

Abstract
Objective:
To analyze a glioblastoma tumor specimen with 3 different platforms and compare potentially actionable calls from each.
Methods:
Tumor DNA was analyzed by a commercial targeted panel. In addition, tumor-normal DNA was analyzed by whole-genome sequencing (WGS) and tumor RNA was analyzed by RNA sequencing (RNA-seq). The WGS and RNA-seq data were analyzed by a team of bioinformaticians and cancer oncologists, and separately by IBM Watson Genomic Analytics (WGA), an automated system for prioritizing somatic variants and identifying drugs.
Results:
More variants were identified by WGS/RNA analysis than by targeted panels. WGA completed a comparable analysis in a fraction of the time required by the human analysts.
Conclusions:
The development of an effective human-machine interface in the analysis of deep cancer genomic datasets may provide potentially clinically actionable calls for individual patients in a more timely and efficient manner than currently possible.
ClinicalTrials.gov identifier:
The clinical application of next-generation sequencing technology to cancer diagnosis and treatment is in its early stages.1–3 An initial implementation of this technology has been in targeted panels, where subsets of cancer-relevant and/or highly actionable genes are scrutinized for potentially actionable mutations. This approach has been widely adopted, offering high redundancy of sequence coverage for the small number of sites of known clinical utility at relatively low cost.
However, recent studies have shown that many more potentially clinically actionable mutations exist both in known cancer genes and in other genes not yet identified as cancer drivers.4,5 Improvements in the efficiency of next-generation sequencing make it possible to consider whole-genome sequencing (WGS) as well as other omic assays such as RNA sequencing (RNA-seq) as clinical assays, but uncertainties remain about how much additional useful information is available from these assays.
Aside from cost, a challenge of WGS or whole-transcriptome data is the expertise and time required to interpret the full spectrum of somatic mutations. To address this challenge, Watson for Genomics (Watson Genomic Analytics [WGA]), a cancer analytic tool, uses standard variant call files (VCFs), copy number variant (CNV), and differential gene expression data to return a list of recommended cancer drugs. Here, we present the results of a targeted cancer panel along with WGS and RNA-seq in a patient with glioblastoma (GBM). We also compare results of expert interpretation of the tumor genome by bioinformaticians and oncologists at New York Genome Center (NYGC) and at collaborating institutions with those generated by WGA.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by multiple institutional review boards (IRBs), including Rockefeller University IRB and Biomedical Research Alliance of New York IRB. The study was registered in ClinicalTrials.gov (NCT02725684). Informed written consent was obtained from the participant.
Participant.
This report describes the first participant in a multi-institutional study. NYGC-GBM-01 was a 76-year-old man with GBM. DNA and RNA were extracted from snap-frozen tissue. DNA from blood was obtained for comparison. The samples were analyzed by WGS and RNA-seq.
Single nucleotide variants and INDELs.
Whole-genome libraries were prepared using the Illumina TruSeq Nano DNA Sample Prep Kit and were sequenced on Illumina HiSeq X instruments (Illumina, San Diego, CA). Paired-end 2 × 150 bp reads were aligned to the GRCh37 human reference (BWA aln v.0.7.8)6 and processed using a pipeline that includes marking of duplicate reads using Picard tools and realignment around INDELs and base recalibration using Genome Analysis Toolkit (GATK) version 2.7.4.7 muTect v1.1.4,8 LoFreq v2.0.0,9 Strelka v1.0.13,10 Pindel,11 and Scalpel12 were used to return the union of variant calls. Variants were filtered out if they were at >1% frequency in the 1000 Genomes or ExAC data sets, had more than 2 alleles to remove artifacts, raw frequency in the tumor was lower than that in the normal, or matched a custom “blacklist” of known systematic errors generated by comparing normal germline replicates. Remaining single nucleotide variants (SNVs) and INDELs were annotated via snpEff,13 snpSift,13 and GATK VariantAnnotator using annotation from ENSEMBL,14 COSMIC,15 Gene Ontology,16 and 1000 Genomes.17
Structural variation.
Structural variants (SVs), such as CNVs and complex genomic rearrangements, were detected by NBIC-seq,18 Delly,19 CREST,20 and BreakDancer.21 We prioritized SVs in the intersection of callers and those with additional split-read evidence via SplazerS.22 SVs with split-read support in the matched normal or annotated as known germline variants (1000 Genomes call set, Database of Genomic Variants) were removed as likely germline variants. The predicted somatic SVs were annotated with gene overlap (RefSeq, Cancer Gene Census) including prediction of potential effect on resulting proteins.
Tumor purity and ploidy.
Tumor purity was calculated from WGS data using Titan.23 In addition, purity and ploidy were calculated from the Illumina OMNI 2.5M Array using ASCAT.24
RNA sequencing.
We used the Illumina TruSeq stranded messenger RNA protocol and sequenced 100 million reads. Reads were aligned using STAR25 and Gencode genes were quantified using featureCounts.26 Ninety-five percent of reads mapped the reference genome. We normalized the counts with DESeq2 and adjusted the quantification to account for GC bias27 and batch effects28 between The Cancer Genome Atlas (TCGA) GBM RNA-seq and our sample. The normalized expression data are used to identify GBM subtypes.29
Therapeutic targets and drug recommendations.
The NYGC uses the custom clinical Tier classification system for SNVs. Tier 1 variants are clinically important variants in the cancer type being studied (e.g., epidermal growth factor receptor [EGFR] T790M is known to be clinically important in lung cancer30). The same variant observed in a cancer unknown to manifest this variant is classified as Tier 2 (e.g., the clinical importance of EGFR T790M is unknown in GBM). Tier 3 variants are in targetable genes; however, the specific variant is not known to be targetable (e.g., an unknown mutation in EGFR). Tier 4 variants are in genes cataloged by COSMIC cancer census and not included in Tiers 1–3.15 All other variants are in Tier 5 and considered variants of uncertain significance (VUS). Variants in Tiers 1–4 are considered potentially targetable. Variants were matched to potential treatments by identifying the most aberrant genes from a combination of SNV, INDEL, SV, and RNA-seq data and by searching the NYGC drug-to-gene database. Prioritization of potential treatments was based on further manual assessment including criteria such as strength of data supporting variants detected, FDA approval of drug in GBM or in another cancer type, current GBM trial for a drug, and successful use of the drug to target the variant identified to treat GBM or other cancer types.
Watson Genomic Analytics.
WGA, an IBM research proof-of-concept environment of Watson for Genomics,31 is a cognitive system built on several different predictive models to analyze up to whole-genome scale molecular data. VCFs, CNV, and gene expression data are input to WGA. The VCF file provided to WGA contains the union from 3 calling algorithms each for SNVs and INDELs specified in the Methods section. CNV data are inputted as copy number log2 (T/N) ratio values per gene. Modified Z-scores of RNA-seq normalized expression data per gene are used as proxy for differential gene expression. Modified z-score per gene is calculated by subtracting the median transcripts per million (TPM) value (over the TCGA GBM cohort) to this sample's TPM and dividing by the TCGA SD. With this input, WGA leverages a comprehensive database of structured (20+ sources include DrugBank, NCI, COSMIC, ClinVar, and 1000 Genomes) and unstructured (evidence extracted from literature using Natural Language Processing [NLP]) biological and medical data. To date, WGA processed abstracts from PubMed and where possible, began analyzing full-text articles. In addition, the NLP engine is being trained to understand the approximate 5,600 clinical trials at ClinicalTrials.gov. It is from the unstructured sources that WGA maintains a current repository of drug-disease associations and biomarkers for prognosis and therapeutics, as well as matching patients to relevant clinical trials based on molecular criteria. WGA identifies gene alterations most likely to be important in cancer and then identifies relevant treatments that directly or indirectly target the variant. WGA also identifies VUS, resistive or sensitizing markers for the drug of interest, and relevant clinical trials.
RESULTS
Case report.
NYGC-GBM-01 was a 76-year-old man who presented with headache and difficulty with ambulation. CT of the brain revealed a mass in the left parietal region. He underwent initial resection for which pathology revealed a GBM, negative for the following: EGFR amplification by in situ hybridization fluorescence, EGFRvIII RNA expression, IDH1 R132H by immunostaining, 1p36/19q13 deletion by fluorescent in situ hybridization analysis, and MGMT methylation. Postsurgically, he had right-sided hemineglect and right/left confusion. He became somnolent and required a re-resection and ventriculoperitoneal shunt placement. Two months after initial resection, he completed radiation therapy with 40 Gy over 3 weeks with concurrent temozolomide 75 mg/m2 daily. He then completed 3 cycles of adjuvant temozolomide at 5 months after initial resection, after which progression was seen on MRI. He considered multiple options, but experienced functional decline, and was no longer trial eligible. Instead, he started on the first dose of bevacizumab and CCNU, 7 months after initial resection. He further declined and died 1 month later.
A sample from the initial resection was examined with a FoundationOne test, and a snap-frozen sample was received for sequencing in this study. DNA and RNA extraction, sequencing, and analysis required 7 weeks at which time a tumor board meeting was convened, including the treating oncologist, a neuro-oncologist, and bioinformaticians, to discuss the results of the analysis completed by the NYGC. A clinical report of the findings was subsequently issued to the oncologist. This tumor board meeting occurred after the completion of the first cycle of adjuvant temozolomide. The oncologist planned on referring the patient to clinical trials identified by the NYGC, but at the time of progression, he was no longer trial eligible due to functional decline.
Tumor analysis.
The metrics for the WGS are shown in table 1. The sample had an estimated tumor purity of 47%–52% and ploidy of 1.99. Table 2 describes the types and number of variants identified. Specifically, variant-calling pipeline analysis identified 8,449 total somatic mutations (with 150 falling in exonic, protein-coding regions) and a complex landscape of amplifications and deletions. Mutational signatures are an important molecular characterization of the tumor and assessing applicability of immunotherapy,32 for example, RNA-seq identified the sample as the mesenchymal subtype of GBM.28 WGS supported this with evidence of an NF1 mutation and a CDKN2A loss with a gain of Chr 7 and a loss of Chr 10.
Table 1.
Sample whole-genome sequencing metrics
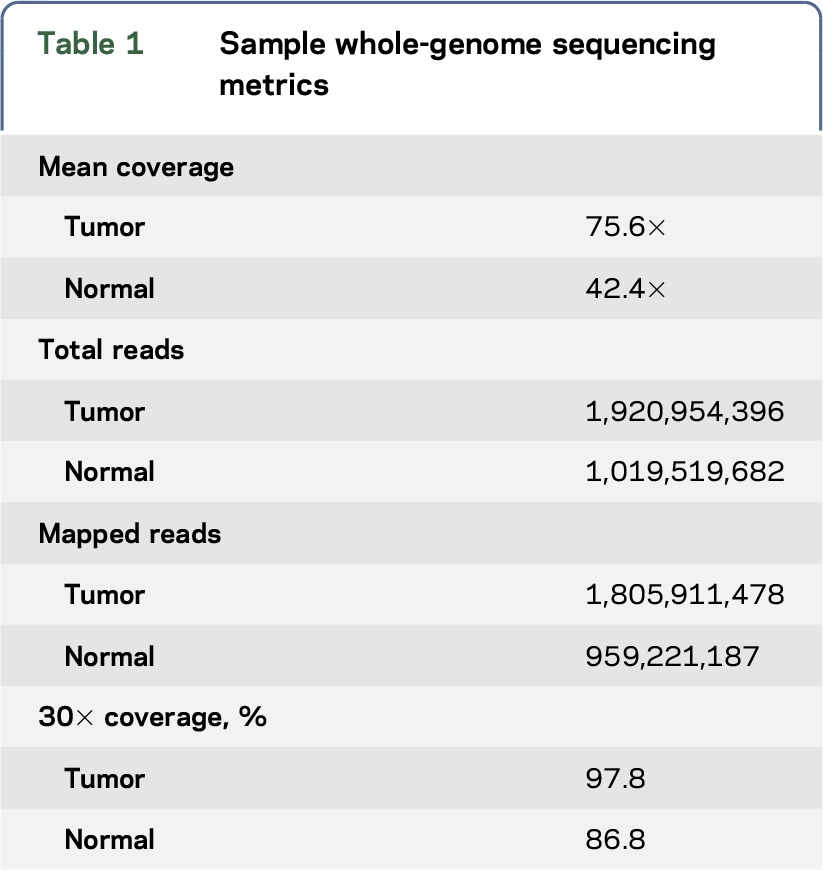
Table 2.
Number of somatic variants identified

The NYGC identified 6 actionable SNVs, of which 2 were Tier 3 variants (MET R755fs and FGFR3 L49V) and 4 were Tier 4 variants (in STAG2, PIK3R1, NF1, and ERG, described in table e-1 at Neurology.org/ng). In addition, 5 CNVs were identified, of which 2 were in genes that had SNVs (table 3). CREST and Pindel identified a 299-bp intragenic deletion at the intron-exon junction of exon 11 in MET, as well as an amplification of MET (log2 CNV 3.64-fold tumor vs normal amplification). RNA-seq confirmed overexpression of MET (z-score 2.23) and an in-frame exon-skipping event (METex11), at an allele frequency of approximately 50% (figure 1). This observation is molecularly analogous to the skipping of exon 14 identified in lung adenocarcinoma and other cancer types.33 Although METex11 is located in the extracellular domain, we hypothesized that this mutation could lead to an overactivation of MET and could be targeted by a tyrosine kinase inhibitor. By analogy with studies of MET(Δ7–8),34 both would lead to the lack of transmembrane localization. We also noted that mislocalization of MET(Δ7–8) renders the variant not targetable using antibodies. However, MET-specific tyrosine kinase inhibitors could efficiently deactivate the kinase.
Table 3.
List of variants identified as actionable by 3 different platforms
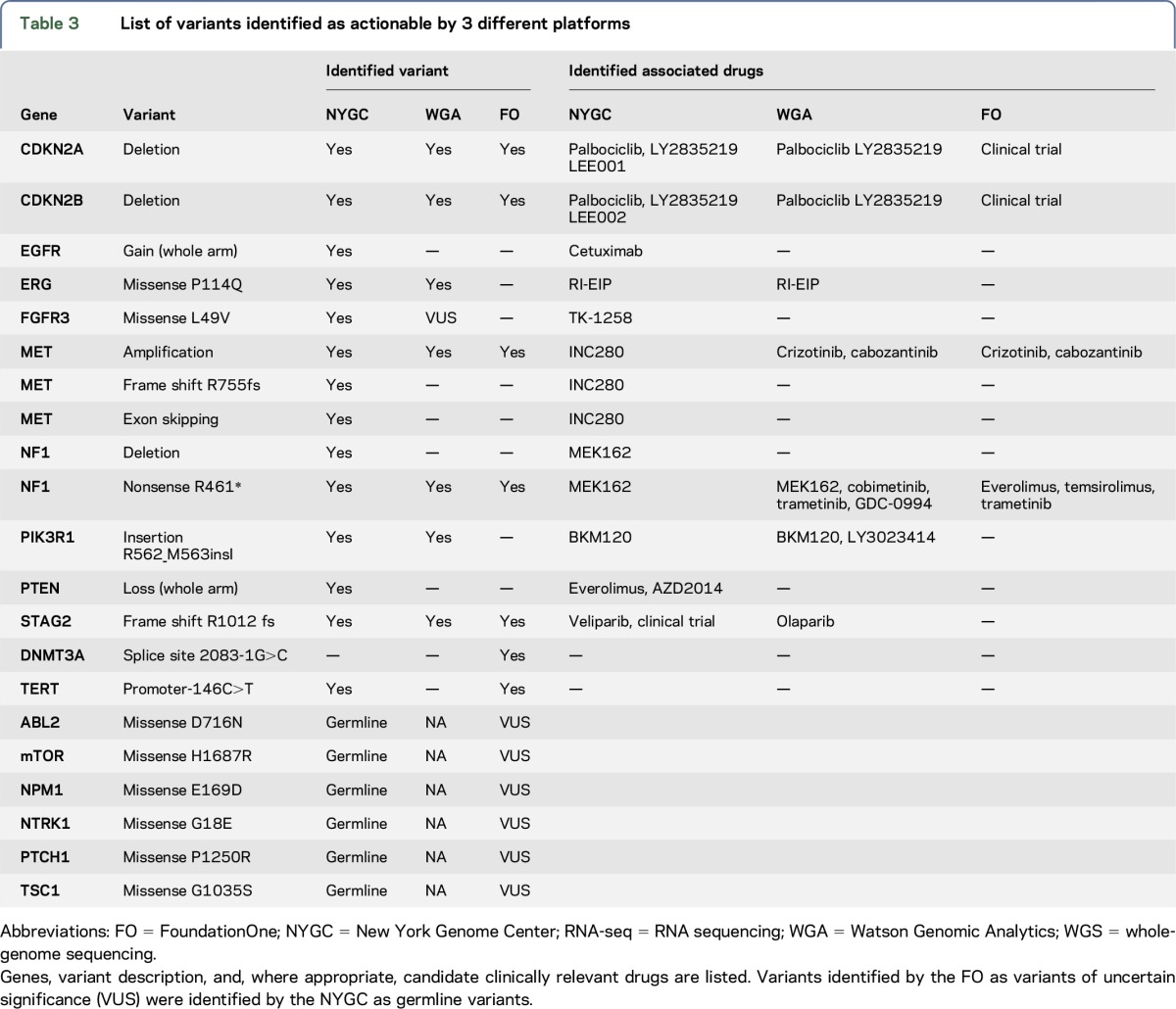
Figure 1. Sashimi plot representing the MET exon–skipping event.

Red lines indicate exon coverage and exon junctions. Numbers in red indicate the number of reads supporting these junctions (for instance, 1,181 reads are split between exons 10 and 12). Only junctions with more than 100 reads are represented here.
We also identified a codon insertion in PIK3R1 (p.R562_M563insIle/c.1686_1688dupTAT), which is a regulatory protein that interacts with and inhibits the functional catalytic protein, PIK3CA. Activating mutations of PIK3CA are known cancer drivers, as are loss-of-function mutations of PIK3R1. Functional studies have shown that PIK3R1 amino acid D560 is involved in hydrogen binding with PIK3CA and is an essential amino acid in regulating the activity of the catalytic subunit. The mutation identified here is in the same helical inhibitory (iSH2) domain of PIK3R1. The variant binds but fails to inhibit PIK3CA, leading to enhanced cell survival, Akt activation, anchorage-independent cell growth, and oncogenesis.35,36 Furthermore, analysis of the crystal structure (figure 2) supports the conclusion that this mutation would inhibit the functional interaction between the 2 proteins, specifically through N345 of PIK3CA, resulting in PIK3CA activation.
Figure 2. Overlay of genomic mutations on crystal structure of PIK3R1.
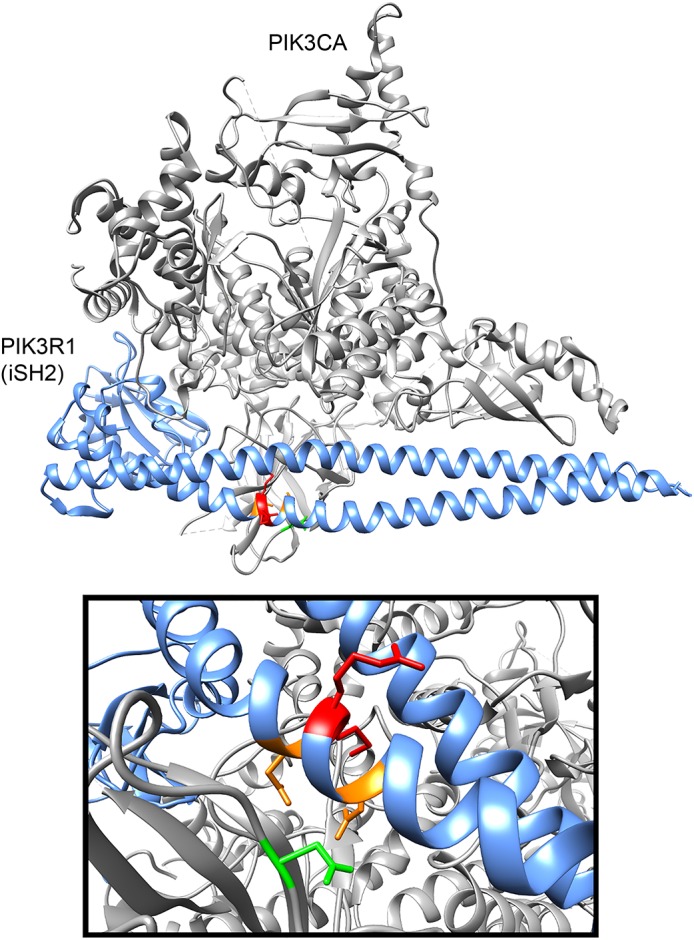
New York Genome Center identified GBM somatic mutations in iSH2 domain of PIK3R1 (red). Known D560Y and N564D (orange) mutants in the PI3K regulatory domain (PIK3R1) fail to inhibit PIK3CA and lead to enhanced cell survival, Akt activation, anchorage-independent cell growth, and oncogenesis.34 The R562-M563 insertion mutation in NYGC-GBM1 potentially alters the functional interaction within this region, specifically through N345 (green) of PIK3CA. The potential inability of catalytic regulation due to PIK3R1 mutation can translate to therapeutic targeting with a PIK3CA inhibitor such as BKM120 and therefore the identification of trial NCT01870726 in combination with MET inhibitor INC280.
A recent cell line study identified a synergistic effect between MET exon 14–skipping variants and a PIK3CA E545K oncogenic variant, in which a combination of an MET inhibitor and a PIK3CA inhibitor showed better sensitivity than single therapy.37 The PIK3CA E545K variant also activates PIK3CA. Taken together, these findings led us to suggest combinatorial INC280 (MET inhibitor) and BKM120 (PIK3CA inhibitor) therapy for potential clinical consideration, and this suggestion would have made the patient eligible for a clinical trial assessing efficacy of this combination (NCT01870726).
Watson for Genomic analysis.
Data for NYGC-GBM-01 were input into WGA, which produced a report summarizing actionable variants and a list of associated drugs, including some based on a pathway target analysis. WGA identified 6 actionable alterations, 14 associated drugs, 9 VUS (including FGFR3), copy number losses in Chr 9 (focal), 10 (armscale), and 11 (focal), and gain on Chr 7 (armscale). Both the NYGC and WGA identified 5 actionable alterations (in genes NF1, MET, CDKN2A, CDKN2B, and PIK3R1; table 3). WGA reported an NF1 SNV and annotated the variant as inactivating but did not deem copy number change to be sufficient for calling this or EGFR and PTEN. A 1-copy gain of EGFR was below WGA's threshold for classification as a targetable variant. Furthermore, this variant was shown to be negative for amplification by in situ hybridization fluorescence. However, the NYGC decided to list it as potentially targetable, given it is a known actionable variant in GBM. Similarly, a 1-copy PTEN loss is reported by the NYGC but not by WGA or by the FoundationOne. The NYGC reported this variant because of its clinical implications; it is associated with resistance to EGFR tyrosine kinase inhibition via AKT/mTOR pathway activation and is linked to cetuximab resistance, but can be targeted by mTOR inhibitors.38 For PIK3R1, the NYGC identified BKM120 as a potential therapeutic option based on additional RNA-seq evidence of overexpression of PIK3CA. WGA identified PIK3R1 as a relevant variant via SNV data by WGS and used RNA-seq information in pathway and drug analysis to also recommend BKM120. The MET amplification and associated drugs are reported by both platforms; however, WGA had 2 drugs for MET amplification, whereas the NYGC prioritized 1 therapeutic option, INC280, based on GBM trial data availability. The NYGC reported 8 clinical trials associated with 5 genes. WGA found 10 clinical trials that may be relevant across 6 actionable alterations.
Comparison with a panel.
Table 3 also compares all variants and drugs identified by the NYGC and FoundationOne. NYGC analysis identified 8 unique variants not found by the FoundationOne, including an exon-skipping event. The NYGC identified drugs for 10 targets, while FoundationOne identified drugs for 4. Furthermore, 6 of the variants reported as of unknown significance occurring in the tumor by FoundationOne were germline variants. One variant (DNMT3A splice site 2083-1G>C) was called by FoundationOne, but the position and base change were different from a nearby variant identified by the NYGC.
DISCUSSION
The NYGC is undertaking a WGS research study in patients with GBM to investigate the efficiency and feasibility of WGS to inform therapeutic options. Here, the results of NYGC WGS and RNA-seq were compared with a clinical panel assay. Also, in collaboration with IBM, the NYGC examined the therapeutic options identified by WGA based on WGS and RNA-seq data. Genomic results from this patient clearly displayed the diversity of driver events typically seen in GBM. Of interest, we identified mutations in targetable genes that were not precise matches to known specific targetable variants, and which nonetheless suggested potential therapeutic options.
Multimodal analysis (WGS and RNA-seq) increased confidence in the identification of the MET mutation; analysis of the literature of prior MET exon–skipping events suggested the plausibility of considering a tyrosine kinase inhibitor that could target MET. Similarly, manual literature search of the PIK3CA E545K oncogenic variant led to the conclusion that this was likely an activating mutation. Moreover, manual database searches resulted in the suggestion of a combinatorial treatment with an MET inhibitor and a PIK3CA inhibitor, which made logical sense and also made the patient eligible for a clinical trial for this combination (NCT01870726).
None of these observations were evident from the panel. This suggests that pursuing a more extensive comparison of panel and deeper sequencing (e.g., WGS and RNA-seq) will be of interest. An added point, previously noted by others,39 is that the sequencing of both germline and tumor DNA not only heightened our sensitivity for what variants might be tumor drivers but was able to rule out a number of germline variants called by the FoundationOne as not likely to be primary drivers of this patient's GBM.
Although we conducted WGS of this sample at roughly twice the cost of WES, the primary analysis was performed on the protein-coding region of the genome. There may be technical advantage to WGS even for assaying targeted regions. WES relies on hybridization capture of specific genes which introduces intrinsic bias for each gene as a function of GC/AT content, while WGS relies more simply on mechanical shearing of DNA prior to sequencing. Previous studies have found that for disorders caused by constitutional mutations,40,41 WGS is more sensitive than WES for variant detection. To assess whether WGS could detect variants not identified by WES to justify the added cost, it would require a direct comparison of the assays on the same sample. We are undertaking a study to address this question.
This patient died approximately 8 months from the time of initial resection falling short of the median survival time for GBM. The oncologist recommended enrollment in a clinical trial targeting PIK3 and MET alterations on recurrence on adjuvant temozolomide. However, the patient's clinical decline eliminated his ability to participate in trials. This highlights one of the challenges of the clinical application of precision medicine technology. The identification of targets and potentially useful drugs in a timely manner is only the first step. Drug and drug trial access is crucial to determine the benefit of this approach in cancer management.
Another key observation was that the WGA analysis vastly accelerated the time to discovery of potentially actionable variants from the VCF files. As previously reported, we found that WGA was able to provide reports of potentially clinically actionable insights within 10 minutes, while human analysis of this patient's VCF file took an estimated 160 hours of person-time. This is critical if sequencing is to be brought out of the research arena and into the scaled, real-world clinical realm. This study is an important step forward promoting human-machine interface as a way to address a key bottleneck in cancer genomics.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful for critical input from many scientific members of the NYGC and for input from the Rockefeller University and BRANY IRBs.
GLOSSARY
- CNV
copy number variant
- EGFR
epidermal growth factor receptor
- GATK
Genome Analysis Toolkit
- GBM
glioblastoma
- IRB
institutional review board
- NLP
Natural Language Processing
- NYGC
New York Genome Center
- RNA-seq
RNA sequencing
- SNV
single nucleotide variant
- SV
structural variant
- TCGA
The Cancer Genome Atlas
- TPM
transcripts per million
- VCF
variant call file
- VUS
variants of uncertain significance
- WGA
Watson Genomic Analytics
- WGS
whole-genome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Kazimierz O. Wrzeszczynski: analysis and interpretation of the data and drafting and revising the manuscript. Mayu O. Frank: design and conceptualization of the study, interpretation of the data, and drafting and revising the manuscript. Takahiko Koyama: analysis and interpretation of the data and revising the manuscript. Kahn Rhrissorrakrai: analysis and interpretation of the data and revising the manuscript. Nicolas Robine: analysis and interpretation of the data and revising the manuscript. Filippo Utro: revising the manuscript. Anne-Katrin Emde, Bo-Juen Chen, Kanika Arora, Minita Shah, Vladimir Vacic, Raquel Norel, Erhan Bilal, Ewa A. Bergmann, Julia L. Moore Vogel, Jeffrey N. Bruce, Andrew B. Lassman, Peter Canoll, Christian Grommes, Steve Harvey, Laxmi Parida, Vanessa V. Michelini, Michael C. Zody, Vaidehi Jobanputra, and Ajay K. Royyuru: revising the manuscript. Robert B. Darnell: design and conceptualization of the study, interpretation of the data, and drafting and revising the manuscript.
STUDY FUNDING
This study was supported by an unrestricted grant from IBM to the NYGC, and from NYGC internal funds.
DISCLOSURE
K.O. Wrzeszczynski reports no disclosures. M.O. Frank has been a consultant for the New York Genome Center and has received research support from the International Society of Nurses in Genetics, Rockefeller University, and NIH CTSA. T. Koyama holds a patent (pending) for Drug scoring utilizing pathway analysis and is an employee of IBM Corporation. K. Rhrissorrakrai owns stock in Johnson & Johnson, Pfizer, and Merck and is an employee of IBM Corporation. N. Robine reports no disclosures. F. Utro is an employee of IBM Corporation. A.-K. Emde, B.-J. Chen, K. Arora, and M. Shah report no disclosures. V. Vacic is an employee of and owns stock options in 23andMe, Inc. R. Norel reports no disclosures. E. Bilal is an employee of IBM Corporation. E.A. Bergmann's husband is an employee of Agenus. J.L. Moore Vogel has been a consultant for the New York Genome Center. J.N. Bruce reports no disclosures. A.B. Lassman has served on the scientific advisory boards of Astra Zeneca, AbbVie, Sapience Therapeutics, Genentech, Bioclinica, VBI Vaccines, Cortice Biosciences, Oxigene, Regeneron, and Novocure; has received travel funding/speaker honoraria from prIME Oncology and from aforementioned scientific advisory boards; has served on the editorial boards of Neuro-Oncology and the Journal of Neurooncology; has been a consultant for WebMD, the American Society of Clinical Oncology, and the American Academy of Neurology; has been a grant reviewer for the Italian Association for Cancer Research; and has received research support from the NCI, the Radiation Therapy Oncology Group Foundation, and the James S. McDonnell Foundation. P. Canoll has served on the editorial boards of PLoS One and GLIA and has received research support from NIH/National Institute of Neurological Disorders and Stroke, NCI, NIDA, and the James F. McDonnell Foundation (Swanson). C. Grommes has received research support from Pharmacyclics. S. Harvey reports no disclosures. L. Parida has served on the advisory board of the NYU Tandon School of Engineering; has served on the editorial boards of BMC Bioinformatics, IEEE/ACM Transactions on Computational Biology and Bioinformatics, the Journal of Computational Biology, and the SIAM Journal of Discrete Mathematics; holds patents for Transductive feature selection with maximum-relevancy and minimum-redundancy criteria, Improved metagenome mapping, Confidence interval estimation of species in metagenomic data, Estimating multiple parents from a matrix of F1 hybrid progeny, Systems and methods for fitting LD distributions at genomic scales, TLASSO: Transductive Lasso for High Dimensional Data Regression Problems, Lossless compression of the enumeration space of founder line crosses, and Efficient sorting of large dimensional data; receives publishing royalties from Chapman & Hall; and is an employee of IBM Research. V.V. Michelini is an employee of, has received research support from, and holds stock/stock options in IBM Corporation. M.C. Zody has received travel funding and teaching honoraria from Fourth Culture, LLC., and teaching honoraria from Harvard CFAR and Cold Spring Harbor Laboratory; has served as a guest member of the editorial board for Annual Review of Genomics and Human Genetics; is supported in part by a grant from the Alfred P. Sloan Foundation; has received research support from IBM, NHGRI/NIH, NCI/NIH, and the Alfred P. Sloan Foundation; holds a patent for Method for inference of HLA types from short read sequencing data; and owns stock in Thermo Fisher and Merck. V. Jobanputra reports no disclosures. A.K. Royyuru holds patents for Mixed polynucleotide and forming method thereof, DNA sequencing using multiple metal layer structure with different organic coatings forming different transient bondings to DNA, Fabrication of tunneling junction for nanopore DNA sequencing, Verification of complex workflows through internal assessment or community based assessment, Field effect based nanosensor for biopolymer manipulation and detection, DNA sequence using multiple metal layer structure with different organic coatings forming different transient bondings to DNA, Integrated nanowire/nanosheet nanogap and nanopore for DNA and RNA-seq, Field effect based nanosensor for biopolymer manipulation and detection, Integrated nanowire/nanosheet nanogap and nanopore for DNA and RNA-seq, Electron beam sculpting of tunneling junction for nanopore DNA sequencing, Charged entities as locomotive to control motion of polymers through a nanochannel, Molecular dispensers (2), Nanopore capture system, Protein structure analysis (2), Method of identifying robust clustering, Hydrophobic moment of multi-domain proteins (3), System and program storage device of object classification utilizing optimized Boolean expressions, Apparatus, method, and product of manufacture for transforming supply chain networks using pair-wise nodal analysis, Method and apparatus for protein structure analysis, Techniques for reconstructing supply chain networks using pair-wise correlation analysis, and Object classification using an optimized Boolean expression; is an employee of IBM Corporation; is a member of the Industry Advisory Board of International Society for Computational Biology; and has received research support from Pfizer and CHDI. R.B. Darnell serves on the Scientific Advisory Board at the New York Genome Center, Roundtable on Translating Genomic-Based Research for Health at National Academy of Medicine, and Stanford Medicine Board of Fellows; holds patent nos. 7,989,203, 6602709, 6,750,029, 7,928,190, and 14/104,581; holds a patent (pending) for Use of BET inhibitors to treat neurodevelopmental disorders and epilepsy; has received honoraria from Lennart Philipson Memorial Lecture, Uppsala University, Sweden; has received research support from IBM, Sohn New York City Collaboration for Pediatric Cancer Research, Starr Cancer Consortium, and NIH; is a Howard Hughes Medical Institute investigator; and owns stock in Amgen, Illumina, and Agios. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Good BM, Ainscough BJ, McMichael JF, Su AI, Griffith OL. Organizing knowledge to enable personalization of medicine in cancer. Genome Biol 2014;15:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffith M, Miller CA, Griffith OL, et al. . Optimizing cancer genome sequencing and analysis. Cell Syst 2015;1:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyman DM, Solit DB, Arcila ME, et al. . Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today 2015;20:1422–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kandoth C, McLellan MD, Vandin F, et al. . Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zack TI, Schumacher SE, Carter SL, et al. . Pan-cancer patterns of somatic copy number alteration. Nat Genet 2013;45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A, Hanna M, Banks E, et al. . The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cibulskis K, Lawrence MS, Carter SL, et al. . Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilm A, Aw PP, Bertrand D, et al. . LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40:11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–1817. [DOI] [PubMed] [Google Scholar]
- 11.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009;25:2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narzisi G, O'Rawe JA, Iossifov I, et al. . Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods 2014;11:1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cingolani P, Platts A, Wang le L, et al. . A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aken BL, Ayling S, Barrell D, et al. . The Ensembl gene annotation system. Database (Oxford) 2016;2016:baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forbes SA, Bindal N, Bamford S, et al. . COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gene Ontology C. The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res 2010;38:D331–D335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genomes Project C, Auton A, Brooks LD, et al. . A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xi R, Hadjipanayis AG, Luquette LJ, et al. . Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci USA 2011;108:E1128–E1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28:i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Mullighan CG, Easton J, et al. . CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 2011;8:652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen K, Wallis JW, McLellan MD, et al. . BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009;6:677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emde AK, Schulz MH, Weese D, et al. . Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics 2012;28:619–627. [DOI] [PubMed] [Google Scholar]
- 23.Ha G, Roth A, Khattra J, et al. . TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res 2014;24:1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Loo P, Nordgard SH, Lingjaerde OC, et al. . Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA 2010;107:16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobin A, Davis CA, Schlesinger F, et al. . STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- 27.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics 2011;12:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–127. [DOI] [PubMed] [Google Scholar]
- 29.Verhaak RG, Hoadley KA, Purdom E, et al. . Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169–181. [DOI] [PubMed] [Google Scholar]
- 31.Rhrissorrakrai K, Koyama T, Parida L. Watson for genomics: moving personalized medicine forward. Trends Cancer 2016;2:392–395. [DOI] [PubMed] [Google Scholar]
- 32.Rizvi NA, Hellmann MD, Snyder A, et al. . Cancer immunology: mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frampton GM, Ali SM, Rosenzweig M, et al. . Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015;5:850–859. [DOI] [PubMed] [Google Scholar]
- 34.Navis AC, van Lith SA, van Duijnhoven SM, et al. . Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol 2015;130:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Backer JM. The regulation of class IA PI 3-kinases by inter-subunit interactions. Curr Top Microbiol Immunol 2010;346:87–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaiswal BS, Janakiraman V, Kljavin NM, et al. . Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 2009;16:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Jia Y, Stoopler MB, et al. . Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol 2016;34:794–802. [DOI] [PubMed] [Google Scholar]
- 38.Frattini M, Saletti P, Romagnani E, et al. . PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 2007;97:1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones S, Anagnostou V, Lytle K, et al. . Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 2015;7:283ra253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belkadi A, Bolze A, Itan Y, et al. . Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA 2015;112:5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner TN, Hormozdiari F, Duyzend MH, et al. . Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet 2016;98:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.