

Summary
Biological aging is associated with a reduction in the reparative and regenerative potential in tissues and organs. This reduction manifests as a decreased physiological reserve in response to stress (termed homeostenosis) and a time‐dependent failure of complex molecular mechanisms that cumulatively create disorder. Aging inevitably occurs with time in all organisms and emerges on a molecular, cellular, organ, and organismal level with genetic, epigenetic, and environmental modulators. Individuals with the same chronological age exhibit differential trajectories of age‐related decline, and it follows that we should assess biological age distinctly from chronological age. In this review, we outline mechanisms of aging with attention to well‐described molecular and cellular hallmarks and discuss physiological changes of aging at the organ‐system level. We suggest methods to measure aging with attention to both molecular biology (e.g., telomere length and epigenetic marks) and physiological function (e.g., lung function and echocardiographic measurements). Finally, we propose a framework to integrate these molecular and physiological data into a composite score that measures biological aging in humans. Understanding the molecular and physiological phenomena that drive the complex and multifactorial processes underlying the variable pace of biological aging in humans will inform how researchers assess and investigate health and disease over the life course. This composite biological age score could be of use to researchers seeking to characterize normal, accelerated, and exceptionally successful aging as well as to assess the effect of interventions aimed at modulating human aging.
Keywords: aging, biological age, biomarkers, score, senescence
‘Time is but the stream I go a‐fishing in. I drink at it; but while I drink I see the sandy bottom and detect how shallow it is. Its thin current slides away, but eternity remains’. – Henry David Thoreau, Walden
.
Introduction
The number of individuals aged 60 or older will increase dramatically in the next three decades. As the fastest growing age‐strata worldwide, the global population over 60 will surpass two billion by 2050: a 12‐fold increase from 1950 (United Nations Department of Economic and Social Affairs Population Division 2013). In the 20th century, decreased mortality and lengthening of average human lifespan shifted the worldwide demographic structure toward the aged. This shift stemmed initially from treatment of infectious diseases and subsequently cardiovascular disorders (Fries, 2005). However, an increase in late‐life disability has accompanied gains in healthy years lived (health span) and longevity (Crimmins et al., 1994). Age represents the primary risk factor for chronic diseases, including cardiovascular, malignant, and neurodegenerative conditions. Extremely aged individuals who survive in good health to the end of the human lifespan are rare, and a fixed limit to human lifespan may exist (Dong et al., 2016).
Biological aging is associated with a reduction in the reparative and regenerative potential in tissues and organs. This reduction manifests as decreased physiological reserve in response to stress (termed homeostenosis) and a time‐dependent failure of complex molecular mechanisms that cumulatively create disorder. Aging inevitably occurs with time in all organisms and emerges on a molecular, cellular, organ, and organismal level with genetic, epigenetic, and environmental modulators (Fig. 1). Individuals with the same chronological age and their organs exhibit differential trajectories of age‐related decline, and it follows that we should assess biological age distinctly from chronological age. Understanding the molecular and physiological phenomena that drive the complex and multifactorial processes underlying biological aging in humans will inform how researchers assess and investigate health and disease over the life course. In this review, we outline mechanisms of aging with attention to well‐described molecular and cellular hallmarks, discuss normal human aging at the organ‐system level, suggest methods to measure biological age, and propose a framework to integrate molecular and physiological data into a composite score that measures biological aging in humans.
Figure 1.

Biological aging is a multifactorial process. The molecular hallmarks of aging and organ‐specific physiological function are both influenced by genetic, epigenetic, and environmental factors. Metastatic aging may contribute to differential aging in remote tissues through a paracrine mechanism.
Search strategy and selection criteria
Findings for this review were identified by searches of MEDLINE, Current Contents, PubMed, and references from relevant articles using the search terms ‘aging’, ‘measurement’, and ‘assessment’. Abstracts and reports from meetings were included only when they related directly to previously published work. With exceptions for historical interest, only articles published in English between 1980 and 2016 were included.
Molecular mechanisms of aging
The heritable contribution to aging is limited for most humans, with genetics accounting for only 20–30% of lifespan variability in human twin and founder population family studies (Mitchell et al., 2001; Kenyon, 2010). However, heritable factors may represent a significantly larger contribution to lifespan at extreme ages, and the exceptionally aged may offer an opportunity to find rare genetic variants associated with longevity (Tan et al., 2010; Sebastiani et al., 2012). Regardless, no single factor or molecular mechanism explains progressive age‐related homeostenosis. Inter‐related molecular and cellular phenomena occur during normal aging, intensify during accelerated or premature aging, and can be mitigated to increase lifespan. López‐Otín and colleagues proposed nine so‐called hallmarks of aging—genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication—that frame mechanisms underlying senescence (Lopez‐Otin et al., 2013). As we discuss in this section, many hallmarks suggest potential therapeutic targets to restore age‐associated functional decline and homeostenosis, although potential therapies are not completely benign. Translation of these hallmarks with surrogate measurements are important to include in a composite biological age score (BAS) because of their direct relationship with the molecular basis of aging (Fig. 2).
Figure 2.
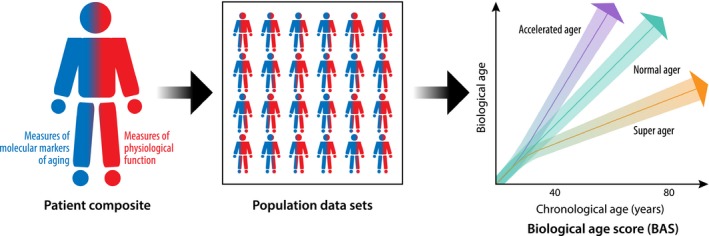
Conceptual derivation of a biological age score (BAS) that combines molecular markers derived from measures of the molecular hallmarks of aging (represented here in blue, e.g., telomere length and gene‐specific DNA methylation) and measures of physiological function (represented here in red, e.g., FEV 1 and e’ velocity) that are longitudinally assessed throughout the life course. Potential mathematical modeling approaches for integrating individual components into a composite BAS include multiple linear regression, principal component analysis, and Klemera and Doubal's method with derivation and validation in population‐based data sets. The BAS graph depicts three hypothetical aging trajectories: 1) normal ager, 2) super ager, and 3) accelerated ager with colored areas representing confidence intervals and demonstrating overlap at young ages. The aging lines are depicted hypothetically as straight lines, but the trajectory of biological aging is not known.
Mouse and human data implicate genomic instability in accelerated aging (Burtner & Kennedy, 2010) with myriad exogenous agents (e.g., radiation and xenobiotic compounds) as well as endogenous processes (e.g., DNA replication errors and reactive oxygen species [ROS]) causing damage to DNA (Hoeijmakers, 2009). Cumulative genomic damage disturbs homeostasis and impacts health span (Moskalev et al., 2013). Genome maintenance represents a potential therapeutic target, as augmenting mitotic regulators involved with chromosomal segregation such as BubR1 improves health span in mice (Baker et al., 2013). Although malignant transformation as a consequence of genomic manipulation remains a concern, high‐level expression of BubR1 reduced tumorigenesis in transgenic mice.
Telomeres, the chromatin tips responsible for preventing degradation at chromosomal ends, shorten and become increasingly susceptible to damage with age (Blackburn et al., 2006). Most mammalian somatic cells lack telomerase—a specialized DNA polymerase responsible for repairing telomeres after cell division—which results in replication‐dependent sequence loss at chromosomal ends, often leading to replicative senescence (the Hayflick limit) (Hayflick & Moorhead, 1961; Jiang et al., 2008). Accordingly, telomerase overexpression increases median lifespan in mice (Bernardes de Jesus et al., 2012). However, in vivo evidence of the causal role telomerase plays in aging remains controversial, as telomerase knockout mice demonstrate no overt phenotype and reduced longevity only after multiple generations (Rudolph et al., 1999). In humans, deficiencies in the telomerase complex cause a variety of age‐related pathologies, including prematurely gray hair, pulmonary fibrosis, liver disease, and aplastic anemia (Armanios & Blackburn, 2012). Gene therapy encoding telomerase components may hold promise to halt or even reverse telomere shortening in humans (Ramunas et al., 2015). However, gene therapy remains in its infancy with ongoing concerns regarding oncogenesis (Hacein‐Bey‐Abina et al., 2003).
Epigenetic alterations—heritable changes in phenotype that are independent of DNA sequence mutations—occur with aging and impact cellular function (Sen et al., 2016). Overall, global heterochromatin loss and redistribution occur with an increase in transcriptional noise (Bahar et al., 2006; Tsurumi & Li, 2012). Specifically, H4K16 acetylation and H4K20 and H3K4 trimethylation increase, while H3K9 methylation and H3K27 trimethylation decrease with age (Han & Brunet, 2012). Further, deleting the H3K4 and H3K27 methylation complexes extends lifespan in worms and flies (Greer et al., 2010; Siebold et al., 2010). The role DNA methylation at cytosine‐phospho‐guanine islands plays in aging remains less clear, with age‐associated global gene‐activating hypomethylation but gene‐repressive hypermethylation at tumor suppressor gene loci (Maegawa et al., 2010). Large‐scale genomewide DNA methylation‐based epigenetic analyses using a variety of cell types and tissues have identified an ‘epigenetic clock’ that is closely correlated with healthy aging (Horvath, 2013). Unlike cumulative DNA damage and many other hallmarks of aging, epigenetic alterations are ostensibly reversible and embody promising pharmacologic targets for therapies designed to promote healthy aging. DNA methyltransferase and histone deacetylate inhibitors represent two potential drug classes (So et al., 2011; Wang et al., 2013), although significant refinement in enzyme isoform specificity will be required to limit off‐target effects of these compounds including immunomodulation (Wang et al., 2015).
Protein homeostasis (proteostasis) becomes impaired with aging and enhanced proteostasis can maintain the integrity of the proteome and delay mammalian senescence (Zhang & Cuervo, 2008; Koga et al., 2011). The two key proteolytic pathways responsible for protein quality control—the autophagy–lysosomal system and the ubiquitin–proteasome system—become hypofunctional with aging (Calamini et al., 2012; Tomaru et al., 2012). Therapies aimed at promoting healthy aging could target the proteostasis system (Calamini et al., 2012), as deubiquitylase inhibitors and proteasome activators augment clearance of harmful protein in human cells (Lee et al., 2010). Of note, the proteasome activator characterized by Lee et al. displayed dose‐dependent and drug target‐independent toxicity (hypoproliferation) in cultured cells, suggesting that further pharmacologic refinement of proteasome activators may be needed.
Deregulated nutrient sensing represents an important and potentially druggable hallmark of aging, as anabolic signaling causes accelerated aging and caloric restriction extends lifespan in murine and nonhuman primate models (Colman et al., 2014; López‐Otín et al., 2016; Vermeij et al., 2016). Nutrient sensing systems, including the amino acid‐sensing mammalian target of rapamycin (mTOR) as well as low‐energy state detectors (AMP kinase and the sirtuins), contribute to the aging process (Houtkooper et al., 2010). The sirtuin family of NAD‐dependent protein deacetylases may promote healthy aging in yeast, flies, and mice (Houtkooper et al., 2012), and compounds that raise NAD+ levels may restore lost mitochondrial function and promote longevity (Gomes et al., 2013). In addition, the insulin and insulin‐like growth factor 1 (IGF‐1) signaling (IIS) pathway, which targets the FOXO transcription factors and mTOR complexes, represents an extremely well‐conserved pro‐aging pathway (Kenyon, 2010; Barzilai et al., 2012). Mutations that reduce the level or function of growth hormone, the IGF‐1 receptor, or intracellular pathway components including AKT, mTOR, and FOXO closely associate with longevity in model organisms as well as humans (Arum et al., 2014). However, aging mouse models display decreased IIS activity, which raises the possibility that IIS activity represents a defensive response to systemic damage (Schumacher et al., 2008). Strikingly, mice fed the mTOR inhibitor rapamycin in late life experienced extension of median and maximal lifespan, supporting a role for mTOR signaling and pharmacologic inhibition in lifespan regulation (Harrison et al., 2009). The long‐term effects of rapamycin administration, including sex‐dependent differences in outcomes, remain incompletely defined (Fischer et al., 2015).
Mitochondrial dysfunction in the form of decreased respiratory chain efficiency, resulting electron leak, and diminished ATP production may contribute to senescence (Green et al., 2011). A significant body of data supports ROS as a cause of accelerated aging (Harman, 1965). However, recent evidence suggests that limited oxidative stress may actually be beneficial to health span (Hekimi et al., 2011) and that mitochondrial ROS impart advantageous effects on healthy cellular function (Sena & Chandel, 2012) up to a threshold (Hekimi et al., 2011). Compelling evidence demonstrates that therapies designed to improve fitness could exploit mitohormesis—the concept that repeated low‐level toxic exposures can trigger a beneficial compensatory mitochondrial response that ultimately leads to augmentation in cellular fitness (Haigis & Yankner, 2010).
With aging, there exists a propensity for stable cell cycle arrest known as cellular senescence (Hayflick & Moorhead, 1961; Collado et al., 2007). DNA damage (independent of telomeres) and derepression of the INK4/ARF locus induce cellular senescence and occur with advancing age. INK4/ARF locus expression of p16INK4a and p19ARF acts as a cellular checkpoint that critically prevents propagation of damaged and possibly malignant cells. As a biomarker, protein levels of p16INK4a and p19ARF correlate with chronological age remarkably well in humans and model organisms with a large difference (up to an order of magnitude) comparing young and old tissues (Krishnamurthy et al., 2004; Ressler et al., 2006). The correlation holds for almost all tissues examined. However, from a functional perspective, accumulation of senescent cells may ultimately become deleterious when regenerative capacity grows exhausted and progenitors cannot replace senescent cells. For example, stem cell attrition occurs in multiple tissues with aging, and DNA damage, p16INK4a expression, and telomere shortening cause decreased hematopoietic stem cell proliferation (Sharpless & DePinho, 2007). Therapeutic removal of p16INK4a‐positive cells could extend healthy lifespan (Baker et al., 2016).
Senescent cells display a characteristic secretory profile—the senescence‐associated secretory phenotype—that contributes to low‐level systemic inflammation (so‐called inflammaging) and likely facilitates the spread of pro‐senescence signals through tissues and systemically alters the extracellular matrix in parallel (Kuilman et al., 2010). These factors enable a senescence messaging system (SMS) and include interleukins, IGFBP3, plasminogen activator inhibitor‐1 (PAI‐1), and transforming growth factor‐β (Kuilman & Peeper, 2009; Capell et al., 2016; Ozcan et al., 2016). In addition to paracrine signaling that results from the senescence‐associated secretory phenotype, accumulation of tissue damage and failure of the immune system to clear damaged proteins, pathogens, and compromised or malignant cells promote inflammaging (Senovilla et al., 2012). Metastatic aging—in which aging in one tissue accelerates aging in other tissues (Lavasani et al., 2012)—may occur via gap junctions and ROS signaling and via miRNAs secreted into the blood that promote senescence in remote tissues (Grillari & Grillari‐Voglauer, 2010; Nelson et al., 2012). Finally, heterochronic parabiosis and knockout murine experiments provide additional supportive evidence of a distinct proteomic signature of senescence with alterations in circulating systemic factors (e.g., GDF‐11, CCL11, Klotho, β2 microglobulin); however, emerging evidence questions the role GDF‐11 plays in aging mice (Conboy et al., 2005; Villeda et al., 2011; Eren et al., 2014; Laviano, 2014; Sinha et al., 2014; Brun & Rudnicki, 2015; Smith et al., 2015). Therapeutic administration or blockade of SMS components could ameliorate inflammaging in humans, although significant concerns persist regarding off‐target effects including skeletal and cardiac muscle wasting (Harper et al., 2016).
The interplay of the above mechanisms and pathways contribute to aging on an organismal level and offer potential parameters to include in a composite assessment of biological age as well as targets for therapies aimed to counter age‐related functional decline and morbidity. In addition to targeted interventions, understanding the molecular mechanisms underpinning senescence could translate into biomarkers of biological age (Table 1).
Table 1.
Biomarkers of aging derived from the hallmarks of aging (Lopez‐Otin et al., 2013)
| Hallmark of aging | Biomarkers |
|---|---|
| Telomere attrition |
Blood leukocyte‐derived telomere length Telomere dysfunction‐induced proteins |
| Epigenetic alterations |
H4K16 acetylation; H4K20 and H3 K4, K9, and K27 methylation DNA methylation patterns Noncoding RNA patterns (e.g., microRNA expression profiles) |
| Loss of proteostasis |
Proteomics Amyloid‐β‐derived diffusible ligands (ADDLs) |
| Deregulated nutrient sensing |
Insulin‐like growth factor‐1 (IGF‐1) Metabolomics |
| Mitochondrial dysfunction |
Number of mitochondria Mitochondrial DNA copy number Mitochondrial protein levels |
| Cellular senescence and pro‐inflammatory cytokines (altered intercellular communication) |
SMS: PAI‐1, IGFBP‐3, IL‐6, TGF‐β IL‐1β, TNF‐α p16INK4a and p19ARF p53, p21 Senescence‐associated β‐galactosidase (SABG) Youth‐associated (GDF‐11) and age‐associated (CCL11, Klotho, β2 microglobulin) circulating factors |
Organ‐level physiological changes of aging
Physiological aging involves a progressive detrimental change in maximal organ function with differential trajectories across organ systems (Fig. 3) (Shock, 1956). Importantly, multiple factors including genetics, environmental conditions, and developmental programming determine maximal organ function, which varies significantly between individuals (Lange et al., 2015). Aging affects all organ systems and must be assessed through a variety of physiological measures, as aging varies greatly organ‐to‐organ and person‐to‐person and results in impaired reserve capacity and limited ability to respond to stress. While there appears to be an organ‐specific or organ‐differential resilience and vulnerability of aging, frailty refers to the cumulative decline and increasing homeostatic imbalance that precedes the ultimate consequence of aging: death (Fries, 2005).
Figure 3.
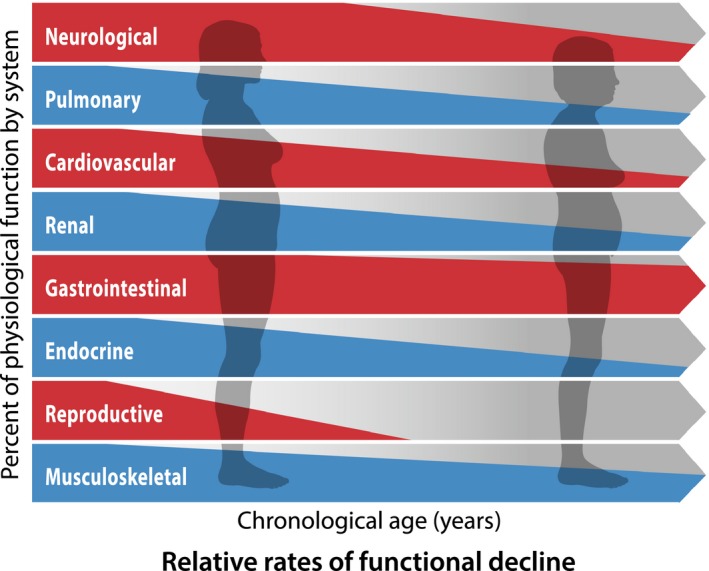
Relative rates of decline of organ‐specific physiological function. Different organ systems may carry a specific vulnerability to age (i.e., the cardiovascular system appears to suffer biological aging more rapidly than the gastrointestinal system).
Cardiovascular
The aging cardiovascular system displays decreased compliance of the aorta and large vessels (Vaitkevicius et al., 1993). This increased arterial stiffness results in a widened pulse pressure with raised systolic blood pressure (due to increased resistance to blood ejection from the left ventricle [LV]) and lowered diastolic pressure (due to a more rapid pressure decrease in diastole). Subsequent changes include increases in LV afterload, mass, wall thickness, and LV end‐diastolic volume. Further alterations in calcium influx cause reduced LV compliance and delayed LV relaxation or decreased diastolic function as assessed by Doppler echocardiography parameters (e.g., E‐wave/A‐wave velocity ratio, septal and lateral e’‐wave velocity depth) (Sun et al., 2004). Intrinsic heart rate declines due to both atrial pacemaker cellular loss (50–75% by age 50) and His bundle fibrosis (Cheitlin, 1989). Fibrosis and calcification occur at the aortic valve cusp bases, annular valvular rings, and fibrous trigones. Finally, aged individuals demonstrate decreased responsiveness to β‐adrenergic receptor stimulation in cardiomyocytes, decreased reactivity to baroreceptor and chemoreceptor output, and increased circulating catecholamines resulting in reduced exercise tolerance and decreased cardiac output (Davies et al., 1996). These changes increase the heart's vulnerability to development of age‐related cardiovascular pathology including hypertension, congestive heart failure, atrioventricular block, and aortic stenosis. Additionally, atherosclerosis is linked to premature biological aging with senescent cells identified in coronary artery disease plaques (Wang & Bennett, 2012).
Pulmonary
Lung function represents one of the few consistently reliable physiological markers of aging. With advancing age, peak aerobic capacity falls with a greater than 20% decline per decade after age 70 (Fleg et al., 2005). The lungs lose elastic tissue, which causes decreased surface area available for gas exchange (increased anatomic dead space) as alveolar ducts enlarge (Gillooly & Lamb, 1993). Chest wall compliance decreases and dominates the increase in lung compliance; functional residual capacity decreases as a result of the fall in total respiratory system compliance. Forced vital capacity (FVC) declines 0.15‐0.30 L per decade in nonsmoking men, and the forced expiratory volume in one‐second (FEV1) falls 0.20–0.30 L/s per decade with a steeper decline in the 7th and 8th decades (Xu et al., 1995). Physiological restriction may result for some individuals, although population mean total lung capacity does not change significantly with age. Residual volume increases by about 10% per decade due to an increased closing volume: the lung volume at which small airways in dependent lung zones begin to collapse during exhalation. Ventilation–perfusion mismatching increases with age, as airways in the better‐perfused dependent lung zones have an increased likelihood of closure during exhalation. Diffusion capacity decreases around 5% per decade, although hypoxemia does not typically develop. Further, advancing age is associated with a diminished central drive to the respiratory muscles in response to hypoxemia, hypercapnia, and mechanical load; exercise training may attenuate this hyporesponsiveness. Collectively, the above changes in combination with decreased respiratory muscle strength and reduced efficacy of mucociliary clearance result in increased susceptibility to pneumonia (Enright et al., 1994).
Renal
The kidneys develop a diffuse glomerulosclerosis with age (up to 30% by age 75) (Nyengaard & Bendtsen, 1992); remaining glomeruli display impaired filtering ability. Only 3% of donor kidneys from 18–29‐year‐olds and over 70% from 70 to 77‐year‐olds contain nephrosclerosis. Creatinine clearance falls 7.5–10 mL per decade on average with a large variance. Serum creatinine, however, may remain constant due to decreased production with age. Cystatin C may therefore represent a more accurate renal function marker in the elderly (Christensson & Elmstahl, 2011). Aged individuals maintain fluid and electrolyte balance in the absence of a significant challenge; however, stress can impair maximal diluting and concentrating ability in older individuals (Christensson & Elmstahl, 2011). In addition, the aging kidney demonstrates a decreased ability to acidify urine and excrete an acid load. Renal plasma flow decreases with age due in part to increased local vasodilating prostaglandin concentration (Ungar et al., 2000). The renal vascular system undergoes spiraling of the afferent arterioles and intimal fibrosis (Tracy et al., 1988) as well as shunt development between afferent and efferent arterioles.
Immune and hematologic systems
The aging immune system displays progressive changes collectively described as immunosenescence; these changes result in increased susceptibility to infection, malignancy, and autoimmunity. The adaptive and innate immune systems both exhibit functional decline with aging, although innate immunity appears better preserved (Weiskopf et al., 2009). Pro‐B‐cell production declines with less striking changes in T‐cell precursors. Regulatory T cells lose their suppressive function (Tsaknaridis et al., 2003) and accumulate in visceral adipose tissue. Indeed, age‐associated chronic inflammation is associated with an inflammatory signature within visceral adipose tissue (Lumeng et al., 2011).
The aging immune system carries a greater likelihood of clonal expansion and hematologic malignancy (Jaiswal et al., 2014). Bone marrow mass decreases and undergoes fatty replacement with a resultant decrease in total bone marrow hematopoietic tissue (Geiger & Rudolph, 2009). This decrease in bone marrow mass leads to a loss of functional reserves, reduced hematopoiesis with hypoproliferative hematopoietic stem cells, and increased incidence of anemia and myeloid diseases. However, iron flux, red cell lifespan, total white blood cell count, and blood volume do not decline with age. Platelet responsiveness increases as do multiple coagulation factor levels (Franchini, 2006).
Neurologic function
Cognitive decline with aging is multifactorial and related to changes in structure as well as synaptic plasticity. Cerebral tissue atrophy and diminished cerebral perfusion result in significant white matter loss, but neuronal dropout varies by brain region with little or no loss in some regions (Bertoni‐Freddari et al., 1996). In addition, dopaminergic signaling demonstrates a progressive decrease in signaling via the D2 receptor (Roth & Joseph, 1994). Functional MRI studies demonstrate less‐coordinated activation in brain regions focused on higher‐order cognitive functions, which suggests a global loss of integrative function with aging (Andrews‐Hanna et al., 2007). Gene expression profiling studies show that significant changes in synaptic gene expression contribute to altered higher‐order integration (Jiang et al., 2001). These alterations in synaptic plasticity and loss as well as impaired neurogenesis may predispose aged individuals to neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease (Loerch et al., 2008).
Other organ systems
Aging modifies the digestive, hepatic, and endocrine systems to varying degrees. The digestive system undergoes only modest changes with time, and normal aging does not cause malnourishment. Micronutrient absorption in the small intestine may decrease with age but not to a level that impairs homeostasis. Liver mass decreases 20–40% with age, and hepatic blood flow declines (Zoli et al., 1989). Serum albumin may fall slightly, but routine liver chemistries do not change with time (Rahmioglu et al., 2009). The aging liver displays decreased vitamin K‐dependent clotting factor synthesis (Froom et al., 2003). Alterations in metabolism influence lifespan in experimental models and potentially embody high‐yield translational targets. Insulin resistance and physiological declines in circulating insulin‐like growth factor characterize the aging process (Barzilai et al., 2012). Further, aging results in decreased β‐cell regeneration in pancreatic islets (Sartori et al., 2014). Metabolomics approaches have identified a potential longevity signature characterized by increased levels of circulating citric acid cycle intermediates (Cheng et al., 2015).
Finally, the musculoskeletal, integumentary, sensory, and behavioral systems undergo a multitude of changes with aging. Muscle mass and contractile force decrease and may limit mobility (Delbono, 2011). Age‐related loss of muscle mass (sarcopenia) occurs along with qualitative changes in muscle characterized by fat and connective tissue infiltration. Findings from the AGES‐Reykjavik study suggest that muscle composition may be associated with mortality risk (Reinders et al., 2015, 2016). Skin changes include epidermal thinning, decreased dermal elasticity, and diminution of subdermal fat that result in increased susceptibility to trauma and infection (Elewa et al., 2015). Progressive miosis, decreased corneal transparency, and increased lens rigidity cause presbyopia and decreased visual acuity (Salvi et al., 2006). Sensory cell loss and cochlear neuron dropout lead to presbycusis (Gates & Mills, 2005). Finally, healthy behavior change becomes less likely with age, seemingly as a result of alterations in social networks among older adults (Tucker et al., 2004).
Comprehensive assessement of biological aging
Chronological age offers limited information regarding the complex processes driving biological aging. Individuals with the same chronological age vary greatly in health and in disease and disability prompting the utility of defining a ‘biological age’. The conceptualization of such a biological age distinct from chronological age has been proposed by many researchers with measures as crude as functional ‘frailty’ and as sophisticated as patterns of DNA methylation (Borkan & Norris, 1980; Ravindrarajah et al., 2013; Weidner et al., 2014; Marioni et al., 2015). While much research has focused on quantification of biological aging, a comprehensive and integrative score incorporating molecular biomarkers and physiological functional parameters is lacking. Current strategies to assess systemic biological age carry significant limitations as individual parameters to accurately reflect an individual's global homeostenosis have been elusive. Further, biological plausibility suggests that no single biomarker is likely to suffice given the underlying multisystem nature of the aging process with changes occurring on a molecular and organ‐based level underscoring the utility of an aggregate score of biological aging. Scoring systems require careful integration of molecular markers (surrogates of the hallmarks of aging) with longitudinal physiological functional measures, yet little consensus exists regarding optimal methods for creation and evaluation of a composite biological age score (BAS).
In human populations, identifying and characterizing successful agers who remain disease‐free at advanced age with physiological function significantly above their age cohort represent a promising approach to derive and validate a BAS (Fig. 2). Centenarians exemplify an exceptional survival phenotype who delay disability and disease until an average age of 93 years (Andersen et al., 2012). However, centenarians are rare, making it difficult to enroll meaningful sample sizes and answer important research questions. Moreover, longevity and healthy aging may not be synonymous, making lifespan a complicated phenotype to study in addition to the burden of cost and time using this approach. Data from the Leiden Longevity Study and the Long Life Family Study suggest that compression of morbidity into later years accelerates with the age of the cohort (Westendorp et al., 2009; Newman et al., 2011). The result is an oftentimes lengthy period of health before age‐associated morbidity develops at old age. Therefore, in any cohort, identifying subclinical disease can help ensure that data sets accurately classify successful aging, delayed morbidity, and increased longevity rather than age‐associated disease states (Fries, 2005).
Investigators have proposed composite scoring systems in epidemiologic studies such as the National Health and Nutrition Examination Survey (NHANES). The derivation cohort for the NHANES‐based measure of biological age contained more than 9000 participants aged 30–75 years at baseline and integrated physiological and biochemical markers at a single time point (Levine, 2013). The Dunedin Study birth cohort, which identified differences in the pace of aging in young adults over a decade, utilized the NHANES score (Belsky et al., 2015). The Healthy Aging Index in the Cardiovascular Health Study and the Modified Physiological Index in the Health, Aging, and Body Composition Study, represent additional examples of cohort‐derived scores that correlate with chronological age yet lack integration of existing novel markers derived from the molecular hallmarks of aging, which connote a true biological age (Ludwig & Smoke, 1980; Baker & Sprott, 1988). Several limitations of existing scores impede current investigations, including small sample sizes, limited testing of variables, omission of novel molecular markers, lack of participant‐level longitudinal follow‐up with repeated measures in the same individual, and dearth of population‐level replication and validation in clinical settings for predictive ability.
We propose a conceptual framework for a composite BAS, which integrates available molecular measurements based on the hallmarks of aging (Table 1) and functional organ physiology measurements (Table 2) across the life course. Comprehensive and repeated assessments over time of existing and emerging molecular biomarkers and organ‐specific functional measures in longitudinal epidemiologic cohorts in parallel with the use of sophisticated bioinformatics methodologies are needed to derive a global BAS. Conceptually, the BAS represents an integrated biomarker signature that assesses systemic aging on a population level (Cohen et al., 2015). Criteria for inclusion of molecular and physiological parameters into a composite score require that the individual parameters be independently associated with aging and provide additive information when combined. In general, components of the BAS should be i) highly correlated with chronological age, ii) predict organ‐system and global age‐related decline, and iii) be minimally invasive, readily observable, and reliably measured. Investigation into the optimal parameters used to derive a BAS will require collection and analysis of data sets that include successful agers without morbidity as well as accelerated agers with genetic progeroid syndromes, inflammatory pathologic conditions (e.g., human immunodeficiency virus, autoimmune disease states, chronic kidney disease), and disease‐related morbidity. Derivation and validation of a BAS will require multiple types of study designs including observational prospective population‐based cohorts, leveraging large sample sizes with repeated measures over several decades as well as case–control studies and family‐based studies to incorporate less common phenotypes of interest, including successful agers and accelerated agers with nongenetic and genetic conditions.
Table 2.
Measures of organ‐specific changes in physiological function
| Organ system | Measures of organ‐specific function |
|---|---|
| Cardiovascular |
Brachial pulse pressure LV mass Relative wall thickness Echocardiographic parameters (E/A, e’) Pulse wave velocity Augmentation index Aortic valve calcification Heart rate variability |
| Respiratory |
Peak aerobic capacity Spirometry (FEV1, FVC, FEV1/FVC ratio) Lung volumes (TLC, FRC, RV) DLCO Quantitative ventilation–perfusion scanning |
| Renal |
Cystatin C Creatinine clearance |
| Immune | Immune risk profile (assessment of T‐cell proliferation in response to mitogens, B‐cell numbers, CD4:CD8 T‐cell ratio, and CMV serologic status) |
| Bone marrow | Hemoglobin |
| Neurocognitive |
Mini‐mental status examination Cognitive battery Functional MRI |
| Digestive and hepatic | Vitamin K‐dependent clotting factor levels |
| Endocrine |
Thyroid biochemical tests Fasting glucose Insulin Circulating estrogen and testosterone levels |
| Musculoskeletal |
Hand grip strength Unipedal stance test of balance Grooved pegboard test of fine motor coordination SF‐36 physical functioning scale |
| Integumentary |
Skin elasticity Thickness Wrinkle parameter |
| Sensory |
Visual acuity Auditory test Retinal microvascular damage (arteriovenous ratio) |
Optimal mathematical modeling using various methodologies such as multiple linear regression, principal component analysis, and Klemera and Doubal's method to derive scoring systems will require head‐to‐head comparisons (Takeda et al., 1982; Nakamura et al., 1988; Klemera & Doubal, 2006). The use of regression equations will be helpful in initially identifying individual components to be included in the BAS. However, given that aging is a systemic process composed of interdependent processes, redundancies in selected aging parameters may exist. The use of principal component analysis will be critical to determine the number of components or biomarkers to include to create the most parsimonious model and exclude overlap in contribution of molecular markers and physiological parameters. Standardizing the process of creating and evaluating longevity phenotypes with a BAS will accelerate research that endeavors to define healthy aging mechanisms, identify interventions to promote health span, and allow translation and validation of therapies that promote healthy aging.
Future directions
Humans are mortal, and natural limits to lifespan will inevitably persist. While aging cannot be escaped (Gompertz, 1825), postponing senescent changes and disease onset offers the potential to extend fitness, vitality, and years lived free of morbidity and frailty. While there is evidence for modulation of lifespan in preclinical models and animal species via genetic and pharmacologic interventions, translation of these findings in human populations is needed. Here, we discuss the limited scientific database regarding mechanisms and physiological changes of aging and describe a framework to capture the complexity of the aging process in a novel integrative score of biological aging. A comprehensive view of biological aging is particularly germane at this time when randomized clinical trials are being discussed and planned with the intention of testing the therapeutic benefit of drugs such as metformin on human aging (Albert Einstein College of Medicine of Yeshiva University 2015; Longo et al., 2015). Integrating novel molecular assays derived from the hallmarks of aging and physiological measurements will help develop a composite BAS for use as a complex quantitative phenotype to translate mechanistic findings of biological pathways into humans. While conclusive methods to measure biological age remain debatable, deriving a BAS will serve multiple purposes: i) propel aging research at the molecular and cellular level, ii) quantify aging in human cohorts, iii) provide a more robust index of the effects of private gene mutations and specific polymorphisms on aging and longevity, and iv) facilitate efficacy studies of therapies designed to promote healthy aging in humans. Following derivation of a composite BAS, prospective validation will be required to assess its accuracy in predicting development of disease, disability, and death. To that end, longitudinal research efforts such as the United States Precision Medicine Initiative seeking to enroll 1 million participants and the European MARK‐AGE Consortium with 3200 enrolled participants are seeking to define a set of human aging biomarkers (Burkle et al., 2015). Next‐generation sequencing, proteomics, and metabolomics show great promise to advance our understanding of complex biological processes. These technologies carry the potential to identify distinct aging signatures; however, they require additional investigation in epidemiologic studies prior to integration into a contemporary BAS. Although discovery of the fountain of youth remains elusive and unlikely, the outlook to characterize and mitigate age‐related morbidity is optimistic.
Funding sources
NIH R01 HL51387 (DEV), NIH K08 HL128867 (BDS) and F32 HL129695 (SSK); and the Parker B. Francis Research Opportunity Award (BDS). The funding sources did not have any role in the preparation of this manuscript.
Conflict of interests
The authors have no conflicts of interest to declare.
Acknowledgments
We thank Drs. Philip Greenland and Jacob Iasha Sznajder for their review of this manuscript.
References
- Albert Einstein College of Medicine of Yeshiva University (2015) Metformin in Longevity Study (MILES). In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US) 2000‐ [2015 Nov 25]. URL https://clinicaltrials.gov/ct2/show/NCT02432287?term=metformin+aging&rank=1 NLM Identifier: NCT02432287.ed^eds). [Google Scholar]
- Andersen SL, Sebastiani P, Dworkis DA, Feldman L, Perls TT (2012) Health span approximates life span among many supercentenarians: compression of morbidity at the approximate limit of life span. J. Gerontol. A Biol. Sci. Med. Sci. 67, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews‐Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, Buckner RL (2007) Disruption of large‐scale brain systems in advanced aging. Neuron 56, 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH (2012) The telomere syndromes. Nat. Rev. Genet. 13, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arum O, Boparai RK, Saleh JK, Wang F, Dirks AL, Turner JG, Kopchick JJ, Liu JL, Khardori RK, Bartke A (2014) Specific suppression of insulin sensitivity in growth hormone receptor gene‐disrupted (GHR‐KO) mice attenuates phenotypic features of slow aging. Aging Cell 13, 981–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J (2006) Increased cell‐to‐cell variation in gene expression in ageing mouse heart. Nature 441, 1011–1014. [DOI] [PubMed] [Google Scholar]
- Baker GT 3rd, Sprott RL (1988) Biomarkers of aging. Exp. Gerontol. 23, 223–239. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo‐Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM (2013) Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol. 15, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016) Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Huffman DM, Muzumdar RH, Bartke A (2012) The critical role of metabolic pathways in aging. Diabetes 61, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Caspi A, Houts R, Cohen HJ, Corcoran DL, Danese A, Harrington H, Israel S, Levine ME, Schaefer JD, Sugden K, Williams B, Yashin AI, Poulton R, Moffitt TE (2015) Quantification of biological aging in young adults. Proc. Natl. Acad. Sci. U. S. A. 112, E4104–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, Blasco MA (2012) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 4, 691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni‐Freddari C, Fattoretti P, Paoloni R, Caselli U, Galeazzi L, Meier‐Ruge W (1996) Synaptic structural dynamics and aging. Gerontology 42, 170–180. [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Greider CW, Szostak JW (2006) Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12, 1133–1138. [DOI] [PubMed] [Google Scholar]
- Borkan GA, Norris AH (1980) Assessment of biological age using a profile of physical parameters. J. Gerontol. 35, 177–184. [DOI] [PubMed] [Google Scholar]
- Brun CE, Rudnicki MA (2015) GDF11 and the Mythical Fountain of Youth. Cell Metab. 22, 54–56. [DOI] [PubMed] [Google Scholar]
- Burkle A, Moreno‐Villanueva M, Bernhard J, Blasco M, Zondag G, Hoeijmakers JH, Toussaint O, Grubeck‐Loebenstein B, Mocchegiani E, Collino S, Gonos ES, Sikora E, Gradinaru D, Dolle M, Salmon M, Kristensen P, Griffiths HR, Libert C, Grune T, Breusing N, Simm A, Franceschi C, Capri M, Talbot D, Caiafa P, Friguet B, Slagboom PE, Hervonen A, Hurme M, Aspinall R (2015) MARK‐AGE biomarkers of ageing. Mech. Ageing Dev. 151, 2–12. [DOI] [PubMed] [Google Scholar]
- Burtner CR, Kennedy BK (2010) Progeria syndromes and ageing: what is the connection? Nat. Rev. Mol. Cell Biol. 11, 567–578. [DOI] [PubMed] [Google Scholar]
- Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, Balch WE, Morimoto RI (2012) Small‐molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 8, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, Natale C, Ridky TW, Adams PD, Berger SL (2016) MLL1 is essential for the senescence‐associated secretory phenotype. Genes Dev. 30, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheitlin MD (1989) The spectrum of cardiovascular disease in the elderly. Int. J. Cardiol. 22, 283–288. [DOI] [PubMed] [Google Scholar]
- Cheng S, Larson MG, McCabe EL, Murabito JM, Rhee EP, Ho JE, Jacques PF, Ghorbani A, Magnusson M, Souza AL, Deik AA, Pierce KA, Bullock K, O'Donnell CJ, Melander O, Clish CB, Vasan RS, Gerszten RE, Wang TJ (2015) Distinct metabolomic signatures are associated with longevity in humans. Nat. Commun. 6, 6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensson A, Elmstahl S (2011) Estimation of the age‐dependent decline of glomerular filtration rate from formulas based on creatinine and cystatin C in the general elderly population. Nephron Clin. Pract. 117, c40–50. [DOI] [PubMed] [Google Scholar]
- Cohen AA, Milot E, Li Q, Bergeron P, Poirier R, Dusseault‐Belanger F, Fulop T, Leroux M, Legault V, Metter EJ, Fried LP, Ferrucci L (2015) Detection of a novel, integrative aging process suggests complex physiological integration. PLoS ONE 10, e0116489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130, 223–233. [DOI] [PubMed] [Google Scholar]
- Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM (2014) Caloric restriction reduces age‐related and all‐cause mortality in rhesus monkeys. Nat. Commun. 5, 3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA (2005) Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764. [DOI] [PubMed] [Google Scholar]
- Crimmins EM, Hayward MD, Saito Y (1994) Changing mortality and morbidity rates and the health status and life expectancy of the older population. Demography 31, 159–175. [PubMed] [Google Scholar]
- Davies CH, Ferrara N, Harding SE (1996) Beta‐adrenoceptor function changes with age of subject in myocytes from non‐failing human ventricle. Cardiovasc. Res. 31, 152–156. [PubMed] [Google Scholar]
- Delbono O (2011) Expression and regulation of excitation‐contraction coupling proteins in aging skeletal muscle. Curr Aging Sci. 4, 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Milholland B, Vijg J (2016) Evidence for a limit to human lifespan. Nature 538, 257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elewa RM, Abdallah MA, Zouboulis CC (2015) Age‐associated skin changes in innate immunity markers reflect a complex interaction between aging mechanisms in the sebaceous gland. J. Dermatol. 42, 467–476. [DOI] [PubMed] [Google Scholar]
- Enright PL, Kronmal RA, Manolio TA, Schenker MB, Hyatt RE (1994) Respiratory muscle strength in the elderly. Correlates and reference values. Cardiovascular Health Study Research Group. Am. J. Respir. Crit. Care Med. 149, 430–438. [DOI] [PubMed] [Google Scholar]
- Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales‐Nebreda L, Urich D, Quaggin SE, Budinger GR, Mutlu GM, Miyata T, Vaughan DE (2014) PAI‐1‐regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc. Natl. Acad. Sci. U. S. A. 111, 7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KE, Gelfond JA, Soto VY, Han C, Someya S, Richardson A, Austad SN (2015) Health Effects of Long‐Term Rapamycin Treatment: the Impact on Mouse Health of Enteric Rapamycin Treatment from Four Months of Age throughout Life. PLoS ONE 10, e0126644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleg JL, Morrell CH, Bos AG, Brant LJ, Talbot LA, Wright JG, Lakatta EG (2005) Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation 112, 674–682. [DOI] [PubMed] [Google Scholar]
- Franchini M (2006) Hemostasis and aging. Crit. Rev. Oncol. Hematol. 60, 144–151. [DOI] [PubMed] [Google Scholar]
- Fries JF (2005) The compression of morbidity. 1983. Milbank Q. 83, 801–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froom P, Miron E, Barak M (2003) Oral anticoagulants in the elderly. Br. J. Haematol. 120, 526–528. [DOI] [PubMed] [Google Scholar]
- Gates GA, Mills JH (2005) Presbycusis. Lancet 366, 1111–1120. [DOI] [PubMed] [Google Scholar]
- Geiger H, Rudolph KL (2009) Aging in the lympho‐hematopoietic stem cell compartment. Trends Immunol. 30, 360–365. [DOI] [PubMed] [Google Scholar]
- Gillooly M, Lamb D (1993) Airspace size in lungs of lifelong non‐smokers: effect of age and sex. Thorax 48, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell 155, 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompertz B (1825) On the Nature of the Function Expressive of the Law of Human Mortality. Philos. Trans. R. Soc. Lond. 1, 513–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy‐inflammation‐cell death axis in organismal aging. Science 333, 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A (2010) Members of the H3K4 trimethylation complex regulate lifespan in a germline‐dependent manner in C. elegans. Nature 466, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillari J, Grillari‐Voglauer R (2010) Novel modulators of senescence, aging, and longevity: small non‐coding RNAs enter the stage. Exp. Gerontol. 45, 302–311. [DOI] [PubMed] [Google Scholar]
- Hacein‐Bey‐Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana‐Calvo M, Fischer A (2003) A serious adverse event after successful gene therapy for X‐linked severe combined immunodeficiency. N. Engl. J. Med. 348, 255–256. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Yankner BA (2010) The aging stress response. Mol. Cell 40, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Brunet A (2012) Histone methylation makes its mark on longevity. Trends Cell Biol. 22, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D (1965) The Free Radical Theory of Aging: effect of Age on Serum Copper Levels. J. Gerontol. 20, 151–153. [DOI] [PubMed] [Google Scholar]
- Harper SC, Brack A, MacDonnell S, Franti M, Olwin BB, Bailey BA, Rudnicki MA, Houser SR (2016) Is Growth Differentiation Factor 11 a Realistic Therapeutic for Aging‐Dependent Muscle Defects? Circ. Res. 118, 143–1150. discussion 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. [DOI] [PubMed] [Google Scholar]
- Hekimi S, Lapointe J, Wen Y (2011) Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH (2009) DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Williams RW, Auwerx J (2010) Metabolic networks of longevity. Cell 142, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL (2014) Age‐related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang CH, Tsien JZ, Schultz PG, Hu Y (2001) The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc. Natl. Acad. Sci. U. S. A. 98, 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Schiffer E, Song Z, Wang J, Zurbig P, Thedieck K, Moes S, Bantel H, Saal N, Jantos J, Brecht M, Jeno P, Hall MN, Hager K, Manns MP, Hecker H, Ganser A, Dohner K, Bartke A, Meissner C, Mischak H, Ju Z, Rudolph KL (2008) Proteins induced by telomere dysfunction and DNA damage represent biomarkers of human aging and disease. Proc. Natl. Acad. Sci. U. S. A. 105, 11299–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ (2010) The genetics of ageing. Nature 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Klemera P, Doubal S (2006) A new approach to the concept and computation of biological age. Mech. Ageing Dev. 127, 240–248. [DOI] [PubMed] [Google Scholar]
- Koga H, Kaushik S, Cuervo AM (2011) Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al‐Regaiey K, Su L, Sharpless NE (2004) Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 114, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS (2009) Senescence‐messaging secretome: SMS‐ing cellular stress. Nat. Rev. Cancer 9, 81–94. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev. 24, 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange P, Celli B, Agusti A, Boje Jensen G, Divo M, Faner R, Guerra S, Marott JL, Martinez FD, Martinez‐Camblor P, Meek P, Owen CA, Petersen H, Pinto‐Plata V, Schnohr P, Sood A, Soriano JB, Tesfaigzi Y, Vestbo J (2015) Lung‐function trajectories leading to chronic obstructive pulmonary disease. N. Engl. J. Med. 373, 111–122. [DOI] [PubMed] [Google Scholar]
- Lavasani M, Robinson AR, Lu A, Song M, Feduska JM, Ahani B, Tilstra JS, Feldman CH, Robbins PD, Niedernhofer LJ, Huard J (2012) Muscle‐derived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat. Commun. 3, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviano A (2014) Young blood. N. Engl. J. Med. 371, 573–575. [DOI] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D (2010) Enhancement of proteasome activity by a small‐molecule inhibitor of USP14. Nature 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME (2013) Modeling the rate of senescence: can estimated biological age predict mortality more accurately than chronological age? J. Gerontol. A Biol. Sci. Med. Sci. 68, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerch PM, Lu T, Dakin KA, Vann JM, Isaacs A, Geula C, Wang J, Pan Y, Gabuzda DH, Li C, Prolla TA, Yankner BA (2008) Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 3, e3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Antebi A, Bartke A, Barzilai N, Brown‐Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, Ingram DK, Johnson TE, Kennedy BK, Kenyon C, Klein S, Kopchick JJ, Lepperdinger G, Madeo F, Mirisola MG, Mitchell JR, Passarino G, Rudolph KL, Sedivy JM, Shadel GS, Sinclair DA, Spindler SR, Suh Y, Vijg J, Vinciguerra M, Fontana L (2015) Interventions to Slow Aging in Humans: are We Ready? Aging Cell 14, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Otín C, Galluzzi L, Freije José MP, Madeo F, Kroemer G (2016) Metabolic control of longevity. Cell 166, 802–821. [DOI] [PubMed] [Google Scholar]
- Ludwig FC, Smoke ME (1980) The measurement of biological age. Exp. Aging Res. 6, 497–522. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Liu J, Geletka L, Delaney C, Delproposto J, Desai A, Oatmen K, Martinez‐Santibanez G, Julius A, Garg S, Yung RL (2011) Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 187, 6208–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP (2010) Widespread and tissue specific age‐related DNA methylation changes in mice. Genome Res. 20, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, Martin NG, Montgomery GW, Feinberg AP, Fallin MD, Multhaup ML, Jaffe AE, Joehanes R, Schwartz J, Just AC, Lunetta KL, Murabito JM, Starr JM, Horvath S, Baccarelli AA, Levy D, Visscher PM, Wray NR, Deary IJ (2015) DNA methylation age of blood predicts all‐cause mortality in later life. Genome Biol. 16, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell BD, Hsueh WC, King TM, Pollin TI, Sorkin J, Agarwala R, Schaffer AA, Shuldiner AR (2001) Heritability of life span in the Old Order Amish. Am. J. Med. Genet. 102, 346–352. [DOI] [PubMed] [Google Scholar]
- Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H, Fraifeld VE (2013) The role of DNA damage and repair in aging through the prism of Koch‐like criteria. Ageing Res. Rev. 12, 661–684. [DOI] [PubMed] [Google Scholar]
- Nakamura E, Miyao K, Ozeki T (1988) Assessment of biological age by principal component analysis. Mech. Ageing Dev. 46, 1–18. [DOI] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin‐Ruiz C, von Zglinicki T (2012) A senescent cell bystander effect: senescence‐induced senescence. Aging Cell 11, 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AB, Glynn NW, Taylor CA, Sebastiani P, Perls TT, Mayeux R, Christensen K, Zmuda JM, Barral S, Lee JH, Simonsick EM, Walston JD, Yashin AI, Hadley E (2011) Health and function of participants in the Long Life Family Study: a comparison with other cohorts. Aging (Albany NY). 3, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyengaard JR, Bendtsen TF (1992) Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat. Rec. 232, 194–201. [DOI] [PubMed] [Google Scholar]
- Ozcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G, Galderisi U (2016) Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 8, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmioglu N, Andrew T, Cherkas L, Surdulescu G, Swaminathan R, Spector T, Ahmadi KR (2009) Epidemiology and genetic epidemiology of the liver function test proteins. PLoS ONE 4, e4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramunas J, Yakubov E, Brady JJ, Corbel SY, Holbrook C, Brandt M, Stein J, Santiago JG, Cooke JP, Blau HM (2015) Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 29, 1930–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindrarajah R, Lee DM, Pye SR, Gielen E, Boonen S, Vanderschueren D, Pendleton N, Finn JD, Tajar A, O'Connell MD, Rockwood K, Bartfai G, Casanueva FF, Forti G, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Lean ME, Punab M, Wu FC, O'Neill TW, European Male Aging Study G (2013) The ability of three different models of frailty to predict all‐cause mortality: results from the European Male Aging Study (EMAS). Arch. Gerontol. Geriatr. 57, 360–368. [DOI] [PubMed] [Google Scholar]
- Reinders I, Murphy RA, Koster A, Brouwer IA, Visser M, Garcia ME, Launer LJ, Siggeirsdottir K, Eiriksdottir G, Jonsson PV, Gudnason V, Harris TB (2015) Muscle Quality and Muscle Fat Infiltration in Relation to Incident Mobility Disability and Gait Speed Decline: the Age, Gene/Environment Susceptibility‐Reykjavik Study. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders I, Murphy RA, Brouwer IA, Visser M, Launer L, Siggeirsdottir K, Eiriksdottir G, Gudnason V, Jonsson PV, Lang TF, Harris TB, Age GESRS (2016) Muscle Quality and Myosteatosis: Novel Associations With Mortality Risk: the Age, Gene/Environment Susceptibility (AGES)‐Reykjavik Study. Am. J. Epidemiol. 183, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter‐Kochanek K, Jansen‐Durr P, Wlaschek M (2006) p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 5, 379–389. [DOI] [PubMed] [Google Scholar]
- Roth GS, Joseph JA (1994) Cellular and molecular mechanisms of impaired dopaminergic function during aging. Ann. N. Y. Acad. Sci. 719, 129–135. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA (1999) Longevity, stress response, and cancer in aging telomerase‐deficient mice. Cell 96, 701–712. [DOI] [PubMed] [Google Scholar]
- Salvi SM, Akhtar S, Currie Z (2006) Ageing changes in the eye. Postgrad. Med. J. 82, 581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori DJ, Wilbur CJ, Long SY, Rankin MM, Li C, Bradfield JP, Hakonarson H, Grant SF, Pu WT, Kushner JA (2014) GATA factors promote ER integrity and beta‐cell survival and contribute to type 1 diabetes risk. Mol. Endocrinol. 28, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, van der Pluijm I, Moorhouse MJ, Kosteas T, Robinson AR, Suh Y, Breit TM, van Steeg H, Niedernhofer LJ, van Ijcken W, Bartke A, Spindler SR, Hoeijmakers JH, van der Horst GT, Garinis GA (2008) Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 4, e1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani P, Solovieff N, Dewan AT, Walsh KM, Puca A, Hartley SW, Melista E, Andersen S, Dworkis DA, Wilk JB, Myers RH, Steinberg MH, Montano M, Baldwin CT, Hoh J, Perls TT (2012) Genetic signatures of exceptional longevity in humans. PLoS ONE 7, e29848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Shah Parisha P, Nativio R, Berger Shelley L (2016) Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso‐Santano M, Shen S, Marino G, Criollo A, Boileve A, Job B, Ladoire S, Ghiringhelli F, Sistigu A, Yamazaki T, Rello‐Varona S, Locher C, Poirier‐Colame V, Talbot M, Valent A, Berardinelli F, Antoccia A, Ciccosanti F, Fimia GM, Piacentini M, Fueyo A, Messina NL, Li M, Chan CJ, Sigl V, Pourcher G, Ruckenstuhl C, Carmona‐Gutierrez D, Lazar V, Penninger JM, Madeo F, Lopez‐Otin C, Smyth MJ, Zitvogel L, Castedo M, Kroemer G (2012) An immunosurveillance mechanism controls cancer cell ploidy. Science 337, 1678–1684. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA (2007) How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 8, 703–713. [DOI] [PubMed] [Google Scholar]
- Shock NW (1956) Some physiological aspects of aging in man. Bull. N. Y. Acad. Med. 32, 268–283. [PMC free article] [PubMed] [Google Scholar]
- Siebold AP, Banerjee R, Tie F, Kiss DL, Moskowitz J, Harte PJ (2010) Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. U. S. A. 107, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, Hirshman MF, Lebowitz J, Shadrach JL, Cerletti M, Kim MJ, Serwold T, Goodyear LJ, Rosner B, Lee RT, Wagers AJ (2014) Restoring systemic GDF11 levels reverses age‐related dysfunction in mouse skeletal muscle. Science 344, 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, Gontier G, Wabl R, Plambeck KE, Udeochu J, Wheatley EG, Bouchard J, Eggel A, Narasimha R, Grant JL, Luo J, Wyss‐Coray T, Villeda SA (2015) beta2‐microglobulin is a systemic pro‐aging factor that impairs cognitive function and neurogenesis. Nat. Med. 21, 932–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY, Jung JW, Lee S, Kim HS, Kang KS (2011) DNA methyltransferase controls stem cell aging by regulating BMI1 and EZH2 through microRNAs. PLoS ONE 6, e19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JP, Popovic ZB, Greenberg NL, Xu XF, Asher CR, Stewart WJ, Thomas JD (2004) Noninvasive quantification of regional myocardial function using Doppler‐derived velocity, displacement, strain rate, and strain in healthy volunteers: effects of aging. J. Am. Soc. Echocardiogr. 17, 132–138. [DOI] [PubMed] [Google Scholar]
- Takeda H, Inada H, Inoue M, Yoshikawa H, Abe H (1982) Evaluation of biological age and physical age by multiple regression analysis. Med Inform (Lond). 7, 221–227. [DOI] [PubMed] [Google Scholar]
- Tan Q, Zhao JH, Li S, Kruse TA, Christensen K (2010) Power assessment for genetic association study of human longevity using offspring of long‐lived subjects. Eur. J. Epidemiol. 25, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, Ono A, Ohara J, Baba T, Murata S, Tanaka K, Kasahara M (2012) Decreased proteasomal activity causes age‐related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 180, 963–972. [DOI] [PubMed] [Google Scholar]
- Tracy RE, Velez‐Duran M, Heigle T, Oalmann MC (1988) Two variants of nephrosclerosis separately related to age and blood pressure. Am. J. Pathol. 131, 270–282. [PMC free article] [PubMed] [Google Scholar]
- Tsaknaridis L, Spencer L, Culbertson N, Hicks K, LaTocha D, Chou YK, Whitham RH, Bakke A, Jones RE, Offner H, Bourdette DN, Vandenbark AA (2003) Functional assay for human CD4 + CD25 + Treg cells reveals an age‐dependent loss of suppressive activity. J. Neurosci. Res. 74, 296–308. [DOI] [PubMed] [Google Scholar]
- Tsurumi A, Li WX (2012) Global heterochromatin loss: a unifying theory of aging? Epigenetics 7, 680–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker JS, Klein DJ, Elliott MN (2004) Social control of health behaviors: a comparison of young, middle‐aged, and older adults. J. Gerontol. B Psychol. Sci. Soc. Sci. 59, P147–150. [DOI] [PubMed] [Google Scholar]
- Ungar A, Cristofari C, Torrini M, Di Serio C, Cantini C, Vallotti B, La Cava G, Castellani S, Masotti G (2000) Changes in renal autacoids in aged human hypertensives. J. Physiol. Pharmacol. 51, 619–630. [PubMed] [Google Scholar]
- United Nations Department of Economic and Social Affairs Population Division . (2013). World Population Ageing 2013.
- Vaitkevicius PV, Fleg JL, Engel JH, O'Connor FC, Wright JG, Lakatta LE, Yin FC, Lakatta EG (1993) Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation 88, 1456–1462. [DOI] [PubMed] [Google Scholar]
- Vermeij WP, Dolle ME, Reiling E, Jaarsma D, Payan‐Gomez C, Bombardieri CR, Wu H, Roks AJ, Botter SM, van der Eerden BC, Youssef SA, Kuiper RV, Nagarajah B, van Oostrom CT, Brandt RM, Barnhoorn S, Imholz S, Pennings JL, de Bruin A, Gyenis A, Pothof J, Vijg J, van Steeg H, Hoeijmakers JH (2016) Restricted diet delays accelerated ageing and genomic stress in DNA‐repair‐deficient mice. Nature 537, 427–431. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard‐Despres S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss‐Coray T (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Bennett M (2012) Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 111, 245–259. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen T, Yan H, Qi H, Deng C, Ye T, Zhou S, Li FR (2013) Role of histone deacetylase inhibitors in the aging of human umbilical cord mesenchymal stem cells. J. Cell. Biochem. 114, 2231–2239. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu Y, Han R, Beier UH, Bhatti TR, Akimova T, Greene MI, Hiebert SW, Hancock WW (2015) FOXP3 + regulatory T cell development and function require histone/protein deacetylase 3. J. Clin. Invest. 125, 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jockel KH, Erbel R, Muhleisen TW, Zenke M, Brummendorf TH, Wagner W (2014) Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 15, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D, Weinberger B, Grubeck‐Loebenstein B (2009) The aging of the immune system. Transpl. Int. 22, 1041–1050. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, van Heemst D, Rozing MP, Frolich M, Mooijaart SP, Blauw GJ, Beekman M, Heijmans BT, de Craen AJ, Slagboom PE, Leiden Longevity Study G (2009) Nonagenarian siblings and their offspring display lower risk of mortality and morbidity than sporadic nonagenarians: The Leiden Longevity Study. J. Am. Geriatr. Soc. 57, 1634–1637. [DOI] [PubMed] [Google Scholar]
- Xu X, Laird N, Dockery DW, Schouten JP, Rijcken B, Weiss ST (1995) Age, period, and cohort effects on pulmonary function in a 24‐year longitudinal study. Am. J. Epidemiol. 141, 554–566. [DOI] [PubMed] [Google Scholar]
- Zhang C, Cuervo AM (2008) Restoration of chaperone‐mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Iervese T, Abbati S, Bianchi GP, Marchesini G, Pisi E (1989) Portal blood velocity and flow in aging man. Gerontology 35, 61–65. [DOI] [PubMed] [Google Scholar]