, Winter JM. Cancer J. 2012;18:530–538. doi: 10.1097/PPO.0b013e31827654ea. [DOI] [PubMed] [Google Scholar]
- 5.Ashworth TR. Aust Med J. 1869;14:146–149. [Google Scholar]
- 6.Plaks V, Koopman CD, Werb Z. Science. 2013;341:1186–1188. doi: 10.1126/science.1235226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LWMM, Hayes DF. N Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]; (b) Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen LWMM, Meropol NJ. J Clin Oncol. 2008;26:3213–3221. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]
- 8.(a) den Toonder J. Lab Chip. 2011;11:375–377. doi: 10.1039/c0lc90100h. [DOI] [PubMed] [Google Scholar]; (b) Parkinson D, Dracopoli N, Petty B, Compton C, Cristofanilli M, Deisseroth A, Hayes D, Kapke G, Kumar P, Lee J, Liu M, McCormack R, Mikulski S, Nagahara L, Pantel K, Pearson-White S, Punnoose E, Roadcap L, Schade A, Scher H, Sigman C, Kelloff G. J Transl Med. 2012;10:138. doi: 10.1186/1479-5876-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Greene B, Hughes A, King M. Front Oncol. 2012;2:69. doi: 10.3389/fonc.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sheng WA, Chen T, Tan WH, Fan ZH. ACS Nano. 2013;7:7067–7076. doi: 10.1021/nn4023747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dong Y, Skelley AM, Merdek KD, Sprott KM, Jiang C, Pierceall WE, Lin J, Stocum M, Carney WP, Smirnov DA. J Mol Diagn. 2013;15:149–157. doi: 10.1016/j.jmoldx.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kurkuri MD, Al-Ejeh F, Shi JY, Palms D, Prestidge C, Griesser HJ, Brown MP, Thierry B. J Mater Chem. 2011;21:8841–8848. [Google Scholar]; (b) Hyun KA, Jung HI. Lab Chip. 2014;14:45–56. doi: 10.1039/c3lc50582k. [DOI] [PubMed] [Google Scholar]
- 11.Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Jr, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Proc Natl Acad Sci U S A. 2010;107:18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman MJ, Xega K, Contino G, Alagesan B, Brannigan BW, Milos PM, Ryan DP, Sequist LV, Bardeesy N, Ramaswamy S, Toner M, Maheswaran S, Haber DA. Nature. 2012;487:510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, Ulman M, Smas ME, Lord JB, Brannigan BW, Trautwein J, Bander NH, Wu CL, Sequist LV, Smith MR, Ramaswamy S, Toner M, Maheswaran S, Haber DA. Cancer Discovery. 2012;2:995–1003. doi: 10.1158/2159-8290.CD-12-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S. Cell. 2014;158:1110–1122. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Sheng W, Chen T, Tan W, Fan ZH. ACS Nano. 2013;7:7067–7076. doi: 10.1021/nn4023747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao W, Cui CH, Bose S, Guo D, Shen C, Wong WP, Halvorsen K, Farokhzad OC, Teo GSL, Phillips JA, Dorfman DM, Karnik R, Karp JM. Proc Natl Acad Sci U S A. 2012;109:19626–19631. doi: 10.1073/pnas.1211234109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, Liu K, Liu J, Yu ZTF, Xu X, Zhao L, Lee T, Lee EK, Reiss J, Lee YK, Chung LWK, Huang J, Rettig M, Seligson D, Duraiswamy KN, Shen CKF, Tseng HR. Angew Chem, Int Ed. 2011;50:3084–3088. doi: 10.1002/anie.201005853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Sheng W, Chen T, Tan W, Fan ZH. ACS Nano. 2013;7:7067–7076. doi: 10.1021/nn4023747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang B, Weldon AL, Kumnorkaew P, Xu B, Gilchrist JF, Cheng X. Langmuir. 2011;27:11229–11237. doi: 10.1021/la2015868. [DOI] [PubMed] [Google Scholar]
- 19.Chen GD, Fachin F, Fernandez-Suarez M, Wardle BL, Toner M. Small. 2011;7:1061–1067. doi: 10.1002/smll.201002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon HJ, Kim TH, Zhang Z, Azizi E, Pham TM, Paoletti C, Lin J, Ramnath N, Wicha MS, Hayes DF, Simeone DM, Nagrath S. Nat Nanotechnol. 2013;8:735–741. doi: 10.1038/nnano.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Reátegui E, Aceto N, Lim EJ, Sullivan JP, Jensen AE, Zeinali M, Martel JM, Aranyosi AJ, Li W, Castleberry S, Bardia A, Sequist LV, Haber DA, Maheswaran S, Hammond PT, Toner M, Stott SL. Adv Mater. 2015;27:1593–1599. doi: 10.1002/adma.201404677. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ao Z, Parasido E, Rawal S, Williams A, Schlegel R, Liu S, Albanese C, Cote RJ, Agarwal A, Datar RH. Lab Chip. 2015;15:4277–4282. doi: 10.1039/c5lc01024a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hou S, Zhao H, Zhao L, Shen Q, Wei KS, Suh DY, Nakao A, Garcia MA, Song M, Lee T, Xiong B, Luo S-C, Tseng H-R, Yu H-h. Adv Mater. 2013;25:1547–1551. doi: 10.1002/adma.201203185. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim K, Jo MC, Jeong S, Palanikumar L, Rotello VM, Ryu JH, Park MH. Nanoscale. 2016;8:11949–11955. doi: 10.1039/c6nr00362a. [DOI] [PubMed] [Google Scholar]; (e) Park H, Yang S, Park MH. Toxicol Environ Health Sci. 2014;6:238–243. [Google Scholar]
- 22.(a) Shah AM, Yu M, Nakamura Z, Ciciliano J, Ulman M, Kotz K, Stott SL, Maheswaran S, Haber DA, Toner M. Anal Chem. 2012;84:3682–3688. doi: 10.1021/ac300190j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Labib M, Green B, Mohamadi RM, Mepham A, Ahmed SU, Mahmoudian L, Chang IH, Sargent EH, Kelley SO. J Am Chem Soc. 2016;138:2476–2479. doi: 10.1021/jacs.5b10939. [DOI] [PubMed] [Google Scholar]; (c) Li W, Reátegui E, Park MH, Castleberry S, Deng JZ, Hsu B, Mayner S, Jensen AE, Sequist LV, Maheswaran S, et al. Biomaterials. 2015;65:93–102. doi: 10.1016/j.biomaterials.2015.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Wan Y, Mahmood MAI, Li N, Allen PB, Kim YT, Bachoo R, Ellington AD, Iqbal SM. Cancer. 2012;118:1145–1154. doi: 10.1002/cncr.26349. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ivanov I, Stojcic J, Stanimirovic A, Sargent E, Nam RK, Kelley SO. Anal Chem. 2013;85:398–403. doi: 10.1021/ac3029739. [DOI] [PubMed] [Google Scholar]; (c) Sekine J, Luo SC, Wang S, Zhu B, Tseng HR, Yu H. Adv Mater. 2011;23:4788–4792. doi: 10.1002/adma.201102151. [DOI] [PubMed] [Google Scholar]; (d) Chen W, Weng S, Zhang F, Allen S, Li X, Bao L, Lam RH, Macoska JA, Merajver SD, Fu J. ACS Nano. 2013;7:566–575. doi: 10.1021/nn304719q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong R, Han G, Fernandez JM, Kim BJ, Forbes NS, Rotello VM. J Am Chem Soc. 2006;128:1078–1079. doi: 10.1021/ja056726i. [DOI] [PubMed] [Google Scholar]
- 25.(a) Wang S, Wang H, Jiao J, Chen KJ, Owens GE, Kamei K, Sun J, Sherman DJ, Behrenbruch CP, Wu H, Tseng HR. Angew Chem, Int Ed. 2009;48:8970–3. doi: 10.1002/anie.200901668. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim ST, Kim DJ, Kim TJ, Seo DW, Kim TH, Lee SY, Kim K, Lee KM, Lee SK. Nano Lett. 2010;10:2877–83. doi: 10.1021/nl100942p. [DOI] [PubMed] [Google Scholar]
- 26.Wang B, Weldon AL, Kumnorkaew P, Xu B, Gilchrist JF, Cheng X. Langmuir. 2011;27:11229–11237. doi: 10.1021/la2015868. [DOI] [PubMed] [Google Scholar]
- 27.(a) Estrela JM, Ortega A, Obrador E. Crit Rev Clin Lab Sci. 2006;43:143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]; (b) Rahman I, Kode A, Biswas SK. Nat Protoc. 2007;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 28.Chompoosor A, Han G, Rotello VM. Bioconjugate Chem. 2008;19:1342–1345. doi: 10.1021/bc8000694. [DOI] [PubMed] [Google Scholar]
- 29.Misawa N, Yamamura S, Yong-Hoon K, Tero R, Nonogaki Y, Urisu T. Chem Phys Lett. 2006;419:86–90. [Google Scholar]
- 30.(a) Prang N, Preithner S, Brischwein K, Göster P, Wöppel A, Müller J, Steiger C, Peters M, Baeuerle P, Da Silva A. Br J Cancer. 2005;92:342–349. doi: 10.1038/sj.bjc.6602310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, Gires O. Nat Cell Biol. 2009;11:162–U117. doi: 10.1038/ncb1824. [DOI] [PubMed] [Google Scholar]
- 31.Orr FW, Buchanan MR, Weiss L. Microcirculation in Cancer Metastasis. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 32.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, Sgroi D, Kapur R, Shioda T, Ting DT, Ramaswamy S, Getz G, Iafrate AJ, Benes C, Toner M, Maheswaran S, Haber DA. Science. 2014;345:216–220. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker LC, Harris GC, Holloway AJ, McKenzie GW, Wells JE, Robinson BA, Morris CM. Cancer Genet Cytogenet. 2007;178:94–103. doi: 10.1016/j.cancergencyto.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Pandey V, Wu ZS, Zhang M, Li R, Zhang J, Zhu T, Lobie PE. Breast Cancer Res. 2014;16:429. doi: 10.1186/s13058-014-0429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walia V, Yu Y, Cao D, Sun M, McLean JR, Hollier BG, Cheng J, Mani SA, Rao K, Premkumar L, Elble RC. Oncogene. 2012;31:2237–2246. doi: 10.1038/onc.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.(a) Zhou W, Pan H, Xia T, Xue J, Cheng L, Fan P, Zhang Y, Zhu W, Xue Y, Liu X, Ding Q, Liu Y, Wang S. J Biomed Sci. 2014;21:97. doi: 10.1186/s12929-014-0097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tanaka M, Ichikawa-Tomikawa N, Shishito N, Nishiura K, Miura T, Hozumi A, Chiba H, Yoshida S, Ohtake T, Sugino T. BMC Cancer. 2015;15:53. doi: 10.1186/s12885-015-1059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You F, Roberts LA, Kang SP, Nunes RA, Dias C, Iglehart JD, Solomon NA, Friedman PN, Harris LN. J Hematol Oncol. 2008;1:2. doi: 10.1186/1756-8722-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA. Nat Protoc. 2010;5:516–535. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.