

Abstract
The emergence of antibiotic resistance is a defining challenge, and Escherichia coli is recognized as one of the leading species resistant to the antimicrobials used in human or veterinary medicine. Here, we analyse the distribution of 2172 antimicrobial-resistance (AMR) genes in 4022 E. coli to provide a population-level view of resistance in this species. By separating the resistance determinants into ‘core’ (those found in all strains) and ‘accessory’ (those variably present) determinants, we have found that, surprisingly, almost half of all E. coli do not encode any accessory resistance determinants. However, those strains that do encode accessory resistance are significantly more likely to be resistant to multiple antibiotic classes than would be expected by chance. Furthermore, by studying the available date of isolation for the E. coli genomes, we have visualized an expanding, highly interconnected network that describes how resistances to antimicrobials have co-associated within genomes over time. These data can be exploited to reveal antimicrobial combinations that are less likely to be found together, and so if used in combination may present an increased chance of suppressing the growth of bacteria and reduce the rate at which resistance factors are spread. Our study provides a complex picture of AMR in the E. coli population. Although the incidence of resistance to all studied antibiotic classes has increased dramatically over time, there exist combinations of antibiotics that could, in theory, attack the entirety of E. coli, effectively removing the possibility that discrete AMR genes will increase in frequency in the population.
Keywords: antibiotics resistance evolution
Abbreviations
AMR, antimicrobial resistance; AMRF, antimicrobial resistance gene family; CARD, Comprehensive Antibiotic Resistance Database; HSP, high-scoring pair; SSS, sequence similarity score.
Data Summary
1. Information on Escherichia coli genome sequences that we believe are mislabelled in GenBank has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434776 (url – https://dx.doi.org/10.6084/m9.figshare.4434776).
2. A figure showing the phylogenetic position of genomes that we consider too distantly related to E. coli to be included in our analysis has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434779 (url – https://dx.doi.org/10.6084/m9.figshare.4434779).
3. A list of genome sequences used in this study has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434782 (url – https://dx.doi.org/10.6084/m9.figshare.4434782).
4. Our curation of the antimicrobial-resistance (AMR) determinants from the Comprehensive Antibiotic Resistance Database has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434788 (url – https://dx.doi.org/10.6084/m9.figshare.4434788).
5. The sequence similarity score data underlying the analyses in our study has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434794 (url – https://dx.doi.org/10.6084/m9.figshare.4434794).
6. A text-based representation of the graphs we used to generate antibiotic-resistance gene families has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434797 (url – https://dx.doi.org/10.6084/m9.figshare.4434797).
7. A table of the genes we identified in the core resistome of E. coli has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434800 (url – https://dx.doi.org/10.6084/m9.figshare.4434800).
8. A table highlighting the presence of core antibiotic-resistance determinants in the horizontally transferred genes of E. coli MG1655 has been deposited in Figshare; DOI: 10.6084/m9.figshare.4595371 (url – https://dx.doi.org/10.6084/m9.figshare.4595371).
9. A table of the resistance gene families we identified in the accessory resistome of E. coli has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434803 (url – https://dx.doi.org/10.6084/m9.figshare.4434803).
10. A visualization of our data on the distribution of accessory resistance in E. coli has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434809 (url – https://dx.doi.org/10.6084/m9.figshare.4434809).
11. A figure that shows it is unlikely that bias inherent in the sequenced E. coli resource has affected our analysis has been deposited in Figshare; DOI: 10.6084/m9.figshare.4595389 (url – https://dx.doi.org/10.6084/m9.figshare.4595389).
12. A figure that shows antibiotic resistance and phylogenetic distance are not correlated has been deposited in Figshare; DOI: 10.6084/m9.figshare.4595377 (url – https://dx.doi.org/10.6084/m9.figshare.4595377).
13. A visualization of the increase in the abundance of resistance to antibiotic classes in E. coli over time has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434815 (url – https://dx.doi.org/10.6084/m9.figshare.4434815).
14. An animated visualization of the co-association of AMR in E. coli over time has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434773 (url – https://dx.doi.org/10.6084/m9.figshare.4434773).
15. Data on the combinations of antibiotics for which all E. coli lack combined resistance has been deposited in Figshare; DOI: 10.6084/m9.figshare.4434818 (url – https://dx.doi.org/10.6084/m9.figshare.4434818).
Impact Statement
Exposure of bacteria to antibiotics in clinical and veterinary settings has placed an immense selective pressure on bacteria that has accelerated the emergence of resistances. Using Escherichia coli as an exemplar, we identify the spectrum of accessory resistance determinants across the entire population of E. coli genome sequences available via the National Center for Biotechnology Information and profile the distribution of resistances. This profiling demonstrates the preponderance of multiple resistances among E. coli strains. Using this data, we model the potential for novel antibiotic combinations to modulate the emergence of resistant populations. These observations indicate that further consideration of population-level genome analyses should be incorporated into implementation of stratagems for managing antimicrobial-resistant pathogens.
Introduction
The spread of antimicrobial resistance (AMR) in pathogenic bacteria is one of the key public-health concerns and national security risks of the modern era [1]. The diminution of efficacious treatments for infections has reached crisis point, not only in impoverished areas of the world, where mortality due to bloodstream infections by Gram-negative bacteria is more than double that caused by malaria [2], but also in affluent countries where bacteria with AMR are associated with many thousands of avoidable deaths each year [3, 4]. Resistance has emerged to almost every class of antimicrobial agent that has been developed [5], and the rapid evolution of resistance to these drugs has led to the fear of an end to the antibiotic era, and a return to a situation where even common ailments and injuries may be sufficient to kill [6].
Bacteria of the species Escherichia coli are among the leading causes of serious intestinal and extra-intestinal disease worldwide [7]. Of still greater concern, these bacteria are recognized as amongst the most resistant to antimicrobial agents that have a tradition of use in human or veterinary medicine [8]. E. coli demonstrates intrinsic resistance to a wide range of antimicrobial and toxic compounds, conferred by a combination of proteins that serve as multidrug efflux pumps, including TolC, AcrAB, AcrEF, EmrKY and MdtABC, amongst others [9–14]. Other mechanisms of resistance can include genetic polymorphisms that render the antibiotic's target less sensitive to inhibition [15–18]. However, high-level resistance to specific classes of antibiotics usually requires additional factors, including proteins that facilitate the export, modification or degradation of the antibiotic molecule [18].
The genes that confer AMR are found not only in isolates of human origin, but also in environmental bacteria, those isolated from domestic and wild animals [19–21], and even natural ecosystems or human populations secluded from modern medical or agricultural interventions [22, 23]. However, although AMR is present in diverse environments, the frequency of AMR can be strongly correlated with anthropogenic activity [24–26]. Furthermore, studies consistently demonstrate a trend for the increasing frequency of AMR and associated determinants over time [27–32].
Although the molecular mechanisms of many AMRs are well characterized, investigation of the population genetics of antibiotic resistance within the E. coli species has received less attention. Understanding the diversity and distribution of AMRs in this species is crucial to understand the evolution of AMR and to explore whether this information can help us extend the useable lifespan of antibiotics in the absence of novel discoveries.
Methods
Acquisition, curation and phylogenetic analysis of E. coli genome sequences
We downloaded the nucleotide sequences of 5788 Escherichia and Shigella genomes from GenBank on the 19th October 2016. Source and date of isolation data and reference information available in the GenBank record was collected at the same time. Four strains were excluded because the strain names and BioSample accession numbers (used for indexing the strains) failed to be retrievable. We selected good quality genome sequences from these records based on the following criteria: (1) sequence contained fewer than 0.1 % ambiguous bases (maximum approximately 5000 ambiguous nucleotides specified by the character), (2) assembly comprised fewer than 500 contigs, (3) greater than 3 Mb and less than 7 Mb sequence length (smaller genomes tended to be incomplete, and larger genomes tended to be mixed samples comprising more than one bacterial genome). By applying these criteria, we identified 4084 genomes that we considered good quality. Next, we queried these genomes to ensure they belonged to genuine genus Escherichia genomes. To do this, we probed these genomes for the presence of 4322 gene sequences from the reference E. coli K12 strain MG1655 (U00096) genome using blast [33], and excluded 11 genomes that shared fewer than 2000 genes in common with this sequence. Aside from the excluded sequence for Escherichia vulneris, the remainder of the genome sequences we excluded we suspected were entirely, or at least heavily contaminated with, non-Escherichia or Shigella DNA (see Data bibliography 1).
To determine which among the remaining 4073 sequences represented true species E. coli genomes, we used our data to determine sequences that would be useful for phylogenetic reconstruction of the population structure of genus Escherichia and Shigella genomes. We identified 16 genes that could be reliably recovered from the genomes of all 4073 strains, 80 genes that could be recovered from the genomes of at least 4072 strains, and 201 genes that could be recovered from the genomes of at least 4071 strains. Subsequently, we extracted the nucleotide sequences for the 201 genes, aligned this individually using Muscle [34], concatenated them to one sequence, removed poorly aligned regions using Gblocks [35], and reconstructed a maximum-likelihood tree under the GTR model using RaxML [36]. The resulting tree was investigated to identify branches that fell outside the major E. coli lineages (for example strains more closely related to Escherichia albertii or Escherichia fergusonii) – 51 strains were removed from further analysis (supporting data under Data bibliography 2 shows the position of these genomes relative to E. coli). This resulted in a final population of 4022 E. coli and Shigella sequences that were used in further analysis. E. coli phylogroups were assigned by grouping genomes into the largest monophyletic group that included all known target phylogroup members, whilst excluding all others. Within the GenBank records, strains isolated from similar sources can have different annotations (i.e. human faecal and Homo sapiens stool). To interrogate the source of isolation for each strain, we reduced the disparate annotations to the shortest list we could devise, using the isolation source data and the reference information in the GenBank record, alongside our own knowledge of widely used reference strains where this information was missing in the GenBank record. Our final source of isolation classification is presented alongside the list of genomes used in this study in Data bibliography 3.
Curation of antibiotic-resistance factors
Data from the Comprehensive Antibiotic Database (CARD) [37] was downloaded on the 19th October 2016. The antibiotic resistance ontology (ARO) designation for each gene present in the CARD homologue model was cross-referenced with the ARO index to determine the resistance profile and mechanism for each gene. We supplemented these resistance profiles with information for resistance to olaquindox as resistance determinants (oqxA, oqxB) were present in the CARD, yet the antimicrobial was not. Other supplementation included assigning ‘multiple drug’ classifications to known mediators and regulators of multidrug efflux. Our supplementary curation is provided in supporting data under Data bibliography 4.
Determining the presence of the CARD determinants within the E. coli population
We queried the presence of the genes contained in the protein homologue model of the CARD within the E. coli population by blast. To recover sequences possibly split across contigs, we recovered high-scoring pairs (HSPs) that matched the reference sequence by greater than 80 % identity, where the database sequence covered more than 40 % of the query sequence. If more than one HSP for a given query sequence was recovered from the database sequence, the query sequence participating in the HSP was mapped against the full-length gene, and this mapping used to reconstruct the full-length gene from the database sequence participating in the HSPs. The mean identity of HSPs participating in the mapping and the percentage coverage of the database sequence over the query sequence was used to calculate a sequence similarity score (SSS), which is the mean of sequence identity and the percentage sequence coverage. A matrix of hits we identified in E. coli is provided in supporting data under Data bibliography 5 – these data underlie our analyses. We defined a gene as being present in a genome when the query sequence matched within the genome at a SSS of greater than 80 %. We then assigned the matched genes into two groups: core determinants – genes present in more than 95 % of the E. coli population; and accessory determinants – genes present in fewer than 95 % of the E. coli population. We eliminated four genes from the accessory determinants: (1) the gene encoding the pesticin receptor fyuA, as we could not find any primary literature to indicate this gene played a role in AMR; and (2) the genes mdtM, ermA and pmrE from MG1655. We excluded these latter genes as MG1655 is generally considered sensitive to antibiotics and so we presumed these are unlikely to confer high-level resistance.
Further treatment of the accessory determinants
To ensure we had accurately determined accessory resistance elements rather than related orthologues that might not provide resistance, we purified the genes we had assigned to be present in the accessory genome by requiring them to have a SSS of 98 % or greater. This resulted in the identification of 1029 homologues within the accessory determinants. Since many of the determinants in the CARD are closely-related factors with slightly modified specificities (for example TEM family β-lactamases), we decomposed the accessory resistance determinants into AMR gene families (AMRFs). To do this, we extracted the genes of the accessory resistance determinants from the CARD and, using blast, retrieved a list of gene pairs where those genes had a SSS of 95 %. We examined these gene pairs to ensure each paired AMR determinant conferred resistance to the same profile of antibiotics. In cases where the AMR profiles differed, the pairs were excluded from the network – these were confined to pairs between AAC(6')-Ib-cr (fluoroquinolone and aminoglycoside resistances) and other similar AAC(6') genes that confer resistance only to aminoglycosides [38]. Remaining gene pairs were then passed to the graph building algorithm MCL [39] to build gene families containing networks of homologues. We used these gene families to collapse the accessory resistance determinants for each strain to one call per family – if a strain matched at least one gene in the family network to at least 98 % we called this gene family as present in this strain. The networks we used to generate the AMRFs are presented in supporting data under Data bibliography 6.
Statistical evaluation of AMR carriage
To investigate whether the number of AMRs found in genomes was different to what would be expected if AMR determinants were randomly distributed in genomes, we employed a resampling algorithm. Here, we randomized the distribution of AMRFs over 100 000 replications, and counted the number of resistances these families provided to each strain. We then compared the actual observed number of resistances with this null distribution. Significance was set at 0.0001.
To test for the possibility of bias within the genome sequences – caused by the selection of strains for sequencing based on their clinical importance, or indeed their antibiotic-resistance profile – first we investigated whether the sequenced genomes in the National Center for Biotechnology Information database reasonably represented the diversity of E. coli; that is to say, how closely related could a newly sequenced isolate of E. coli be expected to be to an existing genome in the sequenced database. To do this, we calculated a pairwise distance matrix from the distances between the tips of the phylogenetic tree using the ‘cophenetic.phylo’ function within the ape package [40] in R. We then randomly drew strains from the list of tree tips over increasing sample sizes (from 2 to 4022), took one of these randomly drawn genomes (a proxy for a newly sequenced strain) and recorded the distance between this sequence and the most closely related genome from the sample (a proxy for existing genomes in the database). We repeated this for 10 000 replications per sample size. Next, to investigate whether the source of isolation of E. coli, or the fact that some genomes have been sequenced because of their AMR profile, overly affected our analysis, we employed several variations of our AMR resampling method on samples of genomes isolated from different sources: all strains listed as isolated from non-human and non-unknown sources; all strains where the words ‘antibiotic’, ‘antimicrobial’, ‘resistance’, or ‘resistant’ did not appear in the reference title; all strains listed as from ‘human.urine’, ‘human.blood’, or ‘human.bodyfluid’; all genomes listed as isolated from ‘farms’, ‘cows’, ‘avian’, or ‘sheep’.
Testing the relationship between clonal-lineage and AMR carriage
To investigate the possibility that phylogenetic distance between strains could be correlated with AMR gene carriage, we calculated two distance matrices – one from the phylogenetic tree describing the population structure of E. coli, and one from the distribution of AMRFs. We then sampled 100 000 identical pairwise distances from these matrices, and compared the phylogenetic distance with the AMR distance calculated for each pair. To investigate whether clonal or lineage-related groups of E. coli tended to be more enriched for AMR genes than more disparate strains, we split the population structure of E. coli into two groups – lineage-related strains or non-lineage-related strains – based on the following criteria: (1) lineage-related strains existed on subtrees with 10 or more tips, and (2) the maximum distance observed between any two genomes of the subtree was no greater than 1 % of the maximum distance observed between all E. coli. These criteria split the population of E. coli into two roughly equal groups. We then repeated our resampling experiment for the evaluation of AMR gene carriage for these two groups.
Estimation of significantly associated resistances
To investigate which antibiotic combinations were found in strains more frequently than would be expected by chance, we used our resampling algorithm. In this case, the AMRFs for the population under investigation (the whole population for ‘all strains’, or the genomes isolated up until and including the stated year for our time-course analysis), we randomized over 10 000 replications. At each iteration, for each pair of AMRs, the number of genomes encoding AMRFs directed at these pairs of AMR were counted. The actual observed number of co-resistances in the population was then compared with this null distribution. We considered a pair of resistances to be significant if less than two of the randomizations yielded a count as large or larger than the actual value. These relationships were visualized using the igraph package [41] within R.
Modelling
To simulate the effect of applying antibiotic combinations on a hypothetical bacterial population, we designed the following model. Firstly 100 random genomes were selected from the E. coli population, and the distribution of AMRFs in these genomes recorded. For 1000 generations, the following steps were followed. (1) One random AMRF was chosen for a chance to spread – if the population contained at least one strain with the chosen AMRF, a randomly selected E. coli was donated this determinant. If the population did not contain the AMRF, nothing changed. If the randomly selected E. coli already has this determinant, nothing changed. (2) A randomly selected E. coli was selected, which was then 'exposed' to the specified combination of antibiotics. If the genome was sensitive to any of the specified antibiotics, the genome was removed from the population. If the genome was resistant to all specified antibiotics, it was placed back into the population. (3) If the strain was removed from the population, a randomly selected genome was duplicated in the population, or if the strain was returned to the population, this genome was duplicated. At each generation, the number of AMRFs in the population and the size of the population relative to the start values was recorded. For the simulations, the model was run for 1000 replicates per antibiotic combination.
Results
Core resistome of E. coli is primarily composed of multidrug efflux pumps
The core resistome of E. coli (defined as: 1, homologues of greater than 80 % identity with the reference sequence present in the CARD; and 2, present in 95 % or more strains) comprises 50 genes (shown in supporting data under Data bibliography 7). Although we expected these genes to be fixed within E. coli genomes and not to be subject to horizontal gene transfer (HGT), we were surprised to find six (evgA, evgS, emrK, emrY, gadE and gadX) listed as putatively horizontally transferred genes in the HGT-DB [42] (Data bibliography 8).
However, we are not clear that all the genes listed in the CARD mediate antibiotic resistance in E. coli. Within species such as Streptococcus pneumoniae, patA encodes part of an efflux pump that has been shown to export fluoroquinolones [43]. However, in E. coli, patA, under the accession number listed in the CARD (NP_417544.5), encodes a putrescine-2-oxoglutaric acid aminotransferase enzyme and we could not find any literature describing this gene relating to AMR in E. coli. Similarly, for the mfd gene, which putatively confers resistance to fluoroquinolones in Campylobacter jejuni [44], we could find no literature describing a similar role in E. coli. It is also uncertain that the genes cysB and alaS influence aminocoumarin resistance in the core resistome of E. coli. Variants in both these genes can cause reduced sensitivity to novobiocin [45], and it is possible that our identity criteria have not discriminated the aminocoumarin-resistant variants from their more sensitive counterparts. However, the SSS for these two genes in our analysis tended to be very high, and E. coli has previously been suggested to be intrinsically insensitive to aminocoumarins [46].
Except for these entries, the remaining genes in the core resistome mediate or modulate the efflux of multiple drugs, including the acr, mdt and emr genes [12]. Other genes in the core resistome included the pmr genes, which confer minor alterations to the structure of cell surface lipopolysaccharide and reduce sensitivity to polymyxin class antibiotics, and bacA, which mediates resistance to peptide antibiotics such as bacitracin.
Accessory resistome of E. coli is non-randomly distributed
Within the CARD homologue model sequences, there were 90 AMRFs that matched to sequences found in the E. coli population (shown in supporting data under Data bibliography 9). These mediate resistances to 18 classes of antibiotic. Table 1 summarizes a selection of reported MICs for certain E. coli strains. These resistances around the phylogenetic tree of E. coli are shown in supporting data under Data bibliography 10.
Table 1. Accessory antibiotic resistance in E. coli.
This table lists 14 antibiotic classes to which E. coli has been reported to be sensitive, and the number of AMRFs from the accessory resistome that are active against each of these classes. These antibiotics are abbreviated as follows: KM (kanamycin), SM (streptomycin), NEO (neomycin), GEM (gentamicin), AMP (ampicillin), VQC (vancomycin-QC14), CAM (chloramphenicol), FOS (fosfomycin), CLIN (clindamycin), ERM (erythromycin), OLA (olaquindox), FLOR (florfenicol), HFU (81.723 hfu), COL (colicin), NAL (nalidixic acid), CIPRO (ciprofloxacin), NOR (norfloxacin), RIF (rifamycin), STRG (streptogramin G), STRF (streptothricin F), NOUR (nourseothricin), SUL (sulfamethoxazole), TET (tetracycline), TRI (trimethoprim).
| Antibiotic class | No. of AMRFs | Typical mode of action | Reported MIC (µg ml−1) | Reference |
|---|---|---|---|---|
| Aminoglycosides | 20 | Protein synthesis inhibitors | 2 (KM) 1.95 (SM) 1 (NEO) 0.2 (GEM) |
[9] [81] [81] [81] |
| β-Lactams | 23 | Peptidoglycan biosynthesis inhibitors | 12.5 (AMP) | [9] |
| Glycopeptides | 1 | DNA damaging agents | 4.5 (VQC) | [82] |
| Chloramphenicols | 6 | Protein synthesis inhibitors | 6.25 (CAM) | [9] |
| Fosfomycins | 2 | Peptidoglycan biosynthesis inhibitors | 32 (FOS) | [83] |
| Lincosamides | 2 | Protein synthesis inhibitors | 100 (CLIN) | [84] |
| Macrolides | 8 | Protein synthesis inhibitors | 50 (ERM) | [9] |
| Olaquindox | 2 | DNA synthesis inhibitor | 9 (OLA) | [85] |
| Phenicols | 1 | Protein synthesis inhibitors | 4 (FLOR) | [86] |
| Pleuromutilin | 1 | Protein synthesis inhibitors | 1 (HFU) | [87] |
| Polymyxin | 1 | Membrane disruption | 0.5 (COL) | [82] |
| Quinolones [Q]/fluoroquinolones [F] | 7 | DNA damaging agents | 3.125 (NAL) [Q] 0.01 (CIPRO) [F] 0.004 (NOR) [F] |
[9] [9] [9] |
| Rifampin | 1 | RNA synthesis inhibitors | 2.4 (RIF) | [88] |
| Streptogramin | 3 | Protein synthesis inhibitors | 500 (STRG) | [89] |
| Streptothricins | 1 | Protein synthesis inhibitors | 8 (STRF) 2 (NOUR) |
[81] [81] |
| Sulfonamides | 3 | Dihydropteroate synthetase inhibitors | 8 (SUL) | [90] |
| Tetracyclines | 4 | Protein synthesis inhibitors | 1.25 (TET) | [9] |
| Trimethoprim | 14 | Dihydrofolate reductase inhibitor | 2 (TRI) | [91] |
Different E. coli strains encode multiple different AMR determinants – a fact clearly illustrated in Data bibliography 8 and 9. We were interested in exploring how the distribution of these AMRs translated to the total number of antibiotics each strain was capable of resisting, and so we calculated the number of separate AMRs conferred by the complement of determinants present in each strain (Fig. 1a). These data revealed that AMR was not evenly spread across the E. coli population, with a large proportion of strains within phylogroups C, D, F and Shigella encoding resistance, while resistance in phylogroups A, B1 and, to a lesser degree, B2 was more sporadic. Phylogroup E strains were marked by a very low level of AMR. Clearly, there are many genomes that encode resistance to a considerable number of antimicrobials. To explore whether the proportion of genomes conferring resistance to different numbers of antibiotics was unusual, we performed a re-sampling analysis whereby, assuming the distribution of resistance determinants was randomly assorted between the genomes, we could calculate how many strains we would expect to encode increasing numbers of AMRs, and compared this with the observed number of strains (Fig. 1b).
Fig. 1.

The abundance of antibiotic resistance in the E. coli population. (a) The distribution of the abundance of accessory AMR around the E. coli population. Clearly there is a wide diversity of AMR, with high-level resistances found in most phylogroups, with particularly high concentrations of resistance in phylogroups C, D and F. Phylogroup E (mainly O157 : H7) displays remarkably low levels of accessory resistance. (b) A violin plot for the strains that would be expected to encode the specified number of AMRs if the distribution of resistance genes was randomly distributed amongst the genomes. This can be contrasted with the observed number, represented as a coloured point. An observed number significantly greater than the expected distribution is represented as a red point, whereas an observed number significantly lower than the expected distribution is represented as a blue point (P<0.0001).
These data showed that the numerical abundance of AMR in E. coli clearly deviated from the range we would expect to find if resistance determinants were randomly distributed between the genomes – this expected distribution is shown as violins within Fig. 1(b). The observed value is shown as a coloured point: a red point indicates an observed value significantly greater than expected, whilst a blue point represents an observed value significantly lower than expected. We were also particularly surprised to find such a large proportion of genomes to encode no accessory resistance determinants – almost 45 % (1809 genomes). This was considerably more than if determinants were randomly distributed across strains, where we would expect on average only 232 strains to lack AMR determinants. By contrast, the number of strains encoding one, two, three or even four AMRs was significantly less than would be expected by chance. However, as the number of resistances rose to five or more, we observed a shift in the significance, whereby the number of strains we observed in these groups was significantly greater than chance – in most instances dramatically so. If resistance determinants were randomly distributed through E. coli we would expect, on average, just 169, 37, 6 and 1 genomes to contain five, six, seven or eight AMRs, respectively, and no strains would encode resistance to nine or more AMRs. However, we observed that 570 genomes encoded five AMRs, 195 encoded six AMRs, 114 encoded seven AMRs, 39 encoded eight AMRs, 24 contained nine AMRs, 11 contained ten AMRs, 14 contained eleven AMRs, 2 contained twelve AMRs and even 1 genome encoded resistance to thirteen separate classes of AMR.
Given the sampling of E. coli selected for sequencing is not random, we investigated the possibility that the loading of the sequence database with clinically important strains, for example from human blood or urinary tract infections, had biased our analyses. To do this, we explored several avenues. Firstly, we found that the samples of E. coli that have been sequenced, although not selected in a random fashion, likely saturate the total diversity of E. coli. In Data bibliography 11(a), we show that a newly sequenced E. coli will, on average, be a phylogenetic distance of just 0.0009 from another, already sequenced isolate. To put this distance in context, this is comparable with the distances calculated between several sequenced O157 : H7 genomes, and only slightly greater than the distances calculated between sequences for different K-12 clones. The relative position of this distance value in an ordered list of unique distance values calculated from the tree is shown in Data bibliography 11(b). This observation may mitigate the effect of a biased selection of strains for sequencing, as it indicates against the possibility that sequenced E. coli represent particularly AMR-enriched sub-lineages within the wider population structure of E. coli, which are otherwise unexplored. Secondly, in Data bibliography 11(c), we could show that the results of our analysis did not change substantially by removing all known E. coli isolated from human (and unknown) sources from the analysed population, nor by removing all genomes for which the reference given in the GenBank record contained words related to antibiotic resistance. Furthermore, by limiting our analysed population to strains isolated from sources that were likely to be considered clinically important, such as human blood or urine, or which were unlikely to be considered clinically important, such as those from agricultural land or farmed animals, we still saw these groups had significantly more genomes with no accessory resistance than would be expected if AMR genes were randomly assorted between strains, along with significantly more strains having large numbers of AMR genes within their genomes, regardless of isolation source.
In some other species of bacteria, AMRs are concentrated into specific clonal or lineage-associated groups [47]. However, even though, superficially, some particularly highly sampled clonal groups of E. coli, such as Shigella or O157 : H7, shared similar profiles of AMR, we could not detect a relationship between phylogenetic distance and similarity in AMR gene carriage (Data bibliography 12(a)). To further test the possibility that clonal or lineage-related strains tended to encode high-levels of AMR genes, we separated the population structure of E. coli into two groups – lineage-related strains and non-lineage-related strains – based on the following criteria: (1) lineage-related strains existed within subtrees of the larger phylogenetic tree that contained ten or more strains, and (2) where the maximum distance between any two of those strains was less than 1 % of the maximum distance between any E. coli. The resulting trees reconstructed from these two groups can be seen in Data bibliography 12(b). As shown in Data bibliography 12(c), when we analysed these strains for significant enrichments in AMR gene carriage, the profiles were remarkably similar for both groups as we observed for the whole population. Together, these data indicated that phylogenetic relatedness between E. coli, perhaps surprisingly, does not play a large role in determining the spectrum of AMR genes an individual strain harbours, and furthermore, the highly resistant strains are not concentrated within specific and closely related clonal, or lineage-related, groups.
Incidence of multidrug resistance in the E. coli population has increased over time
Given that the number of AMRs present in E. coli appears to follow a non-random distribution, we speculated that the trend towards concentration of AMRs may be preserved in the genomic 'fossil record'. From the 4021 genome sequences in our panel, 2925 (72.7 %) of these had their year of isolation recorded in the GenBank record – spanning years between 1885 to 2016. We used this information to count the number of AMRs in strains isolated from the same year, and plotted this data in the box-plot shown in Fig. 2. These data revealed a clear positive correlation between successive years’ isolations and increasing AMRs. Investigation of this trend via the Mann– Kendal test revealed a significant trend towards increasing AMR in isolates over time. In fact, not only were the total number of AMRs found in isolates increasing over time, but also each AMR detected has similarly increased (shown in the supporting data under Data bibliography 10).
Fig. 2.
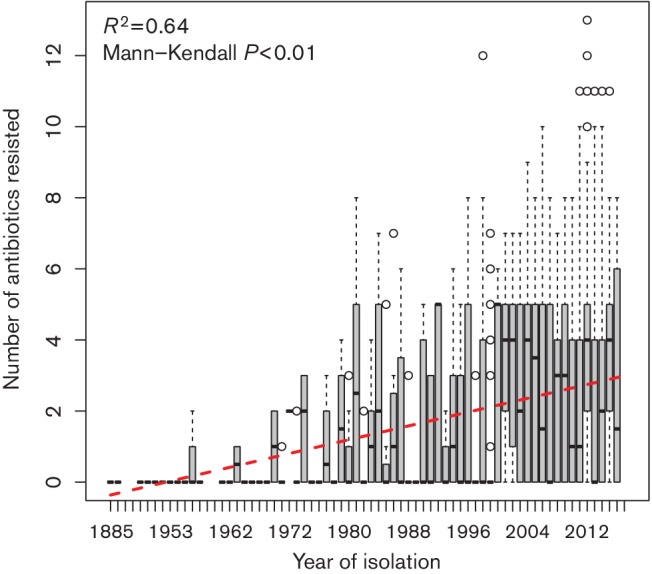
Temporal increase in multi-drug resistance in E. coli. Sequenced E. coli have been collected over time, spanning from 1885 to the present day. The number of antibiotics resisted in strains collected in successive years shows a strong increase over time (Mann–Kendall test P<0.0001). The red dotted trend line is fitted from a linear model (R 2=0.64).
AMR in E. coli is highly interconnected
The concentration of AMR within E. coli strain leads inevitably to the situation whereby treatment of an infection with one antibiotic will probably – if the population of E. coli includes a resistant strain – result in the concomitant population increase in the level of resistance to other, unused antibiotics. This assumption led us to question whether we could detect which antibiotic classes were likely to be encoded together. To do this, we constructed a network of all the co-resistance phenotypes found in the E. coli population. Then, to determine which edges of this network were significant, we randomized the distribution of AMRFs over 10 000 replications and eliminated the edges where at least one of the randomizations resulted in a number of genomes containing both resistances which was as large as – or larger than – the observed value (equivalent to P =0.0001). This analysis revealed a network of AMR genotypes in E. coli that were highly interconnected. Indeed, all 18 classes of AMR found in E. coli were found more often with at least one other class of AMR than would be expected by a random distribution of AMR genes. β-Lactam resistance was the most highly connected AMR, and was significantly associated with 12 other AMR phenotypes, whilst other highly connected AMR phenotypes included trimethoprim (11 connections), sulfonamide (9 connections), aminoglycoside (8 connections) and chloramphenicol (7 connections). A visualization of how this network of antibiotic resistance has evolved over time is presented as an animated picture in supporting data under Data bibliography 14.
Combination of antibiotics reduces the microevolution of resistance in a computer model
As implied in Fig. 3, the use of any antibiotic will likely, when the population contains resistant strains, result in an increase in the level of resistance to multiple antibiotics. What may be needed, therefore, to combat the rise of resistance is to use knowledge of the diversity of E. coli and formulate combinations of antibiotics which, when used together, should be sufficient to kill any E. coli. One of the most pernicious problems with the use of antibiotics is that the effect of the antibiotic is not localized, but is distributed to all resident microbiome bacteria. Ergo, if any of these bacteria encodes resistance to the antimicrobial used, the result will be an increase in the population level of AMR, even if an infection is resolved following the use of antibiotics. By investigating the diversity of AMR in E. coli, we have estimated combinations of three antimicrobials that theoretically should kill all E. coli – with the assumption E. coli are all drawn from the known population (Data bibliography 15). Unsurprisingly, many of these combinations involve those antibiotics where resistance is relatively rare in E. coli.
Fig. 3.

The highly-interconnected network of antibiotic resistance in E. coli. This figure shows a network of AMRs that are more frequently found together in E. coli than would be expected if AMRFs were randomly distributed across genomes. The vertex sizes are proportional to the number of strains that encode the resistance, while the edge widths are proportional to the number of strains that encode both the connected vertices as a function of how many contain either. Edges are coloured for clarity of visual representation of connections .
To demonstrate this effect in a simple simulation, we constructed a model to investigate how the application of antibiotic combinations could change population dynamics in respect of both bacterial and AMR gene numbers. Our model had simple parameters using a sample of strains collected at random from the E. coli population. At each generation, one gene family was randomly selected for the chance to spread. So long as that gene was present in the sampled population, and since our data indicated that antibiotic resistance tends to co-associate, we selected the recipient strain randomly from the subpopulation that already encoded at least one resistance gene. One strain was then chosen as being exposed to a combination of antibiotics. If the strain was resistant to all the selected antibiotics, it was placed back into the population and its genotype was replicated. If the strain was sensitive to at least one antibiotic, it was removed from the population, and a randomly selected genome was chosen to replicate (to prevent the population crashing to zero). We also added an element of openness by allowing, in each generation, a chance for one strain to capture a random AMR gene from outside the population. We linked this chance with the size of the population, so that larger populations had an increased chance of capturing novel genes. At each generation, the mean number of resistance genes found in the population was recorded and calculated as the number of additional resistance determinants in the population compared with the start of the experiment. We also kept track of the number of bacteria in the population, expressed as a fold change from the original population (Fig. 4).
Fig. 4.

Simulated effects of antibiotic combinations on population growth and AMR spread. This figure shows the results of our simulation for commonly used antibiotic combinations (a), and the best [b(ii) and (iii)] and worst [b(iii) and (iv)] performing combinations from our analysis of antibiotic combinations that can kill any E. coli. For commonly used combinations, the population in our simulation rapidly expanded [a (i) and (iii)], and the mean number of resistance determinants in the population quickly increased [a(ii) and (iv)]. For the best performing antibiotic combination in our model (pleuromutilin with polymyxin and glycopeptide), population expansion was minimal [b(i)], while the rate of spread of AMR was low [b(ii)]. For the worst performing combination in our model (streptothricin with macrolide and phenicols), we observed a twofold population increase over 1000 generations [b(iii)], while the mean number of resistances in the population increased substantially [b(iv)].
Fig. 4(a) shows the consequence of two of the most frequently employed combinations of antibiotics – aminoglycosides with β-lactams, and β-lactams with fluoroquinolones – in our model. Since resistance to these antibiotics are so commonly observed together, resistant strains are frequently drawn from the population. This causes the population to quickly grow [Fig 4a (i, iii)]. Furthermore, and likely because these resistances are frequently encoded alongside several other resistances, the mean number of resistances in the population also rapidly expands [Fig 4a (ii, iv)]. The number of antibiotic-resistance genes expands to potentially greater numbers in the β-lactam/fluoroquinolone treatment [Fig. 4a (iv)] than in the aminoglycoside/-lactam treatment [Fig. 4a (iii)].
This can be contrasted with selecting combinations of antibiotics that our previous results indicated should in theory kill any E. coli. We ran our model using all 118 combinations shown in supporting data under Data bibliography 12 to investigate the combinations – per our model parameters – that resulted in the most efficacious killing and the slowest rate of spread of antibiotic-resistance genes (results are shown alongside the combinations used in Data bibliography 12. Our results showed that some combinations of these antibiotics should be more efficacious than others (Fig. 4(b), with one of the most efficacious combinations – pleuromutilin with polymyxin and glycopeptides – outperforming the least effective by a considerable margin in terms of suppressing population growth [compare Fig 4b (i, iii)] and reducing the rate of the spread of resistance genes [compare Fig. 4 b (ii, iv)]. In fact, exposure to pleuromutilin, polymyxin and glycopeptide led to only a marginal population increase over the 1000 generations, indicating the rate at which E. coli gained resistance to all three of these antibiotics in our model was very slow. This was observed alongside the lowest rate of AMR determinant increase for any combination. This is in contrast with the streptothricin, macrolide and phenicol combination, which resulted in at least a twofold increase in the population size at the end of 1000 generations [Fig. 4b (iii)], and approximately six more resistance determinants per strain [Fig. 4b (iv)].
Discussion
Our analyses indicate that all E. coli encode a suite of core resistance genes that presumably facilitate basal levels of non-specific resistance to a variety of antimicrobial compounds. The factors encoded within the core resistome tend to be multidrug efflux pumps and regulators that likely enable E. coli to navigate through environments that contain low levels of a wide variety of toxic molecules, including antibiotics produced by co-resident bacteria and fungi, dyes, free fatty acids, and antimicrobial compounds produced by eukaryotic hosts including bile salts [12–14, 48–50], or even endogenously produced toxic by-products of metabolism [51, 52]. These observations support the assertions put forward by others that the intrinsic resistance of bacteria is due to the activity of multidrug efflux pumps [13, 14].
In addition to the core resistome, many, but not all, E. coli encode accessory resistance determinants. Intriguingly, and despite the perception that E. coli represent amongst the most resistant bacteria to antibiotics commonly employed in medical and veterinary medicine [8], accessory resistance factors are not as frequent in E. coli strains as may be expected, and only slightly more than half of all E. coli encode detectable accessory resistance factors. Phylogroup E isolates tend to encode by far the fewest AMRs and, hence, tend to be resistant to few antibiotic classes. This fact may be explained, at least in part, by phylogroup E representing largely a homogeneous group of E. coli, with a large majority of these isolates drawn from the O157 : H7 genotype, which, it could be argued, is massively oversampled. The general lack of AMRs in the O157 : H7 cluster is contrasted with phylogroup E strains that fall outside this group, such as the O157 : H16 strain Santai (BioSample accession no. SAMN02673556), which encodes resistance to 11 antibiotic classes. Nevertheless, the low abundance of antibiotic resistances in this serotype has also been detected in previous surveys [53]. The low level of resistance in this group of E. coli is accompanied by the knowledge that antibiotic treatment of O157 : H7 infections is known to worsen the disease [54]. However, it is difficult to imagine how this facet alone would lead to a strategy that is evolutionarily successful. Other studies have, however, detected modest [55–57] or even very high [58] levels of antibiotic resistance in O157 E. coli, and so it is possible that antibiotic resistance in this group of E. coli is mediated by cryptic factors that are not represented in the determinants currently present in the CARD. Clearly, the parameters surrounding antibiotic resistance in this group of E. coli remain to be elucidated.
The factors in the accessory genome provide resistance against antibiotic classes that have activity against sensitive E. coli. The antimicrobials present in the CARD homologue model that we could not detect resistance factors for in E. coli are aminococumarins, triclosan, linezolids, elfamycins, fusidic acid, mupirocin and tunicamycin. Most of these antibiotics appear ineffective against E. coli owing to intrinsic insensitivity (aminocoumrin [46] and tunicamycin [59]), efficient efflux (linezolids [60] and fusidic acid [61]) or lack of penetration into the cell (elfamycins [62]). Furthermore, resistance towards mupirocin may be mediated through polymorphisms in the antibiotic target gene [63], which does not appear to be included in the CARD. It is only the antiseptic triclosan that these bacteria do not appear to have yet acquired defined resistance mechanisms, although wild strains of E. coli can vary in their sensitivity to this antimicrobial [64].
The genomes of E. coli that do encode resistance determinants are significantly more likely to contain multiple resistance than would be expected by chance. Indeed, one strain we identified as encoding resistance to 13 of the 18 classes of antibiotic that E. coli may resist. This strain was isolated from the rectum of a pig in China in 2012. Perhaps surprisingly, while it may be expected that this strain would originate within clades recognized for high levels of resistance, such as ST131, this strain belongs to phylogroup A. The massive compliment of AMRFs in this isolate include TEM-91, AAC-6′, ErmB, Oxa-31, CmlA4, FloR, Arr2, Sul2, RmtB, Aph-3′-Ib, MphA, Mrx, Aph-3’−1a, Aac-3-IV, Aac-6′-IB-cr, Sul3, Sul1, DfrA12, Mcr-1, OqxAB, CatB3 and Aph-4-Ia.
The trend for E. coli to accumulate AMR is preserved in the genomes for which the date of isolation has been recorded in GenBank. In many ways, our data showing that AMR has increased over time in E. coli may be unsurprising, as previous studies and meta-analyses of the published literature have shown that the frequency of specific antibiotic resistances and AMRs have been increasing over time in both E. coli and other Gram-negative bacilli [31, 65–71]. However, although it makes sense that a trend for increasing individual resistances would lead to increasing total numbers of resisted antibiotic classes, here we present genomic evidence that the historical increase in antibiotic resistance in E. coli is compounded to make contemporary strains more likely to be MDR strains than their ancestors. The implications of our analysis are dramatic – an average population level increase in resistance to an additional antibiotic every 20 years. E. coli isolated in 2016 are already, on average, resistant to almost three antibiotics, and our trendline predicts that the average E. coli isolated in 20 years are highly likely to be resistant to four agents. This observation supports the contention that high-level multidrug resistance will be an increasing challenge in healthcare over future decades.
Underlying this substantial increase in resistance may be the propensity for resistance determinants to co-associate within genomes, and our data indicates that AMR in E. coli are extensively interconnected. This is consistent with the co-carriage of resistances on plasmids and other mobile elements, although this facet was not a goal in our current investigation. This highly connected network of resistances in E. coli is extremely problematic, since it is indicative that treating a resistant infection with an antibiotic will not just result in the population level increase of resistance to the employed antibiotic, but will also result in the concomitant increase in the level of resistance to several other antibiotics. Furthermore, our visualization of how these relationships have developed over time show an explosion of co-resistances in the early years of the 21st century, and it may not be long before pan-resistance to antimicrobials becomes so frequent that no antibiotic will be useful against many infections. The recent report of a pan-resistant Klebsiella isolate [72] is a salient indication of the potential extent of risk. There is clearly an imperative to address this as indicated in numerous strategy documents from public health organizations and agencies.
Our findings suggest that incorporation of genomic epidemiology and modelling into selecting antibiotics for use in combination therapies presents opportunities to mitigate the spread of antibiotic resistance, as well as increase the probability for the infection to be cured. The use of combinatorial antibiotic therapy is controversial, and meta-analyses on the efficacy of these approaches often show no improvements in mortality rates compared with monotherapies [73–75], other than in high-risk patients [73] or where the infection was caused by Pseudomonas aeruginosa [74], which are characteristically multidrug resistant. Low-risk patients may even have an increased risk of death following combination therapy [73], perhaps due to the reported increase in the risk of complications such as nephrotoxicity [75]. Hence, careful consideration of clinical implications is a further significant issue.
Importantly, what may confound many meta-analyses is information on the appropriateness of the antibiotics used. Inappropriate antibiotic choices (which includes non-susceptibility and lack of timeliness) are associated with worse clinical outcomes, longer hospital stays and higher mortality than appropriate ones [76, 77]. Patients given inappropriate antibiotics can be three times less likely to survive hospitalization following Gram-negative sepsis than those treated appropriately [78]. Appropriate antibiotic choices require understanding of the AMR of individual case isolates, as well as the population genetics of AMR in the pathogen. Several of the meta-analyses showing poor results from combination therapy looked at combinations of β-lactams and aminoglycosides [74, 75], which we find are two of the most commonly resisted – and significantly likely to be resisted together – classes of antibiotic. Of the 2072 E. coli genomes that encode either β-lactam or aminoglycoside resistance over half of all strains – 80 % – encode both. It is possible that poor outcomes in these cases may have resulted from inappropriate antibiotic regimens rather than the failure of the combinatorial approach per se.
Indeed, our model reveals the potential futility of combining drugs such as β-lactams with aminoglycosides or fluoroquinolones, both for controlling infections and for reducing the spread of resistance genes – likely because of the high frequency that these antibiotics are currently co-resisted and in the presence of additional resistance genes – when resistant bacteria multiply following antibiotic challenge this causes the total number of resistance genes in the population to increase massively. Instead, we suggest that using information on the known diversity of AMR in E. coli, we should be choosing combinations of antimicrobials which are rarely, if ever, resisted together. We have identified 118 combinations of antibiotic classes where resistances to all three are not found in E. coli – many of these combinations have complementary targets and modes of action. By applying these combinations in our simulation, we can suggest that some of these combinations may both improve the number of successful treatments and slow the rate at which resistance genes spread in the E. coli population. However, we recognize that the combinations of antibiotics we have indicated may not be clinically practicable.
Combination therapies are not without risk. For example, it has been reported that combination antibiotics may select for broad-spectrum multi-drug resistance, at least in P. aeruginosa where dysregulated efflux pumps appear to drive high levels of resistance following ciprofloxacin (fluoroquinolone) and ceftazidime (β-lactam) treatment [79]. Hence, further evaluation of drug combinations will be necessary to inform any decision-making.
There is a wide diversity of E. coli in the GenBank database. However, and predictably considering the importance of AMR, several studies have sequenced genomes directly associated with resistance. Therefore, the sequenced E. coli population cannot be considered as representing a random sample of the wider population, and the risk of over-estimating AMR in E. coli should be interpreted considering this bias. However, our previous work has indicated that the representation of the diversity of E. coli in sequenced genomes is comprehensive [80], which may mitigate the effect of the bias caused by the selection of AMR strains for sequencing. Irrespective of how representative the 4021 E. coli subjected to this analysis are, the relentless emergence of multi-resistant strains will remain a major healthcare challenge for decades ahead.
Data bibliography
1. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434776 (2016).
2. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434779 (2016).
3. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434782 (2017).
4. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434788 (2016).
5. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434794 (2016).
6. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434797 (2016).
7. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434800 (2016).
8. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4595371 (2017).
9. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434803 (2017).
10. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434809 (2017).
11. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4595389 (2017).
12. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4595377 (2017).
13. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434815 (2017).
14. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434773 (2017).
15. Robert J Goldstone, Figshare https://dx.doi.org/10.6084/m9.figshare.4434818 (2017).
Funding information
The authors acknowledge funding from DEFRA and BBSRC.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
This work used publicly available resources and as such no ethical considerations were necessary.
References
- 1.Smith RD, Coast J. Antimicrobial resistance: a global response. Bull World Health Organ. 2002;80:126–133. [PMC free article] [PubMed] [Google Scholar]
- 2.Cars O, Högberg LD, Murray M, Nordberg O, Sivaraman S, et al. Meeting the challenge of antibiotic resistance. BMJ. 2008;337:a1438. doi: 10.1136/bmj.a1438. [DOI] [PubMed] [Google Scholar]
- 3.de Kraker ME, Davey PG, Grundmann H, on behalf of the BURDEN study group Mortality and hospital stay associated with resistant Staphylococcus aureus and Escherichia coli bacteremia: estimating the burden of antibiotic resistance in Europe. PLoS Med. 2011;8:e1001104. doi: 10.1371/journal.pmed.1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 5.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kåhrström CT. Entering a post-antibiotic era? Nat Rev Microbiol. 2013;11:146. doi: 10.1038/nrmicro2983. [DOI] [Google Scholar]
- 7.Trabulsi LR, Keller R, Tardelli Gomes TA. Typical and atypical enteropathogenic Escherichia coli. Emerg Infect Dis. 2002;8:508–513. doi: 10.3201/eid0805.010385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.FDA US . National Antimicrobial Resistance Monitoring System (NARMS) – Enteric Bacteria: 2008 Executive Report. Rockville, MD: US Food and Drug Administration; 2010. www.fda.gov/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/NationalAntimicrobialResistanceMonitoringSystem/default.htm [Google Scholar]
- 9.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, et al. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother. 2001;45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 2002;184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikaido H, Zgurskaya HI. AcrAB and related multidrug efflux pumps of Escherichia coli. J Mol Microbiol Biotechnol. 2001;3:215–218. [PubMed] [Google Scholar]
- 12.Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178:5853–5859. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikaido H. Multiple antibiotic resistance and efflux. Curr Opin Microbiol. 1998;1:516–523. doi: 10.1016/S1369-5274(98)80083-0. [DOI] [PubMed] [Google Scholar]
- 14.Piddock LJ. Multidrug-resistance efflux pumps — not just for resistance. Nat Rev Microbiol. 2006;4:629–636. doi: 10.1038/nrmicro1464. [DOI] [PubMed] [Google Scholar]
- 15.Sigmund CD, Ettayebi M, Morgan EA. Antibiotic resistance mutations in 16S and 23S ribosomal RNA genes of Escherichia coli. Nucleic Acids Res. 1984;12:4653–4664. doi: 10.1093/nar/12.11.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huovinen P. Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis. 2001;32:1608–1614. doi: 10.1086/320532. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura S, Nakamura M, Kojima T, Yoshida H. gyrA and gyrB mutations in quinolone-resistant strains of Escherichia coli. Antimicrob Agents Chemother. 1989;33:254–255. doi: 10.1128/AAC.33.2.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benveniste R, Davies J. Mechanisms of antibiotic resistance in bacteria. Annu Rev Biochem. 1973;42:471–506. doi: 10.1146/annurev.bi.42.070173.002351. [DOI] [PubMed] [Google Scholar]
- 19.Kozak GK, Boerlin P, Janecko N, Reid-Smith RJ, Jardine C. Antimicrobial resistance in Escherichia coli isolates from swine and wild small mammals in the proximity of swine farms and in natural environments in Ontario, Canada. Appl Environ Microbiol. 2009;75:559–566. doi: 10.1128/AEM.01821-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen SE, Boerlin P, Janecko N, Lumsden JS, Barker IK, et al. Antimicrobial resistance in generic Escherichia coli isolates from wild small mammals living in swine farm, residential, landfill, and natural environments in southern Ontario, Canada. Appl Environ Microbiol. 2011;77:882–888. doi: 10.1128/AEM.01111-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sayah RS, Kaneene JB, Johnson Y, Miller R. Patterns of antimicrobial resistance observed in Escherichia coli isolates obtained from domestic- and wild-animal fecal samples, human septage, and surface water. Appl Environ Microbiol. 2005;71:1394–1404. doi: 10.1128/AEM.71.3.1394-1404.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clemente JC, Pehrsson EC, Blaser MJ, Sandhu K, Gao Z, et al. The microbiome of uncontacted Amerindians. Sci Adv. 2015;1:e1500183. doi: 10.1126/sciadv.1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, et al. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One. 2012;7:e34953. doi: 10.1371/journal.pone.0034953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lescat M, Clermont O, Woerther PL, Glodt J, Dion S, et al. Commensal Escherichia coli strains in Guiana reveal a high genetic diversity with host-dependant population structure. Environ Microbiol Rep. 2013;5:49–57. doi: 10.1111/j.1758-2229.2012.00374.x. [DOI] [PubMed] [Google Scholar]
- 25.Pruden A, Arabi M, Storteboom HN. Correlation between upstream human activities and riverine antibiotic resistance genes. Environ Sci Technol. 2012;46:11541–11549. doi: 10.1021/es302657r. [DOI] [PubMed] [Google Scholar]
- 26.Skurnik D, Ruimy R, Andremont A, Amorin C, Rouquet P, et al. Effect of human vicinity on antimicrobial resistance and integrons in animal faecal Escherichia coli. J Antimicrob Chemother. 2006;57:1215–1219. doi: 10.1093/jac/dkl122. [DOI] [PubMed] [Google Scholar]
- 27.Blaettler L, Mertz D, Frei R, Elzi L, Widmer AF, et al. Secular trend and risk factors for antimicrobial resistance in Escherichia coli isolates in Switzerland 1997-2007. Infection. 2009;37:534–539. doi: 10.1007/s15010-009-8457-0. [DOI] [PubMed] [Google Scholar]
- 28.Kronvall G. Antimicrobial resistance 1979-2009 at Karolinska hospital, Sweden: normalized resistance interpretation during a 30-year follow-up on Staphylococcus aureus and Escherichia coli resistance development. APMIS. 2010;118:621–639. doi: 10.1111/j.1600-0463.2010.02660.x. [DOI] [PubMed] [Google Scholar]
- 29.Mcewen SA, Fedorka‐Cray PJ. Antimicrobial use and resistance in animals. Clin Infect Dis. 2002;34:S93–S106. doi: 10.1086/340246. [DOI] [PubMed] [Google Scholar]
- 30.Jensen VF, Jakobsen L, Emborg HD, Seyfarth AM, Hammerum AM. Correlation between apramycin and gentamicin use in pigs and an increasing reservoir of gentamicin-resistant Escherichia coli. J Antimicrob Chemother. 2006;58:101–107. doi: 10.1093/jac/dkl201. [DOI] [PubMed] [Google Scholar]
- 31.Tadesse DA, Zhao S, Tong E, Ayers S, Singh A, et al. Antimicrobial drug resistance in Escherichia coli from humans and food animals, United States, 1950-2002. Emerg Infect Dis. 2012;18:741–749. doi: 10.3201/eid1805.111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Datta S, Wattal C, Goel N, Oberoi JK, Raveendran R, et al. A ten year analysis of multi-drug resistant blood stream infections caused by Escherichia coli & Klebsiella pneumoniae in a tertiary care hospital. Indian J Med Res. 2012;135:907–912. [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 36.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 37.Mcarthur AG, Waglechner N, Nizam F, Yan A, Azad MA, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, et al. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med. 2006;12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 39.van Dongen SM. Graph clustering by flow simulation. 2000. PhD thesis, University of Utrecht, The Netherlands; [Google Scholar]
- 40.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 41.Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal. 2006:1695. [Google Scholar]
- 42.Garcia-Vallve S, Guzman E, Montero MA, Romeu A. HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003;31:187–189. doi: 10.1093/nar/gkg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boncoeur E, Durmort C, Bernay B, Ebel C, di Guilmi AM, et al. PatA and PatB form a functional heterodimeric ABC multidrug efflux transporter responsible for the resistance of Streptococcus pneumoniae to fluoroquinolones. Biochemistry. 2012;51:7755–7765. doi: 10.1021/bi300762p. [DOI] [PubMed] [Google Scholar]
- 44.Han J, Sahin O, Barton YW, Zhang Q. Key role of Mfd in the development of fluoroquinolone resistance in Campylobacter jejuni. PLoS Pathog. 2008;4:e1000083. doi: 10.1371/journal.ppat.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rakonjac J, Milic M, Savic DJ. cysB and cysE mutants of Escherichia coli K12 show increased resistance to novobiocin. Mol Gen Genet. 1991;228:307–311. doi: 10.1007/BF00282481. [DOI] [PubMed] [Google Scholar]
- 46.Alt S, Mitchenall LA, Maxwell A, Heide L. Inhibition of DNA gyrase and DNA topoisomerase IV of Staphylococcus aureus and Escherichia coli by aminocoumarin antibiotics. J Antimicrob Chemother. 2011;66:2061–2069. doi: 10.1093/jac/dkr247. [DOI] [PubMed] [Google Scholar]
- 47.Doern GV, Brueggemann AB, Blocker M, Dunne M, Holley HP, et al. Clonal relationships among high-level penicillin-resistant Streptococcus pneumoniae in the United States. Clin Infect Dis. 1998;27:757–761. doi: 10.1086/514937. [DOI] [PubMed] [Google Scholar]
- 48.Zgurskaya HI, Nikaido H. Multidrug resistance mechanisms: drug efflux across two membranes. Mol Microbiol. 2000;37:219–225. doi: 10.1046/j.1365-2958.2000.01926.x. [DOI] [PubMed] [Google Scholar]
- 49.Rosenberg EY, Ma D, Nikaido H. AcrD of Escherichia coli is an aminoglycoside efflux pump. J Bacteriol. 2000;182:1754–1756. doi: 10.1128/JB.182.6.1754-1756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 51.Saier MH, Paulsen IT, Sliwinski MK, Pao SS, Skurray RA, et al. Evolutionary origins of multidrug and drug-specific efflux pumps in bacteria. FASEB J. 1998;12:265–274. doi: 10.1096/fasebj.12.3.265. [DOI] [PubMed] [Google Scholar]
- 52.Paulsen IT. Multidrug efflux pumps and resistance: regulation and evolution. Curr Opin Microbiol. 2003;6:446–451. doi: 10.1016/j.mib.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Stanford K, Agopsowicz CA, Mcallister TA. Genetic diversity and antimicrobial resistance among isolates of Escherichia coli O157: H7 from feces and hides of super-shedders and low-shedding pen-mates in two commercial beef feedlots. BMC Vet Res. 2012;8:178. doi: 10.1186/1746-6148-8-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342:1930–1936. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meng J, Zhao S, Doyle MP, Joseph SW. Antibiotic resistance of Escherichia coli O157:H7 and O157:NM isolated from animals, food, and humans. J Food Prot. 1998;61:1511–1514. doi: 10.4315/0362-028X-61.11.1511. [DOI] [PubMed] [Google Scholar]
- 56.Galland JC, Hyatt DR, Crupper SS, Acheson DW, Prevalence ADW. Prevalence, antibiotic susceptibility, and diversity of Escherichia coli O157:H7 isolates from a longitudinal study of beef cattle feedlots. Appl Environ Microbiol. 2001;67:1619–1627. doi: 10.1128/AEM.67.4.1619-1627.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mora A, Blanco JE, Blanco M, Alonso MP, Dhabi G, et al. Antimicrobial resistance of Shiga toxin (verotoxin)-producing Escherichia coli O157:H7 and non-O157 strains isolated from humans, cattle, sheep and food in Spain. Res Microbiol. 2005;156:793–806. doi: 10.1016/j.resmic.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Radu S, Ling OW, Rusul G, Karim MI, Nishibuchi M. Detection of Escherichia coli O157:H7 by multiplex PCR and their characterization by plasmid profiling, antimicrobial resistance, RAPD and PFGE analyses. J Microbiol Methods. 2001;46:131–139. doi: 10.1016/S0167-7012(01)00269-X. [DOI] [PubMed] [Google Scholar]
- 59.Barbosa MD, Yang G, Fang J, Kurilla MG, Pompliano DL. Development of a whole-cell assay for peptidoglycan biosynthesis inhibitors. Antimicrob Agents Chemother. 2002;46:943–946. doi: 10.1128/AAC.46.4.943-946.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schumacher A, Trittler R, Bohnert JA, Kümmerer K, Pagès JM, et al. Intracellular accumulation of linezolid in Escherichia coli, Citrobacter freundii and Enterobacter aerogenes: role of enhanced efflux pump activity and inactivation. J Antimicrob Chemother. 2007;59:1261–1264. doi: 10.1093/jac/dkl380. [DOI] [PubMed] [Google Scholar]
- 61.Ma D, Cook DN, Hearst JE, Nikaido H. Efflux pumps and drug resistance in gram-negative bacteria. Trends Microbiol. 1994;2:489–493. doi: 10.1016/0966-842X(94)90654-8. [DOI] [PubMed] [Google Scholar]
- 62.Hall CC, Watkins JD, Georgopapadakou NH. Effects of elfamycins on elongation factor Tu from Escherichia coli and Staphylococcus aureus. Antimicrob Agents Chemother. 1989;33:322–325. doi: 10.1128/AAC.33.3.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yanagisawa T, Lee JT, Wu HC, Kawakami M. Relationship of protein structure of isoleucyl-tRNA synthetase with pseudomonic acid resistance of Escherichia coli. A proposed mode of action of pseudomonic acid as an inhibitor of isoleucyl-tRNA synthetase. J Biol Chem. 1994;269:24304–24309. [PubMed] [Google Scholar]
- 64.Assadian O, Wehse K, Hübner NO, Koburger T, Bagel S, et al. Minimum inhibitory (MIC) and minimum microbicidal concentration (MMC) of polihexanide and triclosan against antibiotic sensitive and resistant Staphylococcus aureus and Escherichia coli strains. GMS Krankenhhyg Interdiszip. 2011;6:Doc06. doi: 10.3205/dgkh000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Erb A, Stürmer T, Marre R, Brenner H. Prevalence of antibiotic resistance in Escherichia coli: overview of geographical, temporal, and methodological variations. Eur J Clin Microbiol Infect Dis. 2007;26:83–90. doi: 10.1007/s10096-006-0248-2. [DOI] [PubMed] [Google Scholar]
- 66.Johnson L, Sabel A, Burman WJ, Everhart RM, Rome M, et al. Emergence of fluoroquinolone resistance in outpatient urinary Escherichia coli isolates. Am J Med. 2008;121:876–884. doi: 10.1016/j.amjmed.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 67.Zilberberg MD, Shorr AF. Secular trends in gram-negative resistance among urinary tract infection hospitalizations in the United States, 2000-2009. Infect Control Hosp Epidemiol. 2013;34:940–946. doi: 10.1086/671740. [DOI] [PubMed] [Google Scholar]
- 68.Spadafino JT, Cohen B, Liu J, Larson E. Temporal trends and risk factors for extended-spectrum beta-lactamase-producing Escherichia coli in adults with catheter-associated urinary tract infections. Antimicrob Resist Infect Control. 2014;3:39. doi: 10.1186/s13756-014-0039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim HB, Park CH, Kim CJ, Kim EC, Jacoby GA, et al. Prevalence of plasmid-mediated quinolone resistance determinants over a 9-year period. Antimicrob Agents Chemother. 2009;53:639–645. doi: 10.1128/AAC.01051-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wong PH, von Krosigk M, Roscoe DL, Lau TT, Yousefi M, et al. Antimicrobial co-resistance patterns of gram-negative bacilli isolated from bloodstream infections: a longitudinal epidemiological study from 2002-2011. BMC Infect Dis. 2014;14:393. doi: 10.1186/1471-2334-14-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, et al. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J Clin Microbiol. 2007;45:3352–3359. doi: 10.1128/JCM.01284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L, Todd R, Kiehlbauch J, Walters M, Kallen A. Notes from the field: pan-resistant new delhi Metallo-Beta-Lactamase-Producing Klebsiella pneumoniae - Washoe county, Nevada, 2016. MMWR Morb Mortal Wkly Rep. 2017;66:33. doi: 10.15585/mmwr.mm6601a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar A, Safdar N, Kethireddy S, Chateau D. A survival benefit of combination antibiotic therapy for serious infections associated with sepsis and septic shock is contingent only on the risk of death: a meta-analytic/meta-regression study. Crit Care Med. 2010;38:1651–1664. doi: 10.1097/CCM.0b013e3181e96b91. [DOI] [PubMed] [Google Scholar]
- 74.Safdar N, Handelsman J, Maki DG. Does combination antimicrobial therapy reduce mortality in Gram-negative bacteraemia? A meta-analysis. Lancet Infect Dis. 2004;4:519–527. doi: 10.1016/S1473-3099(04)01108-9. [DOI] [PubMed] [Google Scholar]
- 75.Paul M, Lador A, Grozinsky-Glasberg S, Leibovici L. Beta lactam antibiotic monotherapy versus beta lactam-aminoglycoside antibiotic combination therapy for sepsis. Cochrane Database Syst Rev. 2014;1:Cd003344. doi: 10.1002/14651858.CD003344.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raman G, Avendano E, Berger S, Menon V. Appropriate initial antibiotic therapy in hospitalized patients with gram-negative infections: systematic review and meta-analysis. BMC Infect Dis. 2015;15:395. doi: 10.1186/s12879-015-1123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Micek ST, Lloyd AE, Ritchie DJ, Reichley RM, Fraser VJ, et al. Pseudomonas aeruginosa bloodstream infection: importance of appropriate initial antimicrobial treatment. Antimicrob Agents Chemother. 2005;49:1306–1311. doi: 10.1128/AAC.49.4.1306-1311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zilberberg MD, Shorr AF, Micek ST, Vazquez-Guillamet C, Kollef MH. Multi-drug resistance, inappropriate initial antibiotic therapy and mortality in Gram-negative severe sepsis and septic shock: a retrospective cohort study. Crit Care. 2014;18:596. doi: 10.1186/s13054-014-0596-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vestergaard M, Paulander W, Marvig RL, Clasen J, Jochumsen N, et al. Antibiotic combination therapy can select for broad-spectrum multidrug resistance in Pseudomonas aeruginosa. Int J Antimicrob Agents. 2016;47:48–55. doi: 10.1016/j.ijantimicag.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 80.Goldstone RJ, Harris S, Smith DG. Genomic content typifying a prevalent clade of bovine mastitis-associated Escherichia coli. Sci Rep. 2016;6:30115. doi: 10.1038/srep30115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haupt I, Thrum H. Bacterial resistance to streptothricins. J Basic Microbiol. 1985;25:335–339. doi: 10.1002/jobm.3620250508. [DOI] [PubMed] [Google Scholar]
- 82.Yarlagadda V, Manjunath GB, Sarkar P, Akkapeddi P, Paramanandham K, et al. Glycopeptide antibiotic to overcome the intrinsic resistance of Gram-negative bacteria. ACS Infect Dis. 2016;2:132–139. doi: 10.1021/acsinfecdis.5b00114. [DOI] [PubMed] [Google Scholar]
- 83.de Cueto M, López L, Hernández JR, Morillo C, Pascual A. In vitro activity of fosfomycin against extended-spectrum-β-lactamase-producing Escherichia coli and Klebsiella pneumoniae: comparison of susceptibility testing procedures. Antimicrob Agents Chemother. 2006;50:368–370. doi: 10.1128/AAC.50.1.368-370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Douthwaite S. Interaction of the antibiotics clindamycin and lincomycin with Escherichia coli 23S ribosomal RNA. Nucleic Acids Res. 1992;20:4717–4720. doi: 10.1093/nar/20.18.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Suter W, Rosselet A, Knüsel F. Mode of action of quindoxin and substituted quinoxaline-di-N-oxides on Escherichia coli. Antimicrob Agents Chemother. 1978;13:770–783. doi: 10.1128/AAC.13.5.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Salmon SA, Watts JL. Minimum inhibitory concentration determinations for various antimicrobial agents against 1570 bacterial isolates from turkey poults. Avian Dis. 2000;44:85–98. doi: 10.2307/1592511. [DOI] [PubMed] [Google Scholar]
- 87.Drews J, Georgopoulos A, Laber G, Schütze E, Unger J. Antimicrobial activities of 81.723 hfu, a new pleuromutilin derivative. Antimicrob Agents Chemother. 1975;7:507–516. doi: 10.1128/AAC.7.5.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodríguez-Verdugo A, Gaut BS, Tenaillon O. Evolution of Escherichia coli rifampicin resistance in an antibiotic-free environment during thermal stress. BMC Evol Biol. 2013;13:50. doi: 10.1186/1471-2148-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Andremont A, Gerbaud G, Courvalin P. Plasmid-mediated high-level resistance to erythromycin in Escherichia coli. Antimicrob Agents Chemother. 1986;29:515–518. doi: 10.1128/AAC.29.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perreten V, Boerlin P. A new sulfonamide resistance gene (sul3) in Escherichia coli is widespread in the pig population of Switzerland. Antimicrob Agents Chemother. 2003;47:1169–1172. doi: 10.1128/AAC.47.3.1169-1172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aagaard J, Gasser T, Rhodes P, Madsen PO. MICs of ciprofloxacin and trimethoprim for Escherichia coli: influence of pH, inoculum size and various body fluids. Infection. 1991;19:S167–S169. doi: 10.1007/BF01643691. [DOI] [PubMed] [Google Scholar]