

Summary
Perturbed neuronal proteostasis is a salient feature shared by both aging and protein misfolding disorders. The proteostasis network controls the health of the proteome by integrating pathways involved in protein synthesis, folding, trafficking, secretion, and their degradation. A reduction in the buffering capacity of the proteostasis network during aging may increase the risk to undergo neurodegeneration by enhancing the accumulation of misfolded proteins. As almost one‐third of the proteome is synthetized at the endoplasmic reticulum (ER), maintenance of its proper function is fundamental to sustain neuronal function. In fact, ER stress is a common feature of most neurodegenerative diseases. The unfolded protein response (UPR) operates as central player to maintain ER homeostasis or the induction of cell death of chronically damaged cells. Here, we discuss recent evidence placing ER stress as a driver of brain aging, and the emerging impact of neuronal UPR in controlling global proteostasis at the whole organismal level. Finally, we discuss possible therapeutic interventions to improve proteostasis and prevent pathological brain aging.
Keywords: aging, endoplasmic reticulum, endoplasmic reticulum stress, protein misfolding disorders, unfolded protein response
Introduction
As the world population gets older, dementia emerges as a major public health issue worldwide, particularly in middle‐ and middle‐to‐high‐income countries. The prevalence of dementia increases exponentially with age, affecting 5–10% of people over 65, and about 50% of people over 85. In 2011, dementia was estimated to affect 35.6 million people around the world, and it is expected to reach about 135 million by 2050 (Brayne, 2007; World Health Organization and Alzheimer's Disease International 2012). Reduced cognitive function is a common trait present in elderly individuals, which correlates with substantial alterations to functional synapses and normal neuronal physiology at the cellular and molecular level (Leal & Yassa, 2015). Accordingly, a significant percentage of aged individuals will manifest some sort of dementia in the form of a collection of neurodegenerative diseases, transposing the line between normal aging (healthspan) to pathological brain aging (Brayne, 2007). Recently, several interconnected processes have been defined as the hallmarks of aging, where substantial alterations to cellular proteostasis is proposed as one of the major pillars of aging (Lopez‐Otin et al., 2013; Kennedy et al., 2014).
The proteostasis network is decomposed into different subpathways highly conserved across evolution and comprehends a collection of mechanisms related to protein synthesis, folding, trafficking, secretion, and degradation distributed in different compartments inside the cell (Balch et al., 2008; Powers & Balch, 2013). The main players of this network include chaperones and foldases, the ubiquitin–proteasome system, the autophagy pathway, the heat‐shock response, the unfolded protein response (UPR), the integrated stress response, the endoplasmic reticulum (ER)‐associated degradation machinery (ERAD), the mitochondrial UPR, and the mechanisms controlling redox balance (Balch et al., 2008). Those processes are dynamic and tightly coordinated by quality control systems to avoid proteotoxicity and ensure that unfolded or misfolded proteins do not accumulate into cytotoxic aggregates (Labbadia & Morimoto, 2014). Various pathological conditions affecting the nervous system share common molecular features despite presenting different clinical manifestations, highlighting the presence of abnormal protein aggregates in the brain of affected individuals (Walker et al., 2015). These age‐related diseases are classified as protein misfolding disorders (PMDs) and include Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), prion‐related disorders (PrDs), among others (Soto, 2003). Importantly, one of the main nodes of the proteostasis network involved in aging and PMDs is the UPR and the occurrence of abnormal levels of ER stress. Recent advances in model organisms have uncover the significance of the UPR to the control of global proteostasis during aging, where the nervous system has a central role in monitoring alterations in the health of the proteome to adjust the capacity of the cell to cope with ER stress in various peripheral tissues. Here, we discuss new concepts illustrating the functional relevance of the UPR to organismal aging across species and its significance as a risk factor to develop neurodegenerative diseases.
The unfolded protein response
The ER is the main site for the synthesis and folding of around one‐third of the total proteome of a cell (Braakman & Bulleid, 2011). Considered a key component of the proteostasis network, ER‐located proteins regulate folding and quality control through the activity of multiple chaperones, foldases, and co‐factors that assist the folding of nascent proteins as well as degradation pathways, thus preventing abnormal protein aggregation and resultant proteotoxicity (Ellgaard & Helenius, 2003; Kourtis & Tavernarakis, 2011; Hetz et al., 2015). Stressful stimuli such as hypoxia (Badiola et al., 2011), nutrient deprivation (Szegezdi et al., 2006), increased protein oxidation (Santos et al., 2009), and disturbance of the secretory pathway (Badr et al., 2007) may lead to an excessive accumulation of misfolded proteins at the ER, a process termed ER stress (Walter & Ron, 2011; Hetz, 2012). To cope with ER stress, a highly conserved signaling pathway is engaged known as the UPR (Wang & Kaufman, 2016). The UPR is initiated by the activation of at least three types of stress sensors including inositol‐requiring enzyme‐1 (IRE1), PKR‐like ER kinase (PERK), and activating transcription factor 6 (ATF6). IRE1 catalyzes the unconventional splicing of the mRNA encoding X‐box binding protein‐1 (XBP1) (Yoshida et al., 2001; Calfon et al., 2002; Lee et al., 2002), resulting in the expression of an active transcription factor called XBP1s that controls the expression of a cluster of genes related to folding and quality control mechanisms (Hetz et al., 2011). Additionally, IRE1 also degrades several mRNAs, ribosomal RNAs, and microRNAs through a process known as regulated IRE1‐dependent decay (RIDD), having an impact on different processes including inflammation and apoptosis (Maurel et al., 2014). IRE1 also engages distinct stress pathways, including JNK and NF‐κB, through the binding of adapter proteins (Hetz et al., 2015). Activation of PERK leads to the phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2α), which in turn inhibits translation decreasing protein influx into the ER (Ron & Walter, 2007). Paradoxically, some mRNAs, including activating transcription factor 4 (ATF4), are differentially translated, leading to the upregulation of genes related to redox homeostasis, amino acid metabolism, autophagy, and apoptosis control (Harding et al., 2003; Ye & Koumenis, 2009; B'Chir et al., 2014). Moreover, PERK activation has been shown to modulate the activity of nuclear factor erythroid 2‐related factor 2 (NRF2) and forkhead box O (FOXO), linking this pathway to the antioxidant response, insulin responsiveness, and autophagy (Chevet et al., 2015). Under ER stress conditions, ATF6 translocates to the Golgi apparatus where it is cleaved releasing ATF6f, a cytosolic active form. ATF6f exerts its action at the nuclear level as a transcription factor regulating genes associated with ERAD, in addition to enhancing XBP1 mRNA transcription (Yamamoto et al., 2007). Importantly, the control of gene expression by the UPR depends on the cellular context and the stimuli considering that UPR transcription factors can interact with other proteins to drive specific responses, in addition to be regulated by several post‐translational modifications (Hetz et al., 2015). Under chronic ER stress, the UPR triggers apoptosis through different mechanisms that involve the upregulation of CHOP, the induction of oxidative stress, exacerbated RIDD, upregulation of pro‐apoptotic components of the BCL‐2 family, among other mechanisms (Tabas & Ron, 2011; Urra et al., 2013). Thus, under conditions of ER stress, the UPR reprograms the cell toward adaptation, sustaining cell function or the engagement of cell death programs to eliminate irreversibly damaged cells.
ER proteostasis and aging in simple model organisms
During aging, organisms gradually accumulate intracellular aggregates composed by misfolded proteins, an event that is associated with a prominent decline in the buffering capacity of the proteostasis network and a consequent decrease in tissue and cellular function (Fig. 1) (Taylor & Dillin, 2011; Triplett et al., 2015). Several studies in model organisms have uncovered the significance of UPR signaling to the aging process, associated with protection against proteotoxicity (Ben‐Zvi et al., 2009; Labunskyy et al., 2014). For example, caloric restriction has been used as a major strategy to prevent the adverse effects of aging on healthspan (Riera & Dillin, 2015). In yeast, this intervention correlates with increased expression of HAC1, the functional homologue of XBP1s (Choi et al., 2013). Remarkably, genetic ablation of HAC1 abrogates the lifespan extension conferred by caloric restriction (Choi et al., 2013). Other studies indicated that the deletion of distinct UPR‐target genes impact replicative lifespan in yeast, a process dependent on the Ire1p/HAC1 axis (Labunskyy et al., 2014). Furthermore, genetic modifications to improve the activity of the UPR enhance replicative lifespan in Saccharomyces cerevisiae (Cui et al., 2015).
Figure 1.
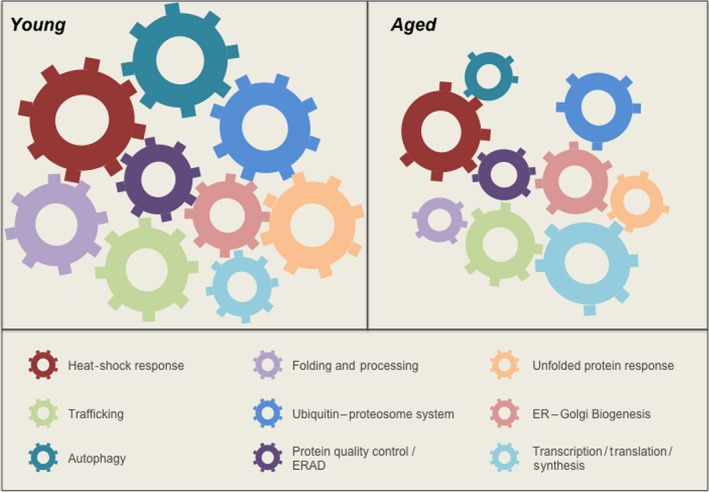
The buffering capacity of the proteostasis network decreases with aging. The aging process is directly associated with a range of specific alterations in distinct components of the proteostasis network. Such alterations disrupt the healthy functioning of cell and may contribute to the emergence of disease.
Studies in Caenorhabditis elegans have revealed a fundamental role of the UPR in adjusting organismal proteostasis during aging through a neuronal control. Exposure of Caenorhabditis elegans to pharmacological inducers of ER stress indicated that the ability of aged worms to respond is significantly reduced compared with young animals (Ben‐Zvi et al., 2009). Interestingly, the same observation was reported when animals were stimulated with heat shock (Ben‐Zvi et al., 2009), suggesting global proteostatic defects during aging. Enforced expression of heat‐shock factor 1 (HSF‐1) or the FOXO‐transcription factor DAF‐16 restores proteostasis of aged worms (Ben‐Zvi et al., 2009). Loss‐of‐function studies in Caenorhabditis elegans demonstrated that lifespan extension conferred by XBP1 expression is dependent on insulin/IGF‐1‐FOXO signaling, a classical pathway associated with aging (Henis‐Korenblit et al., 2010; Douglas et al., 2015). Importantly, lifespan extension was only achieved through the parallel interaction between XBP1 and FOXO‐transcription factor DAF‐16, which acts in conjunction to genes related to longevity (Henis‐Korenblit et al., 2010). According to these observations, Taylor and Dillin demonstrated that the selective overexpression of Xbp1s in neurons or intestine strongly reverts the age‐related susceptibly to ER stress stimulation (Table 1) (Taylor & Dillin, 2013). Remarkably, the overexpression of XBP1s in neurons has a strong impact on lifespan extension, augmenting animal survival up to 30%. A recent study also indicated that loss‐of‐function mutations in distinct subunits of translation initiation factor eIF‐3 confer a 40% extension in the lifespan of Caenorhabditis elegans through a DAF‐16‐dependent and UPR‐independent pathway (Cattie et al., 2016), suggesting that different nodes of the proteostasis network significantly contribute to aging in worms.
Table 1.
Genetic manipulation of the unfolded protein response (UPR) affecting the aging process in different model organisms. Examples of genetic manipulation to UPR components in different species that modifies lifespan with modified genetic backgrounds (Alg12, Bst1, Pmt‐1, or Daf‐2 mutants) or under metabolic stimulus (caloric or dietary restriction)
| Model organism/species | Genetic modification of UPR | Effect on aging | Stimulus/genetic background | References |
|---|---|---|---|---|
| Saccharomyces cerevisiae | Decreased expression of Hac1 | Decreased lifespan | Caloric restriction | Choi et al. (2013) |
| Deletion of Hac1 or Ire1p | Decreased lifespan | Deletion of Alg12 and Bst1 | Labunsky et al. (2014) | |
| Deletion of Hac1 or Ire1p | Decreased replicative lifespan | Deletion of Pmt‐1 | Cui et al. (2015) | |
| Caenorhabditis elegans | Deletion of Ire1 or Xbp1 | Decreased lifespan | Deletion of Daf‐2 | Henis‐Korenblit et al. (2010) |
| Deletion of Atf6 | No effect on lifespan | Deletion of Daf‐2 | ||
| Overexpression of Xbp1 and Daf‐16 | Extended lifespan, through the expression of longevity genes | Deletion of Daf‐2 | ||
| Neuronal overexpression of Xbp1s | Increased lifespan through cell‐nonautonomous mechanism | Basal levels | Taylor & Dillin (2013) | |
| Drosophila melanogaster | Deletion of Perk on intestinal stem cells | Increased lifespan | Basal levels | Wang et al. (2015) |
| Deletion of Ire1 or Xbp1 | Decreased lifespan | Dietary restriction | Luis et al. (2016) |
Studies in flies have also defined contributed to define the relevance of the UPR to the aging process. Intestinal stem cells in Drosophila melanogaster promote a regenerative response upon UPR activation, a process deregulated during aging (Wang et al., 2014). Later, Wang et al. demonstrated that PERK is specifically activated in intestinal stem cells, having a functional role in promoting healthspan in flies. However, chronic engagement of this pathway becomes deleterious during aging in Drosophila melanogaster (Wang et al., 2015). A previous study also indicated that Xbp1 is both sufficient and required to limit intestinal stem cell proliferation (Wang et al., 2014). Recently, the same group reported that engagement of the Ire1/Xbp1 branch also results in lifespan extension in the same model under dietary restriction. The activation of Ire1/Xbp1 pathway in enterocytes under dietary restriction has a positive impact on lifespan of gut cells by regulating lipid synthesis (Luis et al., 2016). Overall, the functional significance of ER stress signaling to lifespan control has been inferred from several studies in simple model organisms.
Cell‐nonautonomous control of organismal aging by the UPR
A novel concept is emerging based on research using fly and worm models of aging, indicating that the ER proteostasis network promotes health and lifespan through cell‐nonautonomous mechanisms, impacting whole organismal proteostasis (Mardones et al., 2015). Studies in Caenorhabditis elegans revealed that besides its importance in individual cells, the UPR acts as a key player in modulating global organism adaptability to stress during aging by integrating information at the level of the nervous system (Martinez et al., 2016a). Accordingly, UPR can be activated on a cell‐nonautonomous manner (Taylor & Dillin, 2013). The ectopic expression of XBP1s in neurons is able to engage a distal UPR activation in the intestine, thus increasing stress resistance and longevity in Caenorhabditis elegans (Taylor & Dillin, 2013). These results suggest that the nervous system may act as a central integrator and adjustor of global proteostasis, with possible major distal effects in the intestine. Importantly, other studies previously demonstrated that neuronal UPR regulates the innate immunity in the gut on a cell‐nonautonomous manner (Martinez & Hetz, 2012; Sun et al., 2012; Aballay, 2013). Chromatin remodeling factors in neurons can also engage ER stress responses through a cell‐nonautonomous mechanism (Kozlowski et al., 2014). Thus, accumulating evidence supports the idea that when an organism is exposed to environmental or pathogenic challenges, the ability of the nervous system to integrate these signals through the activation of the UPR favors the maintenance of homeostasis in various peripheral organs (Mardones et al., 2015). A similar model has been proposed for the heat‐shock response by Morimoto's group, where HSF‐1 in neurons regulates global responses to aging in the gut (Morley & Morimoto, 2004; van Oosten‐Hawle & Morimoto, 2014a; Douglas et al., 2015). Importantly, cell‐nonautonomous control of aging‐related pathways has been extensively described in different model organisms mediated by distinct signaling molecular mediators (Taylor et al., 2014; Leiser et al., 2015; Schinzel & Dillin, 2015). A recent study indicated that PERK is activated in intestinal stem cells by JAK/Stat signaling in response to ER stress in neighboring cells, regulating intestinal homeostasis and lifespan in flies (Wang et al., 2015). A cell‐nonautonomous mechanism has been also described in mammals, where overexpression of XBP1s in the hypothalamus modulates global energy balance through the propagation of signals to the liver and adipose tissue to adjust energy metabolism (Williams et al., 2014). Furthermore, the concept of ‘transcellular chaperone signaling’ was proposed in Caenorhabditis elegans where cells suffering stress from the accumulation of protein aggregates propagate signals to the neighbor tissue to induce adaptive responses and resist further damage (van Oosten‐Hawle & Morimoto, 2014b).
The molecular components that enable communication or signal propagation from neurons to the periphery are still unknown. It also remains to be established whether the activation of the UPR through cell‐nonautonomous mechanisms depends on the induction of ER stress in the target tissue or whether it is actually mediated by signaling events that engage UPR sensors in the absence of stress features (Mardones et al., 2015). This second possibility may be feasible since many examples are available showing that UPR stress sensors can be modulated by post‐translational modifications or protein–protein interactions in the absence of protein misfolding (Hetz, Chevet et al., 2015). Taylor and Dillin showed that the induction of cell‐nonautonomous UPR responses depends on the release of small clear vesicles from synaptic terminals of neurons expressing XBP1s (Taylor & Dillin, 2013). Additionally, XBP1s expression in neurons can promote the expression of brain‐derived neurotrophic factor (BDNF), which may in turn engage IRE1 signaling in a stress‐independent manner (Martinez et al., 2016b). Still, the specific mechanism of global proteostasis control in mammals and the neuronal circuits governing the propagation of UPR signals between cells remain to be determined. Although cell‐nonautonomous control of proteostasis has been validated in simple model organisms during aging, the occurrence of this process in mammals is still an open question. Overall, these examples illustrate how stress signals spread systemically in the organism to induce adaptive programs.
ER stress response in mammalian aging
Although proteostasis impairment and ER stress are proposed as one of the hallmarks of aging, most of the studies addressing the contribution of the UPR to mammalian aging rely mostly on correlative data. For example, the expression of the ER chaperones BiP, calnexin, and PDI has been reported to be downregulated in the hippocampus of aged rats while pro‐apoptotic mediators such as CHOP and the ER‐located caspase‐12 are increased (Paz Gavilan et al., 2006; Gavilan et al., 2009). Additionally, another report demonstrated that levels of both ATF4 and BiP are decreased in this tissue (Hussain & Ramaiah, 2007). In aged brains, basal expression of PERK, GADD34, and total eIF2α is augmented contrasting with reduced levels of eIF2α phosphorylation (Hussain & Ramaiah, 2007). Moreover, young mice under sleep deprivation showed an increase in BiP and eIF2α phosphorylation, which was not observed in aged mice, but there was an upregulation of GADD34, CHOP, and caspase‐12 (Naidoo et al., 2008). Another study demonstrated similar observations in the pancreas of mice (Naidoo et al., 2014). Additionally, aged macrophages exhibit diminished IRE1 activation and increased susceptibility to ER stress‐dependent apoptosis (Song et al., 2013). These findings suggest that the ability to engage the UPR may be disrupted during aging; however, the functional significance of these observations is unknown (Fig. 2).
Figure 2.
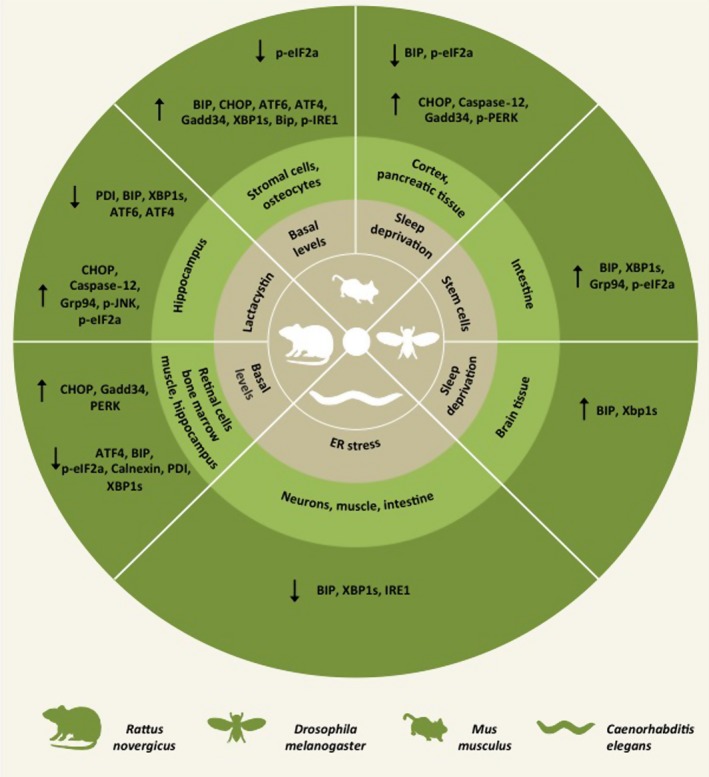
Endoplasmic reticulum (ER) stress in aging across species. The aging process is directly associated with a range of specific alterations in distinct components of the ER proteostasis network in different tissues, highlighting changes in components of the unfolded protein response and the folding machinery.
In contrast, several reports suggest that chronic ER stress is associated with aging in multiple tissues. Increased levels of CHOP, ATF4, and XBP1s were observed in primary osteocytes from aged mice when compared to adult mice exposed to ER stress‐inducing agents (Chalil et al., 2015). Similar results were reported in stromal cells from adipose tissue of aged mice, associated with augmented levels of BIP, CHOP, ATF6, and phosphorylated IRE1 (Ghosh et al., 2015). CHOP was also shown to be induced at baseline in muscular tissue of the hindlimb of aged rats (Baehr et al., 2016). Of interest, gene expression profile studies demonstrated that UPR‐target genes are one of the most affected pathways in bone marrow during aging (Kannan et al., 2016). At the level of the central nervous system, phosphorylation of CHOP, ATF6, GADD34, ATF4, and eIF2α are also upregulated in the retina of aged rats (Lenox et al., 2015).
ER stress signaling components have been shown to interact with classical aging‐related pathways suggesting a functional interconnection. For example, in the context of HD, we showed that XBP1 negatively regulates FOXO1 levels (Vidal et al., 2012). Several reports have also linked ER stress responses with the control of autophagy (reviewed in Vidal et al., 2014), a central pathway involved in proteostasis control (Kaushik & Cuervo, 2015). Insulin signaling is also linked to IRE1 function, as demonstrated in models of diabetes and obesity (Ozcan et al., 2004). Overall, these studies depict a general concept where mammalian aging is directly associated with the occurrence of chronic ER stress. Those findings may be explained by accumulative ER damage rather than an attenuation of UPR responses. Currently, functional analysis is needed to define the actual contribution of ER proteostasis to mammalian aging. Since a variety of mouse models are available to target specific UPR components in various tissues (Cornejo et al., 2013), the means to answer this fundamental question are already available.
Aging as a risk factor to undergo neurodegeneration: a role of ER stress?
Abnormal aggregation of specific proteins is a hallmark of age‐related neurodegenerative diseases. Increasing evidence indicates that despite the fact that PMD‐related proteins distribute in different subcellular locations and have distinct binding partners, a common pathological consequence of their accumulation is the occurrence of ER stress. This mechanistic convergence is explained by the observation that disease‐related proteins actually disrupt the function of one or more components of the proteostasis network, highlighting the inhibition of ERAD, altered vesicle trafficking between the ER and Golgi, perturbed ER calcium homeostasis, autophagy dysregulation, and abnormal interactions with ER chaperones (Hetz & Mollereau, 2014; Vidal et al., 2014; Kaushik & Cuervo, 2015).
Importantly, with the exception of HD, familiar cases of PMDs account for less than 10% of cases, indicating that protein aggregation occurs in the absence of genetic mutations to the affected proteins. This observation suggests that alteration in different components of the proteostasis network during aging may contribute to protein aggregation. The involvement of ER stress in PMDs is highly complex, acting both as protective or detrimental (Hetz & Mollereau, 2014; Scheper & Hoozemans, 2015; Freeman & Mallucci, 2016). Indeed, the activity of the UPR in neurodegenerative diseases could either enhance or reduce neurodegeneration, depending on the process that is modulated by specific ER stress signals and the particular disease studied. A strong correlation between ER stress markers and signs of neurodegeneration has been reported in human postmortem tissue and animal models of PMDs. Remarkably, human neurons derived from induced pluripotent stem cells of AD, PD, and ALS patients revealed that ER stress is a prominent feature of this disease model (Chung et al., 2013; Kondo et al., 2013; Matus et al., 2014; Lee & Huang, 2017). Other studies also suggest that ATF4 may enhance axonal degeneration in AD through cell‐nonautonomous mechanisms (Baleriola et al., 2014; Wei et al., 2015). Importantly, the repressive effects of ER stress over protein synthesis were shown to contribute to the cognitive impairment observed in AD and PrD models by blocking the expression of synaptic proteins (Moreno et al., 2012, 2013; Freeman & Mallucci, 2016). Studies in Drosophila melanogaster (Loewen & Feany, 2010; Casas‐Tinto et al., 2011) and Caenorhabditis elegans (Safra et al., 2013) reported a functional role of XBP1 in neurodegeneration in AD. Interestingly, a polymorphism in the XBP1 promoter previously associated with bipolar disorders and schizophrenia (Kakiuchi et al., 2003; Du et al., 2008; Kim et al., 2009) was also pointed as a risk factor to develop AD in the Chinese population (Liu et al., 2013). In agreement with these findings, a new physiological function of XBP1 was proposed in the hippocampus in the control of learning and memory processes (Martinez et al., 2016b).
Genetic ablation of XBP1 in the nervous system uncovered a dynamic interconnection between the UPR and the autophagy pathway to handle protein aggregation. XBP1 deficiency protects against the development of experimental HD and ALS due to an increase in autophagy levels (Hetz et al., 2009; Vidal et al., 2012). In the context of PD, XBP1 deficiency also provided neuroprotection associated with the basal upregulation of several components of the ER proteostasis network, possibly reflecting the induction of nonlethal stress levels at the substantia nigra (Valdes et al., 2014). The concept of hormesis was proposed as an adaptive mechanism where a mild perturbation to neuronal proteostasis triggers compensatory mechanisms that enhance the capacity of the cell to cope with stress (Mollereau et al., 2016). In fact, treatment of animals with nonlethal doses of the ER stress agent tunicamycin (an inhibitor of N‐glycosylation) provides protection against PD possibly due to the induction of autophagy (Fouillet et al., 2012). Many other functional studies illustrate the therapeutic consequences of enforcing UPR adaptive outputs in ALS, PD, and HD (reviewed in Hetz & Mollereau, 2014; Freeman & Mallucci, 2016; Scheper & Hoozemans, 2015).
Recent evidence suggests that ER stress may underlay the differential neuronal vulnerability observed in neurodegenerative diseases, where most of the advances have been reported in ALS models (Rozas et al., 2016; Ruegsegger & Saxena, 2016). Disruption to the ER folding network is emerging as a key factor underlying the susceptibility of specific neuronal populations to undergo neurodegeneration (Filezac de L'Etang et al., 2015). In addition, genetic evidence has placed the ER proteostasis network in the etiology of ALS as mutations in two‐disulfide isomerase (PDIA1 and ERp57) were proposed as risk factors to develop ALS (Gonzalez‐Perez et al., 2015; Woehlbier et al., 2016). Alterations to the ER folding network may result in abnormal synthesis of synaptic proteins, having a negative effect on the integrity of neuromuscular junctions and neuronal connectivity (Bernard‐Marissal et al., 2012, 2015; Woehlbier et al., 2016). Similarly, genetic inactivation in BiP or its cofactor SIL1 results in spontaneous degeneration, leading to abnormal protein aggregation during aging (Zhao et al., 2005; Jin et al., 2014). Taken together, these studies suggest that alterations to the function of the ER during aging may contribute to synaptic dysfunction and abnormal protein aggregation, increasing the risk to develop neurodegenerative diseases.
Concluding remarks
Imbalance of neuronal proteostasis is one of the pathological hallmarks of aging, and understanding its molecular defects will contribute to develop strategies to intervene age‐associated disorders. Because the nervous system is highly dynamic and plastic, the manifestation of clinical features in patients arises very late, after severe damage has already occurred. Likely, it is predicted that the development of strategies to improve the quality of the aging process will substantially reduce the probability to undergo PMDs. Despite the fact that proteostasis is composed of a complex network of individual interconnected signaling pathways, recent findings suggest that the maintenance of ER physiology is a prominent molecular target to prevent age‐related diseases affecting the nervous system. The involvement of ER stress in the biology of aging is complex as illustrated by most recent advances. The activity of the ER proteostasis network may not only operate as a mechanism to handle abnormal protein aggregation, but it is also proposed as an adjuster of brain function through fine‐tuning synaptic function. Specific neuronal populations are highly vulnerable to perturbations to ER function possibly because their metabolic state depends on the basal activity of the UPR. Furthermore, the UPR may orchestrate repair processes of the nervous system by controlling the expression of neurotrophins such as BDNF, and the regenerative capacity of axons and stem cells pools (Castillo et al., 2015; Martinez et al., 2016b; Onate et al., 2016). Regarding inflammatory reactions, the UPR is known to have important functions in macrophages and dendritic cells by modulating the secretion of pro‐inflammatory cytokines (Bettigole & Glimcher, 2015). In this context, future efforts should address the importance of the UPR to brain inflammation and the activity of astrocytes, microglia, and oligodendrocytes during aging. The fact that the UPR participates in the adjustment of energy and lipid metabolism, an additional layer of complexity, could be also explored to link the UPR with brain aging. Finally, the discovery of cell‐nonautonomous UPR responses and its relation to healthspan control adds a new concept as ER stress‐related signals in the brain may influence the capacity of the whole organism to adapt and cope with ER stress. All those aspects should be considered in future studies aiming to define the relative impact of ER stress on mammalian brain aging and its significance as a risk factor to develop neurodegenerative diseases.
Several novel small molecules are available to target selective UPR components and reduce ER stress levels (Table 2; Hetz et al., 2013; Maly & Papa, 2014; Gallagher & Walter, 2016; Gallagher et al., 2016; Axten, 2017), which promises possible new avenues to intervene the aging process. Importantly, some of these compounds have already been tested in preclinical models of PMDs (Table 2). However, it is important to consider possible side effects as the activity of the UPR has been linked to the physiology of many peripheral organs and the long‐term administration of UPR‐targeting drugs is predicted to induce liver failure, altered immune system function, pancreatic problems, among others maladies (Dufey et al., 2014). In this scenario, gene therapy is emerging as a strategy to locally reduce ER stress by delivering adaptive components of the UPR (i.e., XBP1s, BiP) specifically into the brain regions affected by distinct neurodegenerative diseases (Valenzuela et al., 2016). Overall, although the UPR is emerging as a central and evolutionarily conserved modulator of the normal process of aging, data available in mammalian systems are still correlative and remain to be functionally explored. As the UPR field has greatly evolved in the last five years in terms of generation of animal models and pharmacological tools, it is expected to witness future advances to underscore the significance of the UPR to brain aging and its relation to neurodegenerative diseases.
Table 2.
Pharmacological modulation of the unfolded protein response (UPR). A summary is presented of chemically synthesized compounds to activate or inhibit the different UPR signaling components, including their efficacy in preclinical models of neurodegenerative diseases. Those components may emerge as candidates for lifespan/healthspan extension in the future (Maly & Papa, 2014; Gallagher et al., 2016; Gallagher & Walter, 2016; Plate et al. 2016; Axten, 2017)
| UPR branch related | Drug | Molecular target | Effect | Readout | Model/disease |
|---|---|---|---|---|---|
| PERK | GSK2656157 and GSK2606414 | PERK kinase domain | Inhibitor | Inhibition of eIF2α phosphorylation | Mouse/PrD, Tauopathies |
| Integrated stress response inhibitor (ISRIB) | Guanine nucleotide exchange factor elF2B | Inhibitor | Decreased ATF4 expression | Mouse/PrD — memory and cognition | |
| Salubrinal | Binding GADD34 phosphatase complex | Inhibitor | Repression of translation, decrease in protein misfolding overload | Rat‐mouse/ALS, PD, Prion disease, spinal cord injury, multiple sclerosis, Charcot–Marie–Tooth 1B | |
| Guanabenz and Sephinl1 | PPP1R15A elF2a phosphatase | Inhibitor | Repression of translation, decrease in protein misfolding overload | Mouse/ALS, Charcot–Marie–Tooth 1B | |
| IRE1α | MKC‐3946 SFT‐083010 | RNase active site | Inhibitor | Decrease of mRNA XBPI splicing | Mouse/cancer models |
| Kinase inhibiting RNase attenuators 3 and 6 (Kira 3 and Kira 6) | Kinase domain | Inhibitor | Reduce IRE1α signaling | Mouse/diabetes and retinal damage | |
| ATF6 | Ceapin‐A1 Ceapin‐A7 | Inhibitor | Inhibition of translocation of ATF6 | Cells | |
| Small activators molecules | Activator | Induction of UPR‐regulated genes profile expression | Cells |
Funding
This work was directly funded by FONDAP program 15150012, US Office of Naval Research‐Global (ONR‐G) N62909‐16‐1‐2003, Millennium Institute P09‐015‐F, FONDEF ID16I10223, FONDEF D11E1007, U.S. Air Force Office of Scientific Research FA9550‐16‐1‐0384, CONICYT‐Brazil 441921/2016‐7, FONDECYT no 11160760 (CDA), and FONDECYT no 3150637 (GM). We also thank the support from ALS Therapy Alliance 2014‐F‐059, Muscular Dystrophy Association 382453, Michael J Fox Foundation for Parkinson's Research—Target Validation grant No 9277, FONDECYT no. 1140549, and ALSRP Therapeutic Idea Award AL150111 (CH).
Conflict of interest
Authors declare that they have no conflict of interest.
References
- Aballay A (2013) Role of the nervous system in the control of proteostasis during innate immune activation: insights from C. elegans . PLoS Pathog. 9, e1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axten JM (2017) Protein kinase R(PKR)‐like endoplasmic reticulum kinase (PERK) inhibitors: a patent review (2010–2015). Expert Opin. Ther. Pat. 27, 37–48. [DOI] [PubMed] [Google Scholar]
- Badiola N, Penas C, Minano‐Molina A, Barneda‐Zahonero B, Fado R, Sanchez‐Opazo G, Comella JX, Sabria J, Zhu C, Blomgren K, Casas C, Rodriguez‐Alvarez J (2011) Induction of ER stress in response to oxygen‐glucose deprivation of cortical cultures involves the activation of the PERK and IRE‐1 pathways and of caspase‐12. Cell Death Dis. 2, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr CE, Hewett JW, Breakefield XO, Tannous BA (2007) A highly sensitive assay for monitoring the secretory pathway and ER stress. PLoS One 2, e571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, West DW, Marcotte G, Marshall AG, De Sousa LG, Baar K, Bodine SC (2016) Age‐related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 8, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Baleriola J, Walker CA, Jean YY, Crary JF, Troy CM, Nagy PL, Hengst U (2014) Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B'Chir W, Chaveroux C, Carraro V, Averous J, Maurin AC, Jousse C, Muranishi Y, Parry L, Fafournoux P, Bruhat A (2014) Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell. Signal. 26, 1385–1391. [DOI] [PubMed] [Google Scholar]
- Ben‐Zvi A, Miller EA, Morimoto RI (2009) Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA 106, 14914–14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard‐Marissal N, Moumen A, Sunyach C, Pellegrino C, Dudley K, Henderson CE, Raoul C, Pettmann B (2012) Reduced calreticulin levels link endoplasmic reticulum stress and Fas‐triggered cell death in motoneurons vulnerable to ALS. J. Neurosci. 32, 4901–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard‐Marissal N, Sunyach C, Marissal T, Raoul C, Pettmann B (2015) Calreticulin levels determine onset of early muscle denervation by fast motoneurons of ALS model mice. Neurobiol. Dis. 73, 130–136. [DOI] [PubMed] [Google Scholar]
- Bettigole SE, Glimcher LH (2015) Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 33, 107–138. [DOI] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99. [DOI] [PubMed] [Google Scholar]
- Brayne C (2007) The elephant in the room – healthy brains in later life, epidemiology and public health. Nat. Rev. Neurosci. 8, 233–239. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- Casas‐Tinto S, Zhang Y, Sanchez‐Garcia J, Gomez‐Velazquez M, Rincon‐Limas DE, Fernandez‐Funez P (2011) The ER stress factor XBP1s prevents amyloid‐beta neurotoxicity. Hum. Mol. Genet. 20, 2144–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo V, Onate M, Woehlbier U, Rozas P, Andreu C, Medinas D, Valdes P, Osorio F, Mercado G, Vidal RL, Kerr B, Court FA, Hetz C (2015) Correction: Functional role of the disulfide isomerase erp57 in axonal regeneration. PLoS One 10, e0140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattie DJ, Richardson CE, Reddy KC, Ness‐Cohn EM, Droste R, Thompson MK, Gilbert WV, Kim DH (2016) Mutations in nonessential eIF3k and eIF3 l genes confer lifespan extension and enhanced resistance to ER Stress in Caenorhabditis elegans . PLoS Genet. 12, e1006326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalil S, Jaspers RT, Manders RJ, Klein‐Nulend J, Bakker AD, Deldicque L (2015) Increased endoplasmic reticulum stress in mouse osteocytes with aging alters Cox‐2 response to mechanical stimuli. Calcif. Tissue Int. 96, 123–128. [DOI] [PubMed] [Google Scholar]
- Chevet E, Hetz C, Samali A (2015) Endoplasmic reticulum stress‐activated cell reprogramming in oncogenesis. Cancer Discov. 5, 586–597. [DOI] [PubMed] [Google Scholar]
- Choi KM, Kwon YY, Lee CK (2013) Characterization of global gene expression during assurance of lifespan extension by caloric restriction in budding yeast. Exp. Gerontol. 48, 1455–1468. [DOI] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schule B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, Jaenisch R, Lindquist S (2013) Identification and rescue of alpha‐synuclein toxicity in Parkinson patient‐derived neurons. Science 342, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo VH, Pihan P, Vidal RL, Hetz C (2013) Role of the unfolded protein response in organ physiology: lessons from mouse models. IUBMB Life 65, 962–975. [DOI] [PubMed] [Google Scholar]
- Cui HJ, Liu XG, McCormick M, Wasko BM, Zhao W, He X, Yuan Y, Fang BX, Sun XR, Kennedy BK, Suh Y, Zhou ZJ, Kaeberlein M, Feng WL (2015) PMT1 deficiency enhances basal UPR activity and extends replicative lifespan of Saccharomyces cerevisiae . Age (Dordr) 37, 9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas PM, Baird NA, Simic MS, Uhlein S, McCormick MA, Wolff SC, Kennedy BK, Dillin A (2015) Heterotypic signals from neural HSF‐1 separate thermotolerance from longevity. Cell Rep. 12, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Duan S, Wang H, Chen W, Zhao X, Zhang A, Wang L, Xuan J, Yu L, Wu S, Tang W, Li X, Li H, Feng G, Xing Q, He L (2008) Comprehensive analysis of polymorphisms throughout GAD1 gene: a family‐based association study in schizophrenia. J. Neural. Transm. 115, 513–519. [DOI] [PubMed] [Google Scholar]
- Dufey E, Sepulveda D, Rojas‐Rivera D, Hetz C (2014) Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 307, C582–C594. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191. [DOI] [PubMed] [Google Scholar]
- Filezac de L'Etang A, Maharjan N, Cordeiro Brana M, Ruegsegger C, Rehmann R, Goswami A, Roos A, Troost D, Schneider BL, Weis J, Saxena S (2015) Marinesco‐Sjogren syndrome protein SIL1 regulates motor neuron subtype‐selective ER stress in ALS. Nat. Neurosci. 18, 227–238. [DOI] [PubMed] [Google Scholar]
- Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, Belaidi E, Ovize M, Touret M, Nataf S, Mollereau B (2012) ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman OJ, Mallucci GR (2016) The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 1648, 530–537. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Walter P (2016) Ceapins inhibit ATF6alpha signaling by selectively preventing transport of ATF6alpha to the Golgi apparatus during ER stress. Elife 5, (e11880), 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher CM, Garri C, Cain EL, Ang KK, Wilson CG, Chen S, Hearn BR, Jaishankar P, Aranda‐Diaz A, Arkin MR, Renslo AR, Walter P (2016) Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife 5, (eLife.11878.001), 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavilan MP, Pintado C, Gavilan E, Jimenez S, Rios RM, Vitorica J, Castano A, Ruano D (2009) Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 8, 654–665. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Garg SK, Mau T, O'Brien M, Liu J, Yung R (2015) Elevated endoplasmic reticulum stress response contributes to adipose tissue inflammation in aging. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Perez P, Woehlbier U, Chian RJ, Sapp P, Rouleau GA, Leblond CS, Daoud H, Dion PA, Landers JE, Hetz C, Brown RH (2015) Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 566, 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Henis‐Korenblit S, Zhang P, Hansen M, McCormick M, Lee SJ, Cary M, Kenyon C (2010) Insulin/IGF‐1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc. Natl. Acad. Sci. USA 107, 9730–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. [DOI] [PubMed] [Google Scholar]
- Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 1–19. [DOI] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH (2009) XBP‐1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH (2011) The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 91, 1219–1243. [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP (2013) Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Oakes SA (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SG, Ramaiah KV (2007) Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 355, 365–370. [DOI] [PubMed] [Google Scholar]
- Jin H, Mimura N, Kashio M, Koseki H, Aoe T (2014) Late‐onset of spinal neurodegeneration in knock‐in mice expressing a mutant BiP. PLoS One 9, e112837.25405877 [Google Scholar]
- Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I, Tsujita T, Okazaki Y, Nanko S, Kunugi H, Sasaki T, Kato T (2003) Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet. 35, 171–175. [DOI] [PubMed] [Google Scholar]
- Kannan S, Dawany N, Kurupati R, Showe LC, Ertl HC (2016) Age‐related changes in the transcriptome of antibody‐secreting cells. Oncotarget 7, 13340–13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM (2015) Proteostasis and aging. Nat. Med. 21, 1406–1415. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss‐Coray T, Sierra F (2014) Geroscience: linking aging to chronic disease. Cell 159, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Kim CY, Lee MJ, Joo YH (2009) Preliminary evidence on the association between XBP1‐116C/G polymorphism and response to prophylactic treatment with valproate in bipolar disorders. Psychiatry Res. 168, 209–212. [DOI] [PubMed] [Google Scholar]
- Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, Takahashi K, Asaka I, Aoi T, Watanabe A, Watanabe K, Kadoya C, Nakano R, Watanabe D, Maruyama K, Hori O, Hibino S, Choshi T, Nakahata T, Hioki H, Kaneko T, Naitoh M, Yoshikawa K, Yamawaki S, Suzuki S, Hata R, Ueno SI, Seki T, Kobayashi K, Toda T, Murakami K, Irie K, Klein WL, Mori H, Asada T, Takahashi R, Iwata N, Yamanaka S, Inoue H (2013) Modeling Alzheimer's Disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 12, 487–496. [DOI] [PubMed] [Google Scholar]
- Kourtis N, Tavernarakis N (2011) Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J. 30, 2520–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski L, Garvis S, Bedet C, Palladino F (2014) The Caenorhabditis elegans HP1 family protein HPL‐2 maintains ER homeostasis through the UPR and hormesis. Proc. Natl. Acad. Sci. USA 111, 5956–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI (2014). Proteostasis and longevity: when does aging really begin? F1000Prime Rep. 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labunskyy VM, Gerashchenko MV, Delaney JR, Kaya A, Kennedy BK, Kaeberlein M, Gladyshev VN (2014) Lifespan extension conferred by endoplasmic reticulum secretory pathway deficiency requires induction of the unfolded protein response. PLoS Genet. 10, e1004019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SL, Yassa MA (2015) Neurocognitive aging and the hippocampus across species. Trends Neurosci. 38, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Huang EJ (2017) Modeling ALS and FTD with iPSC‐derived neurons. Brain Res. 1656, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ (2002) IRE1‐mediated unconventional mRNA splicing and S2P‐mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser SF, Miller H, Rossner R, Fletcher M, Leonard A, Primitivo M, Rintala N, Ramos FJ, Miller DL, Kaeberlein M (2015) Cell nonautonomous activation of flavin‐containing monooxygenase promotes longevity and health span. Science 350, 1375–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenox AR, Bhootada Y, Gorbatyuk O, Fullard R, Gorbatyuk M (2015) Unfolded protein response is activated in aged retinas. Neurosci. Lett. 609, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Wang W, Cai ZY, Yao LF, Chen ZW, Wang CY, Zhao B, Li KS (2013) Polymorphism ‐116C/G of human X‐box‐binding protein 1 promoter is associated with risk of Alzheimer's Disease. CNS Neurosci. Ther. 19, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen CA, Feany MB (2010) The unfolded protein response protects from tau neurotoxicity in vivo . PLoS One 5, e13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis NM, Wang L, Ortega M, Deng H, Katewa SD, Li PW, Karpac J, Jasper H, Kapahi P (2016) Intestinal IRE1 is required for increased triglyceride metabolism and longer lifespan under dietary restriction. Cell Rep. 17, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly DJ, Papa FR (2014) Druggable sensors of the unfolded protein response. Nat. Chem. Biol. 10, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardones P, Martinez G, Hetz C (2015) Control of systemic proteostasis by the nervous system. Trends Cell Biol. 25, 1–10. [DOI] [PubMed] [Google Scholar]
- Martinez G, Hetz C (2012) Cell‐nonautonomous control of the UPR. EMBO Rep. 13, 767–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G, Duran‐Aniotz C, Cabral‐Miranda F, Hetz C (2016a) Commentary: XBP‐1 is a cell‐nonautonomous regulator of stress resistance and longevity. Front. Aging Neurosci. 8, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G, Vidal RL, Mardones P, Serrano FG, Ardiles AO, Wirth C, Valdes P, Thielen P, Schneider BL, Kerr B, Valdes JL, Palacios AG, Inestrosa NC, Glimcher LH, Hetz C (2016b) Regulation of memory formation by the transcription factor XBP1. Cell Rep. 14, 1382–1394. [DOI] [PubMed] [Google Scholar]
- Matus S, Medinas DB, Hetz C (2014) Common ground: stem cell approaches find shared pathways underlying ALS. Cell Stem Cell 14, 697–699. [DOI] [PubMed] [Google Scholar]
- Maurel M, Chevet E, Tavernier J, Gerlo S (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254. [DOI] [PubMed] [Google Scholar]
- Mollereau BNM, Rzechorzek BD, Roussel M, Sedru DM, Van dBB, Bailly‐Maitre F, Palladino DB, Medinas PM, Domingos S, Hunot S, Chandran S, Birman T, Baron D, Vivien CB, Duarte HD, Ryoo H, Steller F, Urano E, Chevet G, Kroemer A, Ciechanover EJ, Calabrese R, Kaufman J, Hetz C (2016) Adaptive preconditioning in neurological diseases – therapeutic insights from proteostatic perturbations. Brain Res. 1648, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR (2012) Sustained translational repression by eIF2alpha‐P mediates prion neurodegeneration. Nature 485, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion‐infected mice. Sci. Transl. Med. 5, 206ra138. [DOI] [PubMed] [Google Scholar]
- Morley JF, Morimoto RI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 15, 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo N, Ferber M, Master M, Zhu Y, Pack AI (2008) Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo N, Davis JG, Zhu J, Yabumoto M, Singletary K, Brown M, Galante R, Agarwal B, Baur JA (2014) Aging and sleep deprivation induce the unfolded protein response in the pancreas: implications for metabolism. Aging Cell 13, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onate M, Catenaccio A, Martinez G, Armentano D, Parsons G, Kerr B, Hetz C, Court FA (2016) Activation of the unfolded protein response promotes axonal regeneration after peripheral nerve injury. Sci. Rep. 6, 21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten‐Hawle P, Morimoto RI (2014a) Organismal proteostasis: role of cell‐nonautonomous regulation and transcellular chaperone signaling. Genes Dev. 28, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten‐Hawle P, Morimoto RI (2014b) Transcellular chaperone signaling: an organismal strategy for integrated cell stress responses. J. Exp. Biol. 217, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. [DOI] [PubMed] [Google Scholar]
- Paz Gavilan M, Vela J, Castano A, Ramos B, del Rio JC, Vitorica J, Ruano D (2006) Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 27, 973–982. [DOI] [PubMed] [Google Scholar]
- Plate L, et al. (2016) Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 5, e15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ET, Balch WE (2013) Diversity in the origins of proteostasis networks – a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 14, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, Dillin A (2015) Tipping the metabolic scales towards increased longevity in mammals. Nat. Cell Biol. 17, 196–203. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rozas P, Bargsted L, Martinez F, Hetz C, Medinas DB (2016) The ER proteostasis network in ALS: determining the differential motoneuron vulnerability. Neurosci. Lett. 636, 9–15. [DOI] [PubMed] [Google Scholar]
- Ruegsegger C, Saxena S (2016) Proteostasis impairment in ALS. Brain Res. 1648, 571–579. [DOI] [PubMed] [Google Scholar]
- Safra M, Ben‐Hamo S, Kenyon C, Henis‐Korenblit S (2013) The ire‐1 ER stress‐response pathway is required for normal secretory‐protein metabolism in C. elegans . J. Cell Sci. 126, 4136–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CX, Tanaka LY, Wosniak J, Laurindo FR (2009) Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 11, 2409–2427. [DOI] [PubMed] [Google Scholar]
- Scheper W, Hoozemans JJ (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel R, Dillin A (2015) Endocrine aspects of organelle stress‐cell non‐autonomous signaling of mitochondria and the ER. Curr. Opin. Cell Biol. 33, 102–110. [DOI] [PubMed] [Google Scholar]
- Song Y, Shen H, Du W, Goldstein DR (2013) Inhibition of x‐box binding protein 1 reduces tunicamycin‐induced apoptosis in aged murine macrophages. Aging Cell 12, 794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. [DOI] [PubMed] [Google Scholar]
- Sun J, Liu Y, Aballay A (2012) Organismal regulation of XBP‐1‐mediated unfolded protein response during development and immune activation. EMBO Rep. 13, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Duffy A, O'Mahoney ME, Logue SE, Mylotte LA, O'Brien T, Samali A (2006) ER stress contributes to ischemia‐induced cardiomyocyte apoptosis. Biochem. Biophys. Res. Commun. 349, 1406–1411. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Dillin A (2011) Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3, a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Dillin A (2013) XBP‐1 Is a cell‐nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Berendzen KM, Dillin A (2014) Systemic stress signalling: understanding the cell non‐autonomous control of proteostasis. Nat. Rev. Mol. Cell Biol. 15, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triplett JC, Tramutola A, Swomley A, Kirk J, Grimes K, Lewis K, Orr M, Rodriguez K, Cai J, Klein JB, Perluigi M, Buffenstein R, Butterfield DA (2015) Age‐related changes in the proteostasis network in the brain of the naked mole‐rat: implications promoting healthy longevity. Biochim. Biophys. Acta 1852(Pt A), 2213–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Dufey E, Lisbona F, Rojas‐Rivera D, Hetz C (2013) When ER stress reaches a dead end. Biochem. Biophys. Acta. 1833, 3507–3517. [DOI] [PubMed] [Google Scholar]
- Valdes P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C (2014) Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 111, 6804–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela V, Martinez G, Duran‐Aniotz C, Hetz C (2016) Gene therapy to target ER stress in brain diseases. Brain Res. 1648, 561–570. [DOI] [PubMed] [Google Scholar]
- Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura‐Aguilar J, Cuervo AM, Glimcher LH, Hetz C (2012) Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal RL, Matus S, Bargsted L, Hetz C (2014) Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 35, 583–591. [DOI] [PubMed] [Google Scholar]
- Walker L, McAleese KE, Thomas AJ, Johnson M, Martin‐Ruiz C, Parker C, Colloby SJ, Jellinger K, Attems J (2015) Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 129, 729–748. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. [DOI] [PubMed] [Google Scholar]
- Wang L, Zeng X, Ryoo HD, Jasper H (2014) Integration of UPRER and oxidative stress signaling in the control of intestinal stem cell proliferation. PLoS Genet. 10, e1004568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ryoo HD, Qi Y, Jasper H (2015) PERK limits drosophila lifespan by promoting intestinal stem cell proliferation in response to ER Stress. PLoS Genet. 11, e1005220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, Zhu LQ, Liu D (2015) ATF4: a novel potential therapeutic target for Alzheimer's Disease. Mol. Neurobiol. 52, 1765–1770. [DOI] [PubMed] [Google Scholar]
- Williams KW, Liu T, Kong X, Fukuda M, Deng Y, Berglund ED, Deng Z, Gao Y, Liu T, Sohn JW, Jia L, Fujikawa T, Kohno D, Scott MM, Lee S, Lee CE, Sun K, Chang Y, Scherer PE, Elmquist JK (2014) Xbp1s in pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehlbier U, Colombo A, Saaranen MJ, Perez V, Ojeda J, Bustos FJ, Andreu CI, Torres M, Valenzuela V, Medinas DB, Rozas P, Vidal RL, Lopez‐Gonzalez R, Salameh J, Fernandez‐Collemann S, Munoz N, Matus S, Armisen R, Sagredo A, Palma K, Irrazabal T, Almeida S, Gonzalez‐Perez P, Campero M, Gao FB, Henny P, van Zundert B, Ruddock LW, Concha ML, Henriquez JP, Brown RH, Hetz C (2016) ALS‐linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 35, 845–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization and Alzheimer's Disease International . (2012). Dementia a Public Health Priority. Geneva, London: World Health Organization; Alzheimer's Disease International; 1 online resource (viii, 252 p.). [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13, 365–376. [DOI] [PubMed] [Google Scholar]
- Ye J, Koumenis C (2009) ATF4, an ER stress and hypoxia‐inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr. Mol. Med. 9, 411–416. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Zhao L, Longo‐Guess C, Harris BS, Lee JW, Ackerman SL (2005) Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat. Genet. 37, 974–979. [DOI] [PubMed] [Google Scholar]