

Summary
Cell stress of various kinds can lead to the induction of cell death and a damaging inflammatory response. Hence, a goal of therapeutic cell‐stress management is to develop agents that might effectively regulate undesirable cell death and inflammation. To that end, we developed a synthetic peptide of seven amino acids based on structural mimicry to a functional domain of p53, a key factor in the responses of cells to stressful stimuli. This heptapeptide, which we term Stressin‐1, was found to inhibit both cell death and the secretion of inflammatory mediators by various cell types in response to different stressful agents in vitro. The combined anti‐inflammatory and anti‐apoptotic activities of Stressin‐1 were associated with a cellular signalling cascade that induced activation of Akt kinase and activation of the cAMP response element‐binding protein (CREB) transcription factor. These immediate signalling events led to the inhibition of the signal transducer and activator of transcription and nuclear factor‐κB pathways 24 hr later. Unexpectedly, we found no evidence for a direct involvement of p53 in the effects produced by Stressin‐1. Intraperitoneal administration of 100 μg of Stressin‐1 to lethally irradiated mice significantly protected them from death. These findings show that activating the Akt–CREB axis with Stressin‐1 can counteract some of the undesirable effects of various cell stresses. Stressin‐1 may have clinical usefulness.
Keywords: cell death, endotoxin, inflammation, p53, peptide inhibitor, radioprotection
Abbreviations
- EMSA
electrophoretic mobility shift assay
- LPS
lipopolysaccharide
- MEF
mouse embryo fibroblasts
- NF‐κB
nuclear factor‐κB
- STAT‐1
signal transducer and activator of transcription 1
- Stressin‐1
STress RESponse Specific peptide INhibitor‐1
- TNF
tumour necrosis factor
Introduction
Various forms of cellular stress can induce cell death through activation of signalling pathways that facilitate the elimination of damaged cells.1 Cellular stress and damage are also a cause of inflammation, which may further increase cell loss in stressed tissues.2, 3 Hence, excessive cell death and inflammation in response to cell stress can cause tissue damage and precipitate pathologies.4, 5 Therefore, stress‐induced pathologies might be ameliorated by agents that effectively down‐regulate undesirable cell death and inflammation. A prototypical stress‐induced pathology is the toxicity of ionizing radiation or DNA damaging drugs used for cancer treatment; moreover, stress‐induced pathologies also include neurodegeneration and ischaemic diseases.5, 6, 7
The p53 transcription factor is a central regulator of the cellular response to a variety of stresses,8 and p53‐dependent cell death has been reported to be involved in the occurrence of stress‐induced pathologies due to excessive cell loss.5, 6 However, activation of p53 can result in diverse outcomes that include cell death, but also growth arrest and damage repair.8 How the transcriptional activity of p53 is regulated to produce such diverse outcomes is not entirely clear, but it is assumed that p53 activity is determined by multiplex post‐translational modifications and co‐factor interactions.8 One such activating modulation of p53 has been reported to occur after binding to the C‐terminal regulatory domain of p53 by damaged DNA or the specific monoclonal antibody PAb‐421.9, 10 The p53 C‐terminus seems to recognize both PAb‐421 and DNA based on the structural mimicry between this antibody and DNA.11, 12
Here we set out to derive a small peptide ligand that might interact with the p53 C‐terminal domain and hence regulate p53 activation. However, direct selection of potential peptide ligands from a library based on recognition by p53 is difficult technically. Therefore, we developed a monoclonal antibody (Idi‐2) that functionally mimics the structure of the p53 C‐terminus and used it in place of the actual p53 molecule to identify such a peptide. Our putative p53‐mimic was Idi‐2, a monoclonal antibody that had been selected to recognize both PAb‐421 and DNA, as does the p53 C‐terminal domain (see Supplementary material, Fig. S1).11, 12 We then used Idi‐2 to pan a phage display library of heptapeptides,13 and a peptide was identified by its binding to Idi‐2. We now report that this peptide exhibits potent anti‐apoptotic and anti‐inflammatory activities, both in vitro in cell culture and in vivo in a mouse model of lethal irradiation.
Materials and methods
Peptide selection from phage display library
A Ph.D.‐7 library from New England Biolabs (Ipswich, MA) was screened according to the manufacturer's instructions. Briefly, three rounds of selection by Idi‐2 monoclonal antibody were performed and the predominant consensus peptide sequence was identified by sequencing of phage DNA. A candidate peptide, designated Stressin‐1, was then synthesized by PolyPeptide Laboratories (Torrance, CA) and further studied in the functional assays described below. Various control peptides, which were weakly selected by Idi‐2 in the first two rounds of selection, were used as control peptides in the described assays. None of these control peptides (WWPPRHW, TYWYMTP, LPRNSPV, HSRLVPA, NLPRLYC, MHARTLA, SWYPSFS) produced any significant functional activities (not shown).
Cell culture and mice
Cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. As indicated, cells were stimulated with 50 ng/ml lipopolysaccharide (LPS) from Escherichia coli O55:B5 (Sigma‐Aldrich, Schnelldorf, Germany) or with 10–100 μm Cisplatin (Sigma‐Aldrich). The RAW‐blue reporter cell line was obtained from InvivoGen (Toulouse, France). BALB/c mice were bred and kept at the animal facilities of the Weizmann Institute of Science, Rehovot, Israel. Animal experiments have been conducted according to relevant national and international guidelines and were approved by the Weizmann Institute of Science Institutional Animal Care and Use Committee. Radioprotection was assessed after whole body irradiation with 6·5 Gy. Mice received one intraperitoneal injection of 0·1 mg Stressin‐1 peptide or saline 1 hr after irradiation. Animals were inspected daily and were humanely killed by cervical dislocation when they met a critical clinical condition, as defined by a fall in body weight (20%) and/or deterioration in appearance and behaviour. Animals were co‐housed under specific pathogen‐free conditions at light–dark cycles in groups of three to five per cage with free access to water and chow, or supplied with mashed diet when indicated.
Quantification of cell death and inflammatory mediators
In vitro cell death was quantified with the MTT assay by adding 0·5 mg/ml MTT (Sigma‐Aldrich) to cultured cells. After 2 hr, the supernatant was removed, the tetrazolium dye dissolved in dimethyl sulphoxide was added, and absorbance was measured at 570 nm. The secretion of tumour necrosis factor (TNF) and interleukin‐6 was determined by specific ELISA (R&D Systems, Wiesbaden, Germany) from culture supernatant. Nitric oxide (NO) production was estimated by determining the amounts of the nitric oxide oxidation products nitrite and nitrate with a colorimetric assay at 540 nm based on the formation of an azo dye following addition of Griess reagent.14 Results are shown as the means of quadruple experiments.
Western blot and electrophoretic mobility shift assay
Cell lysates were prepared by lysis in HEPES‐buffered saline with 1% Triton X‐100 containing proteinase/phosphatase inhibitors. Proteins were separated in 12% SDS–polyacrylamide gels and blotted onto nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). After incubation with 5% dry non‐fat milk, membranes were probed with primary antibodies and appropriate horseradish peroxidase‐conjugated secondary antibodies (Cell Signaling, Danvers MA). Protein‐bound antibody was detected with an ECL kit (Roth, Karlsruhe, Germany). Quantification was performed using the software image J (National Institutes of Health, Bethesda, MD); values indicate band intensity relative to baseline samples (untreated or at 0 min) after normalization to actin bands. Electrophoretic mobility shift assay (EMSA) was performed with the LightShift Chemiluminescent EMSA Kit (Pierce, Rockford, IL) using recombinant p53 and the p53‐responsive element consensus sequence oligonucleotide15, 16 prepared in a double‐stranded form.
Statistics
Statistical significance of differences between data sets was tested by Student's t‐test or one‐way analysis of variance with Tukey's post test. Survival differences were analysed by Fisher's exact test. P values < 0·05 were considered significant. Data are represented as mean and standard deviation.
Results
To obtain peptides that might modify the cellular stress response, we screened a heptameric peptide library for peptides that mimic damaged DNA based on binding to Idi‐2, a p53‐mimicking monoclonal antibody that recognizes damaged DNA (see Supplementary material, Fig. S1).11 We performed three rounds of selection and obtained one predominant consensus amino acid sequence (LPPLPYP). This peptide was then tested for its ability to interfere with the cellular stress response in an assay in which apoptosis was induced in mouse embryo fibroblasts (MEF) by incubation with 80 μm cisplatin. The cisplatin‐treated MEF were incubated in the absence or presence of increasing doses of Stressin‐1, the selected candidate peptide (10 μm, 50 μm or 100 μm), and cell survival was assessed by light microscopicy inspection (Fig. 1a); we observed that the viability of the cells increased with the dose of Stressin‐1.
Figure 1.
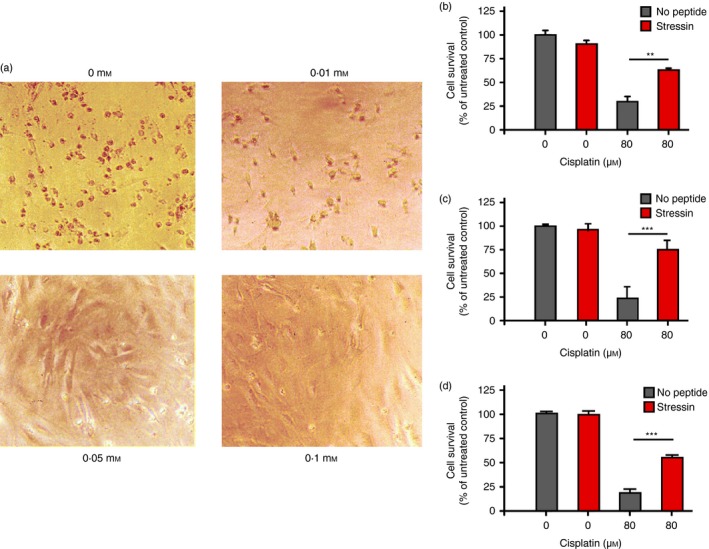
Inhibition of stress‐induced apoptosis by Stressin‐1 peptide. (a) Mouse embryo fibroblasts (MEF) were incubated with 80 μm cisplatin in the absence (upper left panel) or presence of 10, 50 or 100 μm Stressin‐1 as indicated, and cell survival was assessed by light microscopy inspection, revealing a dose‐dependent anti‐apoptotic effect of Stressin‐1. (b) MEF survival induced by 50 μm Stressin‐1 was quantified by an MTT assay, measuring MEF cell vitality in the presence or absence of 80 μm cisplatin. (c) Murine RAW 246.7 macrophage survival induced by 50 μm Stressin‐1 was quantified by an MTT assay, measuring cell vitality in the presence or absence of 80 μm cisplatin. (d) Human foreskin fibroblast cell survival induced by 50 μm Stressin‐1 was quantified by an MTT assay, measuring cell vitality in the presence or absence of 80 μm cisplatin. **P<0·01; ***P<0·001.
To quantify the survival benefit mediated by Stressin‐1, we performed an MTT assay of cisplatin‐treated MEF that were incubated in the absence or presence of peptide (50 μm). We found that Stressin‐1 significantly increased the viability of cisplatin‐treated MEF (29·7% vitality without peptide versus 63% with peptide; P < 0·001) (Fig. 1b). To confirm that the peptide could confer protection from cisplatin‐induced apoptosis, we also performed an MTT assay with the murine macrophage cell line RAW 264.7 (Fig. 1c) and human foreskin fibroblasts (Fig. 1d). We found that Stressin‐1 protected cisplatin‐treated RAW 264.7 cells (22·7% vitality in the absence of Stressin‐1 compared with 75·1% vitality with added peptide; P < 0·0001) (Fig. 1c); cisplatin‐treated human fibroblasts (18·4% vitality without peptide versus 54% with peptide; P < 0·0001) (Fig. 1d) were also significantly protected from induced apoptosis. ‘Stressin‐1’, which stands for STress RESponse Specific peptide INhibitor‐1, was named for its amelioration of the apoptotic response to these stresses.
At the level of the organism, the response to cell stress is inflammation, which includes a large number of immune activities that serve tissue maintenance.2, 3 To learn whether the Stressin‐1 peptide may modify the inflammatory response to cell stress, we studied the response of RAW 264.7 macrophages to the toll‐like receptor 4 (TLR4) ligand LPS, which stimulates the secretion of pro‐inflammatory mediators. The RAW 246.7 cells were stimulated with LPS (50 ng/ml) in the presence or absence of Stressin‐1 peptide (50 μm) and we measured the induction of inflammatory mediators in the supernatant as a measure for macrophage activation after 1 day of culture (Fig. 2). We found that Stressin‐1 treatment significantly inhibited the secretion of inflammatory TNF‐α (456 versus 2077 pg/ml; P < 0·0001) (Fig. 2a), interleukin‐6 (146 versus 513 pg/ml; P < 0·0001) (Fig. 2b), and nitric oxide (4 versus 34 nm; P < 0·0001) (Fig. 2c). Stressin‐1 treatment in the absence of LPS did not induce secretion of these inflammatory mediators (Fig. 2a–c). These findings indicated that the Stressin‐1 peptide seems to be an effective inhibitor of stress‐induced inflammatory macrophage activity.
Figure 2.
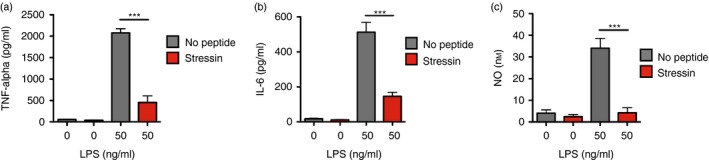
Inhibition of lipopolysaccharide (LPS)‐induced inflammatory mediator induction by Stressin‐1 peptide. RAW 264.7 macrophages were stimulated with LPS (50 ng/ml) in the presence or absence of Stressin‐1 peptide (50 μm) and the induction of inflammatory mediators in the supernatant was measured after 1 day of culture. Stressin‐1 inhibited the production of tumour necrosis factor‐α (TNF‐α) (a), interleukin‐6 (IL‐6) (b), and nitric oxide (c). ***P<0·001.
We then tested whether the Stressin‐1 peptide could also act in vivo as a functional inhibitor of stress‐induced apoptosis and inflammation. To that end, we subjected two groups of 16 and 17 mice each to whole body γ‐irradiation (6·5 Gy), which causes both extensive endothelial apoptosis and inflammation,17, 18, 19 and treated the mice 1 hr after irradiation with saline or Stressin‐1 peptide (0·1 mg/mouse). We found that the treatment with Stressin‐1 peptide conferred significant radioprotection (88% survival versus 44% survival in the saline group; P < 0·05) (Fig. 3). Hence, Stressin‐1 peptide seems to be an effective radioprotectant capable of inhibiting the cellular and inflammatory stress response in vivo.
Figure 3.
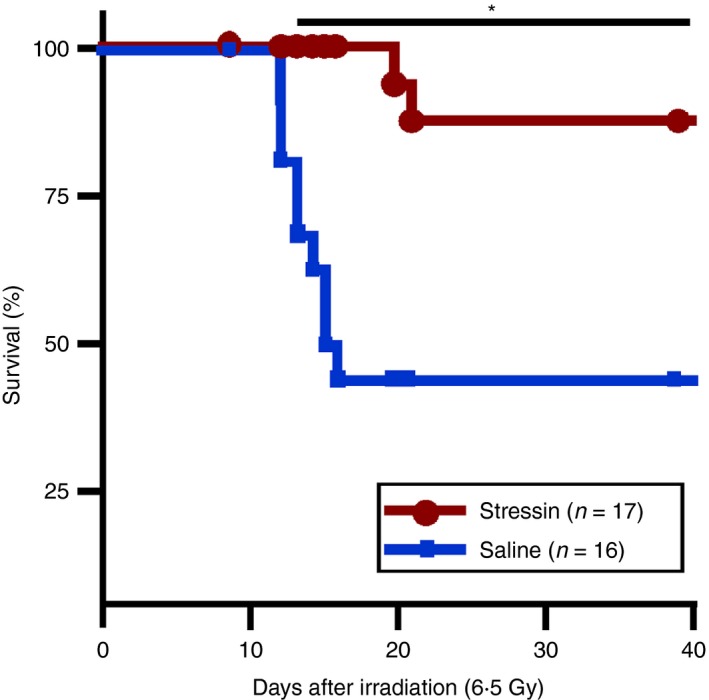
Stressin‐1 treatment of mice confers radioprotection in vivo. Two groups of 16 and 17 mice were subjected to whole body γ‐irradiation (6·5 Gy). One hour after irradiation, mice were treated with saline (blue lines) or Stressin‐1 peptide (0·1 mg/mouse, red lines), and the survival rates were determined. Treatment with Stressin‐1 peptide conferred significant radioprotection. *P<0·05.
To learn how Stressin‐1 might inhibit apoptosis and the secretion of inflammatory mediators at the molecular level, we investigated the signalling pathways stimulated by Stressin‐1. We first tested by EMSA whether Stressin‐1 peptide may directly modify the interaction of recombinant active p53 with the p53‐responsive element (p53RE).15, 16 Addition of Stressin‐1 to the EMSA reaction did not change the band shift of the labelled p53RE nucleotide probe mediated by p53 (Fig. 4a, lane 1 versus lane 2). Moreover, Stressin‐1 did not change the supershift of the labelled probe induced by p53 in the presence of the monoclonal anti‐p53 antibody PAb‐421 (Fig. 4a, lanes 3 versus 4). These findings indicated that Stressin‐1 did not appear to directly modify transcriptional p53 activity.
Figure 4.
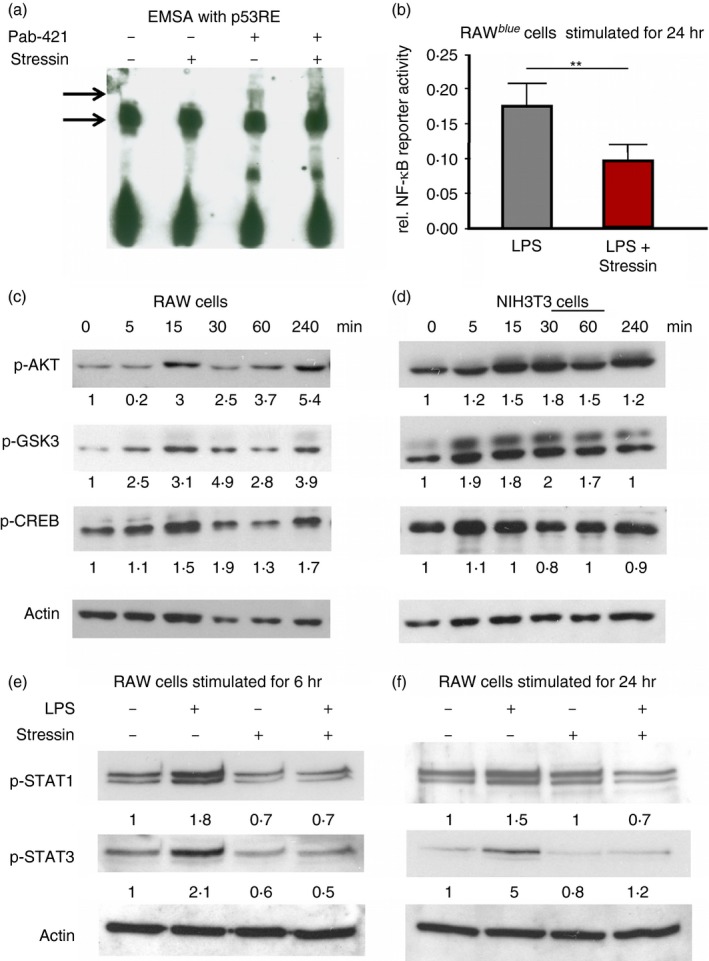
Activation of the Akt–CREB signalling axis by Stressin‐1. (a) An EMSA with a labelled p53‐responsive element probe and recombinant p53 was performed in the presence or absence of Stressin‐1 (lanes 1 and 2), and the presence or absence of the monoclonal anti‐p53 antibody PAb‐421 (lanes 3 and 4). Stressin‐1 did not directly modify the band shift or PAb‐421‐induced supershift of the labelled probe. (b) Lipopolysaccharide (LPS) ‐induced nuclear factor‐κB (NF‐κB) activity of RAW‐blue reporter cells was assessed in the presence or absence of Stressin‐1, indicating that Stressin‐1 inhibited NF‐κB activity, (c, d) Stressin‐1 induced rapid phosphorylation of Akt kinase, GSK‐3 and CREB within minutes both in RAW 264.7 macrophages (c) and in NIH3T3 cells (d). (e,f) Stressin‐1 strongly reduced the LPS‐induced activation of signal transducer and activator of transcription 1 (STAT1) and STAT3 in RAW 264.7 cells, as assessed after 6 hr (e) and 24 hr (f) of LPS incubation. Values given in (c–f) indicate the band intensity relative to baseline samples (untreated or at 0 min) after normalization to actin bands. **P<0·01.
We next tested whether Stressin‐1 might modify the activity of nuclear factor‐κB (NF‐κB); decreased activity of the NF‐κB pathway, which is the major inflammatory pathway in macrophages,20 could explain the inhibited inflammatory response of macrophages induced by Stressin‐1 (Fig. 2). We assayed the effects of Stressin‐1 on the activity of NF‐κB in RAW‐blue reporter cells stimulated with LPS. In the presence of Stressin‐1, NF‐κB reporter activity was significantly reduced (0·09 versus 0·17 relative reporter activity; P < 0·001), indicating that, indeed, the Stressin‐1 peptide seems to inhibit macrophage activity by interference with the NF‐κB pathway.
To explore how Stressin‐1 influences the NF‐κB pathway, we analysed potential upstream signalling molecules that might be targeted by the peptide. We did not find a difference in the extracellular signal‐regulated kinase, c‐Jun N‐terminal kinase or p38 pathways (not shown); however, we observed that Stressin‐1 induced rapid activation of Akt kinase within minutes both in RAW 264.7 macrophages (Fig. 4c) and in NIH3T3 cells (Fig. 4d). This observation is fully compatible with reports showing that Akt activation can confer radioprotection in vitro by inhibiting apoptosis.21, 22, 23 Along with Akt phosphorylation, Stressin‐1 also induced phosphorylation of GSK‐3 and cAMP response element‐binding protein (CREB); however, more so in the macrophages than in the fibroblasts (Figs 4c and d). Note that GSK‐3 is a direct target of Akt and is inhibited by Akt‐induced phosphorylation.24 Akt activation together with GSK‐3 inhibition facilitates the activation (by phosphorylation) of CREB.25 As the transcriptional activity both of CREB and NF‐κB depends largely on recruitment of the co‐activator CREB‐binding protein (CBP), CREB and NF‐κB compete for the limited amounts of CBP present in the nucleus.26, 27 Thus, activation of CREB induced by Stressin‐1 could account for the reduced NF‐κB activity and the inhibition of inflammatory mediators mediated by Stressin‐1 (Figs 2 and 4b). Indeed, it was shown that competition for CBP can inhibit NF‐κB‐induced secretion of TNF‐α.28 Moreover, it was shown that the inhibition of TNF‐α secretion is associated with radioprotection in vivo.29 Hence, activation of the Akt–CREB signalling axis by Stressin can explain both its anti‐apoptotic and anti‐inflammatory activities, as well as its efficacy in protecting mice from lethal irradiation in vivo.
Moreover, CBP is also an important co‐factor for the transcriptional activity of p53 that can facilitate acetylation of histones and the p53 C‐terminal domain.6 Acetylation of p53 and histones is associated with an increase in p53 transcriptional activity.6 Hence, although we did not find a direct effect on p53 activation in an EMSA, Stressin‐1 may indirectly inhibit p53 activity by limiting the amounts of available CBP through CREB activation.
To explore whether the early activation of the Akt–CREB signalling axis by Stressin‐1 might also modify the secondary response of RAW cells to LPS, we assessed the activation of signal transducer and activator of transcription 1 (STAT1) and STAT3 after 6 hr (Fig. 4e) and 24 hr (Fig. 4f) of LPS incubation. At both time‐points, Stressin‐1 strongly reduced the LPS‐induced activation of both STAT1 and STAT3, which adds to the reduced secretion of inflammatory mediators by Stressin‐1‐treated macrophages.
Discussion
Here we report that a proline‐rich heptapeptide, Stressin‐1, was active in vitro in preventing cisplatin‐induced cell death of various cell types (Fig. 1) and in modifying the secretion of inflammatory mediators by macrophages in response to endotoxin (Fig. 2). The findings in vitro were strengthened by finding in vivo showing that a single treatment with Stressin‐1 provided significant radioprotection in lethally irradiated mice (Fig. 3). The anti‐apoptotic and anti‐inflammatory cytokine responses were associated with rapid activation of the Akt signalling axis and delayed inhibition of the STAT and NF‐κB pathways (Fig. 4).
How does Stressin‐1 produce its effects on stress‐induced apoptosis and the inflammatory response? Does Stressin‐1 act as an agonist for a known receptor? Structurally, Stressin‐1 is rich in Proline residues – four of its seven amino acids are Prolines: LPPLPYP. A Proline residue adds rigidity to a polypeptide, and multiple Prolines can create a helical structure.30 Indeed, we selected the Stressin‐1 peptide by its ability to bind to Idi‐2, a monoclonal antibody that also binds DNA and is thought to mimic the structure of the C‐terminus domain of p53 that also binds DNA11 – Stressin‐1 could well mimic a domain of DNA. Based on this reasoning, we tested whether some of the effects of Stressin‐1 might be explained by stimulation of innate pattern‐recognition receptors, such as the TLRs that are activated by nucleic acids and other ligands.31 Indeed, TLR signalling can induce activation of the Akt kinase and, through induction of NF‐κB activation, increase cellular resistance to apoptosis.31 Moreover, TLR stimulation has been found to be radioprotective in vivo.32 However, we were not able to detect Stressin‐induced activation of any specific TLR (not shown). Moreover, as we demonstrated above (Figs 2 and 4), we found that Stressin‐1 inhibited NF‐κB activation and subsequent inflammatory cytokine molecules through the STAT1 and STAT3 transcription factors, which argue against TLR‐signalling. Hence, the effects of Stressin‐1 cannot be explained simply by its possible interaction with known TLR molecules. On the contrary, the unique amino acid composition of the Stressin‐1 peptide could conceivably allow it to enter cells directly; its activation of the Akt–CREB signalling axis could be mediated by some intracellular interactions that by‐pass a membrane‐bound receptor. Indeed, proline‐rich peptide motifs such as that of Stressin‐1 are frequently involved in signalling events due to their recognition by protein‐interaction domains, such as SH3 or WW domains.33 Therefore, Stressin‐1 might plausibly produce its anti‐apoptotic and anti‐inflammatory effects through direct stimulation of, or interference with signalling molecules involved in the cellular stress response. The nature of the putative intracellular receptor(s) activated by Stressin‐1 remains to be characterized. However, it is likely that Stressin‐1 acts on a conserved receptor, as the peptide appeared to function equally in murine and human cells. Such functionally equivalent activity across species seems to indicate that the peptide targets fundamental molecular pathways involved in the control of cell death and inflammatory activation.
In summary, we have discovered a peptide that, through its anti‐apoptotic and anti‐inflammatory activities, can prevent excessive cell loss induced by cell stress. Indeed, the combined inhibition of cell death and inflammation seemed to be effective in preventing radiation‐induced tissue degeneration in vivo. Future work will reveal whether Stressin‐1 peptide treatment and its activation of the Akt–CREB axis might be of benefit in clinical degenerative disorders.
Disclosure
Nothing to disclose.
Supporting information
Figure S1. Schematic representation of structural complementarity and mimicry between DNA, monoclonal antibodies and Stressin‐1 peptide.
Acknowledgements
JH, JS, NE, AWL and IRC conceived experiments. JH and JS performed the experiments. JH, JS and IRC interpreted experimental data. JH and IRC wrote the paper. All authors read and approved the manuscript. We thank Martina Fahl for excellent technical assistance.
Contributor Information
Johannes Herkel, Email: jherkel@uke.de.
Irun R. Cohen, Email: Irun.Cohen@weizmann.ac.il
References
- 1. Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 2008; 9:378–90. [DOI] [PubMed] [Google Scholar]
- 2. Medzhitov R. Origin and physiological roles of inflammation. Nature 2008; 454:428–35. [DOI] [PubMed] [Google Scholar]
- 3. Cohen IR. Real and artificial immune systems: computing the state of the body. Nat Rev Immunol 2007; 7:569–74. [DOI] [PubMed] [Google Scholar]
- 4. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science 2011; 333:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gudkov AV, Komarova EA. Radioprotection: smart games with death. J Clin Invest 2010; 120:2270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 2009; 137:413–31. [DOI] [PubMed] [Google Scholar]
- 7. Frey B, Hehlgans S, Rödel F, Gaipl US. Modulation of inflammation by low and high doses of ionizing radiation: implications for benign and malign diseases. Cancer Lett 2015; 368:230–7. [DOI] [PubMed] [Google Scholar]
- 8. Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2010; 2:a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee S, Elenbaas B, Levine A, Griffith J. p53 and its 14 kDa C‐terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 1995; 81:1013–20. [DOI] [PubMed] [Google Scholar]
- 10. Hupp TR, Sparks A, Lane DP. Small peptides activate the latent sequence‐specific DNA binding function of p53. Cell 1995; 83:237–45. [DOI] [PubMed] [Google Scholar]
- 11. Herkel J, Kam N, Erez N, Mimran A, Heifetz A, Eisenstein M, Rotter V, Cohen IR. Monoclonal antibody to a DNA‐binding domain of p53 mimics charge structure of DNA: anti‐idiotypes to the anti‐p53 antibody are anti‐DNA. Eur J Immunol 2004; 34:3623–32. [DOI] [PubMed] [Google Scholar]
- 12. Herkel J, Mimran A, Erez N, Kam N, Lohse AW, Märker‐Hermann E, Rotter V, Cohen IR. Autoimmunity to the p53 protein is a feature of systemic lupus erythematosus (SLE) related to anti‐DNA antibodies. J Autoimmun 2001; 17:63–9. [DOI] [PubMed] [Google Scholar]
- 13. Ferrer M, Sullivan BJ, Godbout KL, Burke E, Stump HS, Godoy J, Golden A, Profy AT, van Schravendijk MR. Structural and functional characterization of an epitope in the conserved C‐terminal region of HIV‐1 gp120. J Pept Res 1999; 54:32–42. [DOI] [PubMed] [Google Scholar]
- 14. Stuehr DJ, Nathan CF. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med 1989; 169:1543–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. El‐Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet 1992; 1:45–9. [DOI] [PubMed] [Google Scholar]
- 16. Wolkowicz R, Elkind NB, Ronen D, Rotter V. The DNA binding activity of wild type p53 is modulated by blocking its various antigenic epitopes. Oncogene 1995; 10:1167–74. [PubMed] [Google Scholar]
- 17. Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz‐Friedman A, Cordon‐Cardo C, Kolesnick R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001; 293:293–7. [DOI] [PubMed] [Google Scholar]
- 18. Molla M, Panes J. Radiation‐induced intestinal inflammation. World J Gastroenterol 2007; 13:3043–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schaue D, McBride WH. Links between innate immunity and normal tissue radiobiology. Radiat Res 2010; 173:406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gasparini C, Feldmann M. NF‐κB as a target for modulating inflammatory responses. Curr Pharm Des 2012; 18:5735–45. [DOI] [PubMed] [Google Scholar]
- 21. Zheng H, Chen ZW, Wang L, Wang SY, Yan YQ, Wu K, Xu QZ, Zhang SM, Zhou PK. Radioprotection of 4‐hydroxy‐3,5‐dimethoxybenzaldehyde (VND3207) in culture cells is associated with minimizing DNA damage and activating Akt. Eur J Pharm Sci 2008; 33:52–9. [DOI] [PubMed] [Google Scholar]
- 22. Bonnaud S, Niaudet C, Legoux F, Corre I, Delpon G, Saulquin X, Fuks Z, Gaugler MH, Kolesnick R, Paris F. Sphingosine‐1‐phosphate activates the AKT pathway to protect small intestines from radiation‐induced endothelial apoptosis. Cancer Res 2010; 70:9905–15. [DOI] [PubMed] [Google Scholar]
- 23. Qiu W, Leibowitz B, Zhang L, Yu J. Growth factors protect intestinal stem cells from radiation‐induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene 2010; 29:1622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol 2005; 17:150–7. [DOI] [PubMed] [Google Scholar]
- 25. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol 2001; 65:391–6. [DOI] [PubMed] [Google Scholar]
- 26. Zhong H, Voll RE, Ghosh S. Phosphorylation of NF‐κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1998; 1:661–1. [DOI] [PubMed] [Google Scholar]
- 27. Parry GC, Mackman N. Role of cyclic AMP response element‐binding protein in cyclic AMP inhibition of NF‐κB‐mediated transcription. J Immunol 1997; 159:5450–6. [PubMed] [Google Scholar]
- 28. Ollivier V, Parry GCN, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF‐κB‐mediated transcription in human monocytic cells and endothelial cells. J Biol Chem 1996; 271:20828–35. [DOI] [PubMed] [Google Scholar]
- 29. Zhang M, Qian J, Xing X, Kong FM, Zhao L, Chen M, Lawrence TS. Inhibition of the tumor necrosis factor‐α pathway is radioprotective for the lung. Clin Cancer Res 2008; 14:1868–76. [DOI] [PubMed] [Google Scholar]
- 30. Berisio R, Vitagliano L. Polyproline and triple helix motifs in host–pathogen recognition. Curr Protein Pept Sci 2012; 13:855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol 2004; 4:499–511. [DOI] [PubMed] [Google Scholar]
- 32. Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D, Kurnasov OV, Fort FL, Osterman AL, Didonato JA, Feinstein E, Gudkov AV. An agonist of toll‐like receptor 5 has radioprotective activity in mouse and primate models. Science 2008; 320:226–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ball LJ, Kühne R, Schneider‐Mergener J, Oschkinat H. Recognition of proline‐rich motifs by protein–protein interaction domains. Angew Chem Int Ed Engl 2005; 44:2852–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic representation of structural complementarity and mimicry between DNA, monoclonal antibodies and Stressin‐1 peptide.