

Summary
The study aimed to identify an immunoregulatory factor that restores the phosphoantigen response of Vγ9Vδ2+ T cells from HIV‐positive individuals on antiretroviral therapy. It was designed to characterize the effects of interleukin‐18 (IL‐18) on proliferation and effector function in Vγ9Vδ2 T cells from HIV‐negative individuals and test whether exogenous IL‐18 reconstitutes the Vγ9Vδ2 T‐cell response to phosphoantigen from HIV‐positive donors. Vγ9Vδ2 T cells from HIV‐negative individuals responded strongly to phosphoantigen or aminobisphosphonate stimulation of peripheral blood mononuclear cells (PBMC), whereas cells with similar T‐cell receptor profiles from HIV‐positive individuals only responded to aminobisphosphonate. Interleukin‐18 was higher after aminobisphosphonate stimulation due to activation of the inflammasome pathway. Both IL‐18 and IL‐18 receptor levels were measured and the activity of exogenous IL‐18 on HIV‐negative and HIV‐positive PBMC was evaluated in terms of Vγ9Vδ2 T‐cell proliferation, memory subsets, cytokine expression and CD107a expression. Interleukin‐18 stimulation increased proliferation, enhanced the accumulation of effector memory cells, and increased expression of cytotoxic markers in HIV‐negative controls. When Vγ9Vδ2 T cells from HIV‐positive individuals were stimulated with isopentenyl pyrophosphate in the presence of IL‐18, there was increased proliferation, accumulation of memory cells, and higher expression of CD56, NKG2D and CD107a (markers of cytotoxic effector phenotype). Interleukin‐18 stimulation specifically expanded the Vγ9‐JγP+ subset of Vγ9Vδ2 T cells, as was expected for normal responses to phosphoantigen. Interleukin‐18 is a potent stimulator of Vγ9Vδ2 T‐cell proliferation and effector function. Therapies directed at reconstituting Vγ9Vδ2 T‐cell activity in HIV‐positive individuals should include stimulators of IL‐18 or direct cytokine supplementation.
Keywords: γδ T cell, human immunodeficiency virus, inflammasome, interleukin‐18, phosphoantigen, Vγ9Vδ2
Abbreviations
- CD
cluster of differentiation
- DNAM‐1
DNAX accessory molecule‐1
- GGPP
geranyl geranyl pyrophosphate
- HIV
human immunodeficiency virus
- IFN‐γ
interferon‐γ
- IL
interleukin
- IPP
isopentenyl pyrophosphate
- NKG2D
natural killer group 2D
- NK
natural killer
- NLRP3
NOD‐like receptor pyrin containing 3
- PBMC
peripheral blood mononuclear cells
- TCR
T‐cell receptor
- ZOL
zoledronic acid
Introduction
The Vγ9Vδ2 T‐cell receptor (TCR) is required for cellular responses to five carbon pyrophosphate intermediates (phosphoantigens) in the pathway for cholesterol biosynthesis.1, 2 Responses to these phosphoantigens are MHC‐unrestricted and specific to a subset of cells expressing the Vγ9‐JγP rearrangement.3, 4 Because isopentenyl pyrophosphate (IPP) and other stimulatory phosphoantigens are ubiquitous, a constant positive selection amplifies the Vγ9‐JγPVδ2+ subset in blood and lymphoid tissues.5 Hence, a majority of circulating Vγ9Vδ2 T cells in humans have central or effector memory phenotypes due to chronic phosphoantigen exposure and are potently cytotoxic against many tumours and microbially infected cells.6, 7, 8, 9 Phosphoantigen‐responsive Vγ9‐JγPVδ2+ T cells usually comprise > 75% of circulating γδ T cells in HIV‐negative (HIV–) adults, and this population responds so rapidly to cancer or infected cells that it resembles innate immunity.10, 11 Self/non‐self discrimination by Vγ9‐JγPVδ2+ T cells depends mainly on natural killer (NK) or killer inhibitor receptors.12, 13, 14, 15 Immunoglobulin binding to cell surface FcgRIII also increases Vγ9‐JγPVδ2+ T‐cell cytotoxicity.16, 17 In addition to direct effector activities, these γδ T cells also co‐stimulate NK cells for increased tumour cell or dendritic cell killing.18, 19
Rapid loss of Vγ9‐JγPVδ2 T cells is an important part of acquired immunodeficiency disease and is among the earliest T‐cell defects after HIV infection.18, 19, 20, 21, 22, 23, 24 Among all persons with HIV disease only natural virus suppressors (also termed elite controllers) maintain near normal Vγ9Vδ2 levels and the frequencies of CD27– CD45RA– effector cells are similar to those found in HIV– control donors.25, 26, 27 Reconstitution of the Vγ9Vδ2 TCR repertoire occurs after prolonged antiretroviral therapy but these cells remain unresponsive to phosphoantigen stimulation and cannot be amplified in vitro despite having TCR sequences capable of responding to IPP.28, 29, 30 Curiously, these reconstituted cells are responsive to stimulation with aminobisphosphonate drugs including zoledronic acid (ZOL) that increase intracellular IPP in antigen‐presenting cells (APC).30, 31
The mechanism of action for aminobisphosphonate (ZOL) drugs is competitive inhibition of farnesyl diphosphate synthase, which prevents conversion of IPP into downstream farnesyl diphosphate synthase and geranylgeranyl pyrophosphate (GGPP). ZOL is incorporated into APC cells where it increases intracellular IPP, which is presented to Vγ9‐JγPVδ2 T cells by cell surface butyrophilin3A1.31, 32, 33, 34 GGPP is an important negative regulator of the NOD‐like receptor pyrin containing 3 (NLRP3) inflammasome and farnesyl diphosphate synthase inhibitors including ZOL reduce GGPP levels. Consequently, ZOL indirectly activates NLRP3 and increases IPP; these effects are sufficient to stimulate Vγ9Vδ2 T‐cell proliferation, differentiation and effector function.
We postulated that the differences in IPP versus ZOL responses among HIV‐positive (HIV+) individuals might be explained by activity of the NLRP3 inflammasome including release of interleukin‐18 (IL‐18) and/or IL‐1β.33, 34, 35, 36 Insufficient production of IL‐18 and/or IL‐1β in peripheral blood mononuclear cells (PBMC) from HIV+ patients might explain the failed response to IPP.37 Here, we assessed the effects of IL‐18 on Vγ9Vδ2 T‐cell stimulation and tested whether this cytokine could reconstitute the IPP response in PBMC from HIV+ individuals.
Materials and methods
Samples
Venous blood samples were obtained from HIV+ and HIV– individuals. PBMC were purified by Ficoll gradient centrifugation and stored as viable, frozen cells. All HIV+ individuals were being treated with combination antiretroviral therapy and all were suppressed to < 50 copies/ml of plasma viral RNA at the time blood specimens were obtained. Informed written consent was obtained from all patients and this study was approved by the Institutional Review Board of the University of Maryland, Baltimore (Baltimore, MD).
Cell culture
Purified PBMC were re‐suspended in R‐10 medium consisting of RPMI‐1640 supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY), 2 mmol/l l‐glutamine (Invitrogen, Camarillo, CA), 1 U/ml penicillin/streptomycin (Invitrogen) and 100 U/ml recombinant human IL‐2 (Tecin, Biological Resources Branch, NIH, Bethesda, MD). Zoledronic acid at a concentration of 1 μm (zoledronate/Zol; Sigma, St Louis, MO) or IPP (Sigma) at a concentration of 15 μm was added to trigger Vγ9Vδ2+ cell proliferation. Cultures were incubated at 37° in 5% CO2 and replenished every 3 days by adding R‐10 medium containing 100 U/ml IL‐2 as needed. On day 14, cells were rested by shifting to medium with lower IL‐2 (10 U/ml) for 2 days. Phenotyping and functional assays were performed on ex vivo PBMCs or cells harvested 14–16 days after stimulation. An absolute Vγ9Vδ2 T‐cell count on day 14 was calculated as: frequency of Vγ9Vδ2 T cells in culture * (specific cells/μl). Interleukin‐18 inhibition in PBMC cultures was measured with additional compounds GGPP (Sigma) at a concentration of 15 μm, 15D (A438079 HCl; R&D Systems, Minneapolis, MN) at a concentration of 100 μm, and Caspase‐1 (Ac‐YVAD‐cmk; Invivogen, San Diego, CA) at a concentration of 50 μm.
Flow cytometry
Day 14 expanded Vγ9Vδ2 T cells (2 × 105−5 × 105) were stained with monoclonal antibodies for 30 min at 4°. Antibodies included Vδ2, CD8, CD4, CD3, CD56, CD16, CD45RA, CD27, IL‐18R, NKG2D, DNAM‐1, Perforin, CD107a and CCL5/RANTES. Antibodies were purchased from BioLegend (San Diego, CA) unless noted otherwise. After staining for surface markers, cells were washed with RPMI‐1640 and fixed with BD CytoFix/CytoPerm for 20 min at 4°. Samples were read on a BD Accuri C6 Flow Cytometer. Data were analysed by flowjo (TreeStar, San Jose, CA) and graphpad prism (GraphPad Software, La Jolla, CA).
Granule mobilization assay
Day 16 (rested) cells were collected and re‐suspended at 1 × 106 cells/ml in fresh R‐10 medium and re‐stimulated with 15 μm IPP. Cells were plated with anti‐CD107a‐phycoerythrin‐Cy7, Brefeldin A (1 : 1000; BD Biosciences, San Jose, CA), and Monensin (1 : 1500; BD Biosciences). After 6 hr of incubation at 37°, cells were washed with cold PBS and stained for surface marker using anti‐Vδ2‐FITC, followed by fixation/permeabilization (BD Biosciences), and intracellular staining with anti‐Perforin‐phycoerythrin and anti‐CCL5‐Alexafluor647 (Biolegend).
Antigen re‐stimulation
Day 16 (rested) cells were re‐suspended at 1 × 106 cells/ml in fresh R‐10 medium and re‐stimulated with 15 μm IPP (Sigma). After 24 hr of incubation at 37°, cells were pelleted and supernatants were collected. Cell‐free interferon‐γ (IFN‐γ) levels were determined by ELISA. Polystyrene plates (Nunc MaxiSorp; Thermo Fisher, Waltham, MA) were coated with capture antibody in PBS overnight at 25°. The plates were washed with 50 mm Tris–HCl, 0·2% Tween‐20, and then blocked for 90 min at 25° with assay buffer, PBS containing 4% BSA (Sigma). Then 50 μl of sample or standard prepared in assay buffer and incubated at 37° for 2 hr. The plates were washed and 100 μl of biotinylated detecting antibody in assay buffer was added and incubated for 1 hr at 25°. After washing, strepavidin‐peroxidase polymer in casein buffer (RDI, Flanders, NJ) was added and incubated at 25° for 30 min. The plate was washed and 100 μl of commercially prepared substrate (TMB Dako, Agilent Technologies, Santa Clara, CA) was added and incubated at 25° for approximately 10–30 min. The reaction was stopped with 100 μl 2 m HCl and the absorbance at 450 nm (A450) (minus A650) was read on a microplate reader (Molecular Dynamics, GE Healthcare, Uppsala, Sweden). A curve was fitted to the standards using a computer program (softpro; Molecular Dynamics) and cytokine concentration in each sample was calculated from the standard curve equation.
RNA extraction, RT‐PCR, PCR
RNA extraction, reverse transcription, and PCRs were performed as previously described using the following primers for Vγ9 chain: oligo‐Vγ9 (5′‐ATC AAC GCT GGC AGT CC‐3′) and oligo‐Cy‐1 (5′‐GTT GCT CTT CTT TTC TTG CC‐3′).38
Cloning and sequencing
Cloning and sequencing reactions were performed as previously described.39 In summary, Vγ9 coding region PCR products were purified by gel extraction. Purified PCR products were denatured and ligated into the plasmid vector. Ligated vectors were transfected into bacterial colonies and grown overnight. Colonies containing recombinant Vγ9 chain plasmids were amplified in bacterial suspensions. Suspensions were processed and used as DNA templates for amplifying individual Vγ9 chain sequences. PCR was performed using M13 forward primer (5′‐GTA AAA CGA CGG CCA G‐3′) and reverse M13 primer (5′‐CAG GAA ACA GCT ATG AC‐3′). PCR products were purified by size exclusion. Sequencing reactions were performed using M13 Forward or M13 Reverse primers for each target. Sequences were loaded on an automated sequencer and analysed using sequencher (Gene Codes Corp, Ann Arbor, MI) and macclade (Sinauer Associates, Sunderland, MA) software.
Statistical analysis
Statistical analyses were performed using the software graphpad prism. For each measured variable, a D'Agostino and Pearson omnibus normality test (GraphPad Software, La Jolla, CA) was performed to determine if values were normally distributed. Values that were not normally distributed were excluded from further statistical analyses. Differences between normally distributed means were evaluated using a Student's t‐test when comparing two groups.
Results
Inhibiting IL‐18 in PBMC cultures
Zoledronic acid inhibits GGPP production and indirectly stimulates the inflammasome.35 We confirmed that ZOL treatment increased the production of NLRP3 products IL‐18 and IL‐1β via a Caspase‐1‐induced mechanism. Ex vivo PBMC were stimulated for 24 hr with ZOL, IPP or control (no stimulus) in the presence of IL‐2. Low levels of endogenous IL‐18 (10 pg/ml) were detected in both control and IPP cultures even in the absence of ZOL. IL‐18 levels increased to 20 pg/ml in the presence of ZOL after 24 hr (see Supplementary material, Fig. S1a) and were significantly higher than IL‐18 levels in control or IPP‐stimulated cultures. 15D, a P2X7 receptor antagonist that inhibits NLRP3 activity added with GGPP significantly reduced ZOL‐stimulated IL‐18 production but did not reduce cytokine to control levels.36 The Caspase‐1 inhibitor YVAD decreased ZOL‐stimulated IL‐18 production fourfold and decreased endogenous control IL‐18 production twofold.
Since YVAD inhibits Caspase‐1, which processes the precursor forms of both IL‐18 and IL‐1β, this inhibitor was not specific for IL‐18. To test the role for IL‐18 in Vγ9Vδ2 T‐cell activation, we used blocking antibodies against the IL‐18 receptor α chain or neutralized soluble IL‐18 with a monoclonal antibody. A bioassay for IL‐18, measuring IFN‐γ production in KG‐1 cell cultures that were stimulated with exogenous IL‐18, was used to evaluate IL‐18 blocking reagents. A natural inhibitor of IL‐18, IL‐18‐binding protein, was effective at reducing the IFN‐γ response in KG‐1 cells.40 A concentration of 0·8 μg/ml IL‐18‐binding protein blocked > 95% of IL‐18 stimulated IFN‐γ production in KG‐1 cell cultures and was used here as an inhibitor of IL‐18 in PBMC cultures (see Supplementary material, Fig. S1b).
High level IL‐18Ra expression on Vγ9Vδ2 T cells
After confirming that ZOL induces IL‐18 production in PBMC, we next evaluated the distribution of IL‐18 receptor on leucocyte subpopulations. We compared IL‐18Ra levels on NK, CD4, CD8 T and Vγ9Vδ2 T cells. The cell subsets were defined on the basis of surface marker expression as follows: CD14+ CD3− for monocytes, CD20+ CD3− for B cells, CD3+ for T cells, CD3+ CD4+ for CD4 T cells, CD3+ CD8+ for CD8 T cells, CD56+ CD3− for NK cells, CD3+ Vδ2+ for Vγ9Vδ2 T cells, and CD27+ /CD45RA− for central memory T cells. The IL‐18Ra expression was highest on Vγ9Vδ2 T cells and was above the levels seen for NK or CD8 T cells (Fig. 1a). This is important because both NK and CD8 T cells respond directly to IL‐18.40 The circulating Vγ9Vδ2+ T‐cell population was highly skewed towards a central memory phenotype compared with αβ CD4 or CD8 T cells and this might affect differences in IL‐18R density. Even accounting for differences in the proportion of central memory cells, Vγ9Vδ2+ T cells still had higher levels of IL‐18Ra compared with both CD4 and CD8 T cells (Fig. 1b). Next, we assessed the relationships between IL‐18R levels and CD56 expression on NK and Vγ9Vδ2 T cells (Fig. 1c). There was no apparent correlation between CD56 expression and IL‐18R levels for Vγ9Vδ2+ T cells but IL‐18Ra levels were significantly higher among CD56bright NK cells compared with the CD56dim population. In PBMC obtained from HIV+ donors IL‐18R expression was highest on Vγ9Vδ2+ cells among all subsets that were tested (Fig. 1d). In PBMC from HIV+ donors there were insufficient numbers of Vγ9Vδ2 T cells to stratify on the basis of CD56 expression.
Figure 1.
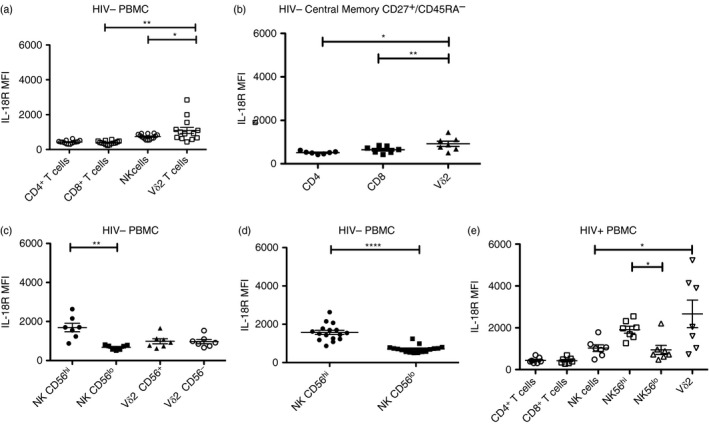
Vγ9Vδ2 T cells express high levels of interleukin‐18 (IL‐18) receptor α. (a) Ex vivo peripheral blood mononuclear cells (PBMC) were stained for CD14, CD20, CD3, CD4, CD8, CD56 and Vδ2. The MFI of IL‐18Ra was measured in each cell population (n = 14). (b) Ex vivo PBMC were stained for CD3, CD4, CD8, Vδ2, CD27 and CD45RA (n = 7). (c) Ex vivo PBMC were stained for CD3, CD56 and Vδ2. Natural killer (NK) cells were separated by CD56bright and CD56dim. Vγ9Vδ2 T cells were separated by CD56+ and CD56–. IL‐18Ra was measured for each subpopulation (n = 7). (d) Ex vivo PBMC were stained for CD3, CD56 and IL‐18R. NK cells were separated by CD56bright and CD56dim (n = 16). (e) Ex vivo PBMCs from HIV+ patients were stained for CD3, CD4, CD8, Vδ2 and CD56. NK cells were separated further by high and low CD56 expression (n = 7). ****P < 0·0001, **P < 0·01, *P < 0·05.
IL‐18 enhances Vγ9Vδ2 T‐cell proliferation
To examine the effects of IL‐18 on Vγ9Vδ2 T‐cell proliferation we treated PBMC with IPP in the presence or absence of IL‐18. PBMC from 15 control (HIV–) donors were stimulated with ZOL or IPP (Fig. 2a). As expected, proliferation responses to ZOL were higher than with IPP. Next, we added 10 or 50 ng/ml of IL‐18 to PBMC that were stimulated with IPP. Adding the cytokine increased Vγ9Vδ2+ T‐cell proliferation to levels comparable to ZOL stimulation (Fig. 2a) and the effect was blocked by adding IL‐18‐binding protein (Fig. 2b). There were no significant differences between 10 and 50 ng/ml IL‐18 doses.
Figure 2.
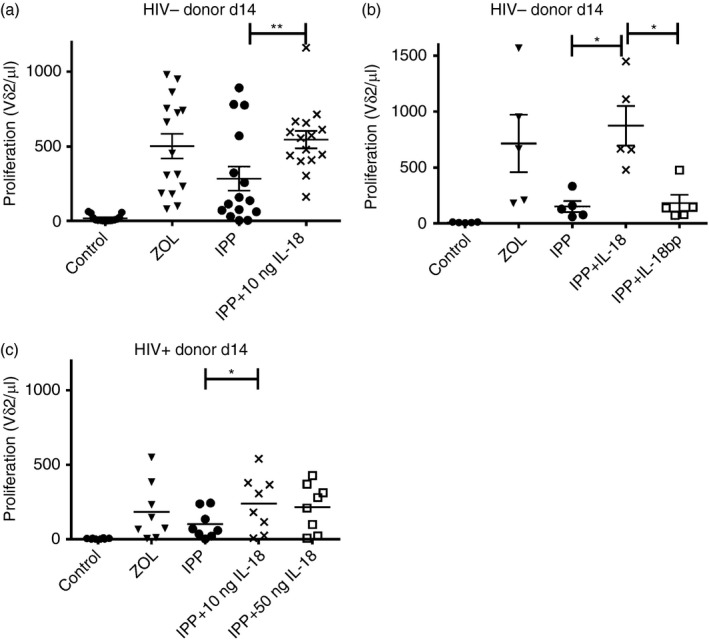
Exogenous interleukin‐18 (IL‐18) enhances proliferation. (a) Peripheral blood mononuclear cells (PBMC) (1·5 × 106/ml) were stimulated with 1 μm zoledronic acid (ZOL) or 15 μm isopentenyl pyrophosphate (IPP) on day 0 in R‐10 medium supplemented with 100 U IL‐2. Exogenous IL‐18 (10 or 50 ng) was added on days 0, 1 and 2. Proliferation was assessed on day 14 by flow cytometry. Cells were stained for Vδ2 and counted (n = 15 HIV– donors). (b) PBMC (1·5 × 106/ml) were stimulated with 1 μm ZOL or 15 μm IPP on day 0 in R‐10 medium supplemented with 100 U IL‐2. Exogenous IL‐18 (10 ng/ml) or IL‐18 bp (0·8 μg/ml) was added on days 0, 1 and 2. Proliferation was assessed on day 14 by flow cytometry. Cells were stained for Vδ2 and counted. n = 5 donors (c) PBMCs (2·0 × 106/ml) were stimulated with 1 μm ZOL or 15 μm IPP on day 0 in R‐10 medium supplemented with 100 U IL‐2. Exogenous IL‐18 (10 ng or 50 ng) was added on days 0, 1 and 2. Proliferation was assessed on day 14 by flow cytometry. Cells were stained for Vδ2 and counted. n = 8 donors. **P < 0·01, *P < 0·05.
We then tested whether IL‐18 could restore proliferative responses to IPP in PBMC from eight HIV+ donors. Interleukin‐18 supplementation increased the proliferative response to IPP in six out of eight HIV+ donors and even exceeded proliferation levels seen with ZOL treatment (Fig. 2c). Two HIV+ donors had minimal responses to exogenous IL‐18.
Memory phenotype and functional markers of IL‐18 expanded Vγ9Vδ2 T cells
Vδ2+ T cells expanded in the presence of IPP plus IL‐18 were assessed to determine the distribution of memory and effector cell phenotypes. For these experiments we used PBMC from HIV– (control) donors. Vγ9Vδ2 T cells were stimulated by IPP plus IL‐18 and cultured for 14 days. Cultures supplemented with IL‐18 had decreased proportions of central memory (CD27+ CD45RA−) cells and increased proportions of effector memory (CD27− CD45RA−) cells compared with IPP alone. The percentage of effector memory cells approached 95% after high dose IL‐18 (50 ng/ml) treatment. Day 14 expanded Vγ9Vδ2 T cells usually have low proportions of naive (CD27+ CD45RA+) or terminally differentiated effector (CD27− CD45RA+) cells, but IL‐18 supplementation decreased the proportion of naive cells even further (Fig. 3a).
Figure 3.
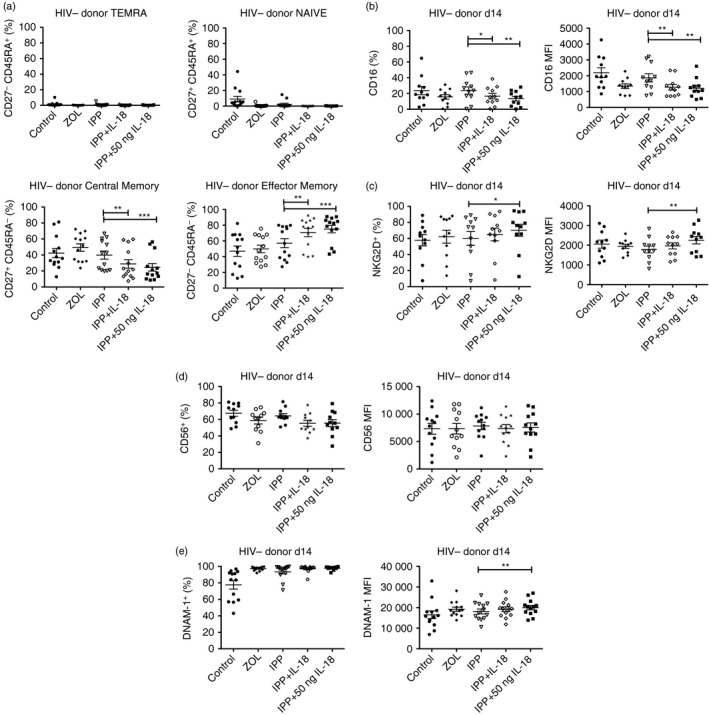
Effect of exogenous interleukin‐18 (IL‐18) on memory and functional markers. (a) Day 14 expanded cells were stained for Vδ2, CD3, CD27 and CD45RA. Populations shown were gated on Vδ2+ CD3+ (n = 13). (b) Day 14 expanded cells were stained for Vδ2, CD16 and NKG2D (n = 11). (c) Day 14 expanded cells were stained for Vδ2, CD56 and DNAM‐1. Populations shown were gated on Vδ2+ CD56+ n = 11, CD56 MFI n = 12, DNAM‐1 n = 13. ***P < 0·001, **P < 0·01, *P < 0·05.
Unexpectedly, IL‐18 supplementation decreased the percentage and the levels of CD16 expression on Vγ9Vδ2+ cells (Fig. 3b). The percentage of cells expressing NKG2D increased with IL‐18 supplementation compared to IPP alone (Fig. 3c). A small decrease in the percentage of CD56+ cells was observed after IL‐18 treatment but the levels of CD56 expression remained unchanged (Fig. 3d). The percentage of DNAM‐1+ cells increased as did the levels of DNAM‐1 expression on Vγ9Vδ2 T cells exposed to IL‐18. The proportion of DNAM‐1+ Vγ9Vδ2 T cells exceeded 99% in the high dose IL‐18 group (Fig. 3e).
Vδ2+ T cells from HIV+ donors were cultured for 14 days and tested for the same phenotypic markers. HIV+ donor Vγ9Vδ2 T cells showed an increase in the proportion of effector memory cells after IL‐18 supplementation compared with stimulation with IPP alone (Fig. 4a). However, this trend was not statistically significant due to donor variability within the IPP treatment group. There were no measurable changes in the proportion of central memory, terminally differentiated or naive Vγ9Vδ2 cells in the IL‐18‐treated group compared with IPP alone. IL‐18 supplementation did not affect CD16 expression (Fig. 4b) but slightly increased NKG2D expression and the percentage of NKG2D+ cells compared with treatment with IPP alone (Fig. 4c). Interleukin‐18 supplementation slightly increased the percentage of CD56+ cells and significantly raised CD56 expression compared with IPP alone (Fig. 4d). The percentage of DNAM‐1+ cells and DNAM‐1 expression were slightly increased with IL‐18 supplementation compared with IPP alone but these results were not significant (Fig. 4e).
Figure 4.
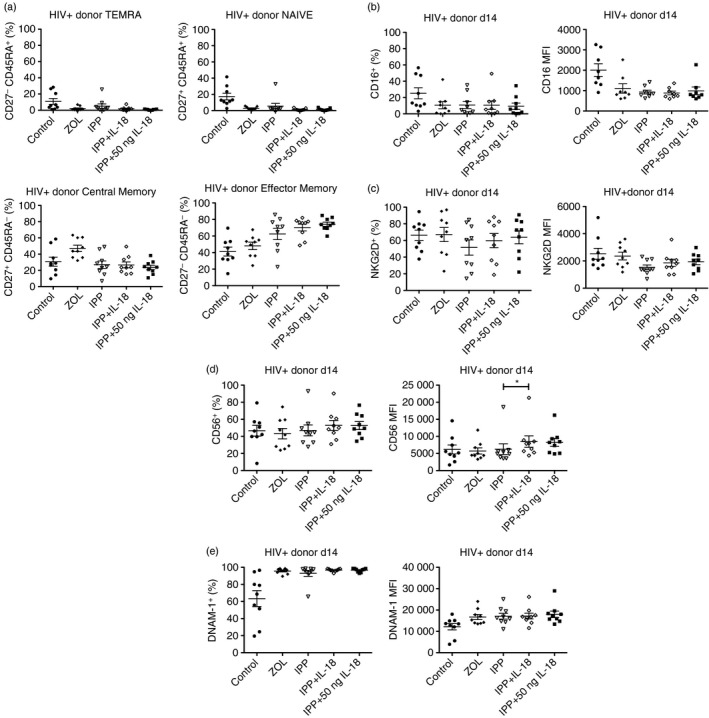
Effect of exogenous interleukin‐18 (IL‐18) on memory and functional markers from HIV+ donors. (a) Day 14 expanded cells were stained for Vδ2, CD3, CD27 and CD45RA. Populations shown were gated on Vδ2+ CD3+ (n = 9). (b) Day 14 expanded cells were stained for Vδ2, CD16 and NKG2D. CD16+ n = 9, CD16 MFI n = 8, NKG2D n = 9. (c) Day 14 expanded cells were stained for Vδ2, CD56 and DNAM‐1. Populations shown were gated on Vδ2+ (n = 9) *P < 0·05.
Recall responses to IPP
One way to assess Vγ9Vδ2+ T‐cell functional responses is to expand the Vγ9Vδ2 population from PBMC with ZOL or IPP treatment for 14 days, then rest with low IL‐2 for 2 days before re‐stimulating with IPP and measuring the short‐term ‘recall’ response. Here, expanded cells (ZOL or IPP) were rested with low IL‐2 (10 U/ml) for 2 days then re‐stimulated with 15 μm IPP and no additional IL‐2. CD107a degranulation, perforin expression and IFN‐γ secretion were measured as markers of effector responses. CD107a degranulation in conjunction with perforin expression was used as a surrogate marker for perforin‐mediated cytotoxicity because we did not have sufficient numbers of cells from all donors for direct cytotoxicity assays. In PBMC from HIV‐negative donors, IL‐18 supplementation yielded slightly higher percentages of CD107a+ Perforin+ Vγ9Vδ2 T cells compared with IPP without IL‐18 (Fig. 5a). Vγ9Vδ2 T cells secreted more IFN‐γ after IL‐18 plus IPP compared with IPP alone (Fig. 5b). In HIV+ donors, we saw similar, non‐significant increases in CD107a+ Perforin+ Vγ9Vδ2 cells from the IL‐18 treatment groups compared with the IPP only group (Fig. 5c). However, there were no differences in IFN‐γ secretion between IL‐18‐supplemented and IPP alone groups (Fig. 5d).
Figure 5.
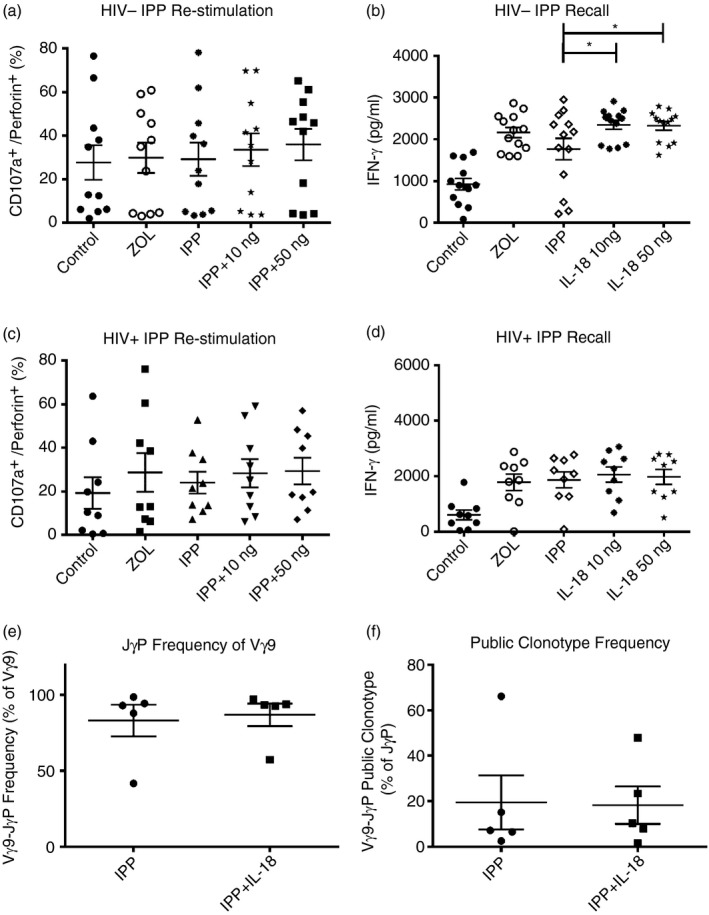
Exogenous interleukin‐18 (IL‐18) improves phosphoantigen response. (a) Day 16 rested Vγ9Vδ2 T cells (1 × 105/100 μl) were stimulated for 6 hr with 15 μm isopentenyl pyrophosphate (IPP) in the presence of monensin, brefeldin and CD107a. Cells were stained for Vδ2, fixed/permeabilized, and then stained for perforin (n = 11). (b) Day 16 rested Vγ9Vδ2 T cells (5 × 105/500 μl) were stimulated with 15 μm IPP for 24 hr, then supernatants were collected for ELISA (n = 13). (c) Day 16 rested Vγ9Vδ2 T cells (1 × 105/100 μl) from HIV+ donors were stimulated for 6 hr with 15 μm IPP in the presence of monensin, brefeldin and CD107a. Cells were stained for Vδ2, fixed/permeabilized, and then stained for Perforin (n = 9). (d) Day 16 rested Vγ9Vδ2 T cells (5 × 105/500 μl) from HIV+ donors were stimulated with 15 μm IPP for 24 hr, then supernatants were collected for ELISA (n = 9). (e) Day 16 rested cells (1 × 106) were cloned and sequenced. Per cent of JγP rearrangements within productive Vγ9 sequences. (f) Day 16 rested cells (1 × 106) were cloned and sequenced. Per cent of public clonotypes within JγP rearrangements. *P < 0·05.
IL‐18 addition does not skew the Vγ9 chain repertoire
The TCR‐γ chain rearrangement Vγ9‐JγP is required for phosphoantigen responses. We tested whether IL‐18 supplementation changed the proportion of Vγ9‐JγP+ cells in PBMC from five HIV+ donors after IPP stimulation and proliferation. A sample of 100 Vγ9 chains were sequenced from PBMC stimulated with IPP only or IPP plus IL‐18 (10 ng/ml group). There were no differences in the frequencies of Vγ9‐JγP rearranged chains between the two groups (Fig. 5e). We also measured the frequency of eight common public clonotypes (predicted from the nucleotype sequences) among all Vγ9‐JγP rearranged γ chains. The frequencies of common public clonotypes were similar in IPP and IPP plus IL‐18 treatment groups (Fig. 5f). Hence, IL‐18 supplementation increased the proliferative response to IPP but did not alter the proportion of Vγ9‐JγP rearrangements or select for a specific subset of public clonotypes.
Discussion
Depletion of Vγ9‐JγPVδ2 T cells and decreased responses to phosphoantigens are hallmarks of HIV disease.21, 22, 23 Only natural virus suppressors (also termed elite controllers) who are capable of controlling viraemia in the absence of antiretroviral therapy have normal Vγ9Vδ2 T‐cell responses to phosphoantigen.27 The absence of phosphoantigen responses even after long‐term therapy, is difficult to explain as molecular analyses of the γ‐chain repertoire showed reconstitution of cells expressing Vγ9‐JγP rearranged chains and predicted a phosphoantigen response.30, 31, 32 However, Vγ9Vδ2 T cells from HIV+ donors did respond to aminobisphosphonate stimulation suggesting that mechanisms other than TCR recognition of antigen might be responsible for the lack of responses in HIV+ donors.32 Here, we identify IL‐18 as a cytokine potentially capable of reversing the HIV‐associated defect in phosphoantigen responses and show that aminobisphosphonate stimulation increased the cytokine levels.
The effects of IL‐18 on Vγ9Vδ2 T cells included increased proliferative responses to phosphoantigen stimulation, a shift toward effector memory subsets, increased expression of CD56 that is associated with Vγ9Vδ2 cell cytotoxicity, and elevated expression of NK receptors that are important for Vγ9Vδ2 T‐cell cytotoxic effector function.41 These effects are over and above the impact of phosphoantigen alone.
It is important to note that IL‐18 supplementation increased the proliferative response to phosphoantigen in PBMC from both control and HIV+ donors. This accounts for a consistent finding that aminobisphosphonate responses usually exceed phosphoantigen responses for most HIV– donors and emphasizes the importance of inflammasome activation for optimal Vγ9Vδ2+ T‐cell responses. In PBMC from HIV+ donors, the response to phosphoantigen alone is low compared with control individuals, and the impact of IL‐18 is immediately obvious. The proportion of memory Vγ9Vδ2+ T cells in HIV+ donors is also low because naive cells constitute a significantly greater proportion of total Vγ9Vδ2+ T cells due to poor phosphoantigen responses in vivo.5 The apparent advantage of IL‐18 for HIV+ donor PBMC reflects the ability of IL‐18 to activate cells and increase the memory subset. Vγ9Vδ2 T cells with a CD27− CD45RA– effector phenotype are depleted during active tuberculosis, progressing HIV infection, and tuberculosis/HIV co‐infection.42, 43, 44 Interleukin‐18, or compounds such as aminobisphosphonates that increase IL‐18 production might be therapeutic for these infections partly by reconstituting the Vγ9Vδ2 T‐cell memory subsets.
Additionally, since Vγ9Vδ2 T cells provide co‐stimulation for NK cells through 41BBL and ICOS, reconstitution of functional Vγ9Vδ2 cells would improve NK cell cytotoxicity against tumour cells and dendritic cells.18, 19 Unfortunately, IL‐18 supplementation decreased the proportion of CD16+ Vγ9Vδ2 T cells in both populations and might limit antibody‐mediated cellular cytotoxicity effector function.45 Interleukin‐18 was seen to increase the proportion of NKG2D+ Vγ9Vδ2 cells in PBMC from HIV+ and control donors that is important for enhancing direct cytotoxicity.14, 15
Based on our understanding of IL‐18 effects on Vγ9Vδ2 T‐cell proliferation, we can postulate that delivering this cytokine may be one part of a broader immunotherapeutic approach targeting Vγ9Vδ2 T cells in HIV+ individuals. Our studies on IL‐18 do not fully explain the failure to reconstitute normal cell levels or phosphoantigen responses after prolonged antiretroviral therapy but hint at insufficient numbers of functional monocytic cells, insufficient inflammasome activation, or even suppression of the inflammasome as possible mechanisms responsible for the failure to reconstitute Vγ9Vδ2 T cells after prolonged antiretroviral therapy. Knowing the importance of inflammasome activity for Vγ9Vδ2+ T‐cell phosphoantigen responses encourages further studies of HIV+ individuals receiving aminobisphosphonate therapy for osteoporosis to learn whether inflammasome activation impacts the function of Vγ9Vδ2+ T cells and reconstitutes this important T‐cell subset.
Disclosures
CDP owns stock in and is employed by American Gene Technologies International, Inc, Rockville, MD (www.americangene.com).
Supporting information
Figure S1. Stimulators and inhibitors of interleukin‐18 production.
Acknowledgements
Funding from NIH/NHLBI 5F31HL128159 (ASM) and 5R21HL126533 (CDP).
References
- 1. Tanaka Y, Brenner MB, Bloom BR, Morita CT. Recognition of nonpeptide antigens by T cells. J Mol Med (Berl) 1996; 74:223–31. [DOI] [PubMed] [Google Scholar]
- 2. Morita CT, Beckman EM, Bukowski JF, Tanaka Y, Band H, Bloom BR et al Direct presentation of nonpeptide prenyl pyrophosphate antigens to human γδ T cells. Immunity 1995; 3:495–507. [DOI] [PubMed] [Google Scholar]
- 3. Fisch P, Malkovsky M, Braakman E, Sturm E, Bolhuis RL, Prieve A et al γδ T cell clones and natural killer cell clones mediate distinct patterns of non‐major histocompatibility complex‐restricted cytolysis. J Exp Med 1990; 171:1567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Evans PS, Enders PJ, Yin C, Ruckwardt TJ, Malkovsky M, Pauza CD. In vitro stimulation with a non‐peptidic alkylphosphate expands cells expressing Vγ2‐ Jγ1.2/Vδ2 T‐cell receptors. Immunology 2001; 104:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parker CM, Groh V, Band H, Porcelli SA, Morita C, Fabbi M et al Evidence for extrathymic changes in the T cell receptor γδ repertoire. J Exp Med 1990; 171:1597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alexander AA, Maniar A, Cummings JS, Hebbeler AM, Schulze DH, Gastman BR et al Isopentenyl pyrophosphate‐activated CD56+ γδ T lymphocytes display potent antitumor activity toward human squamous cell carcinoma. Clin Cancer Res 2008; 14:4232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mattarollo SR, Kenna T, Nieda M, Nicol AJ. Chemotherapy and zoledronate sensitize solid tumour cells to Vγ9Vδ2 T cell cytotoxicity. Cancer Immunol Immunother 2007; 56:1285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poccia F, Agrati C, Martini F, Capobianchi MR, Wallace M, Malkovsky M. Antiviral reactivities of γδ T cells. Microbes Infect 2005; 7:518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bukowski JF, Morita CT, Brenner MB. Recognition and destruction of virus infected cells by human γδ CTL. J Immunol 1994; 153:5133–40. [PubMed] [Google Scholar]
- 10. Bukowski JF, Morita CT, Brenner MB. Human γδ T cells recognize alkylamines derived from microbes, edible plants, and tea: implications for innate immunity. Immunity 1999; 11:57–65. [DOI] [PubMed] [Google Scholar]
- 11. Beetz S, Wesch D, Marischen L, Welte S, Oberg HH, Kabelitz D. Innate immune functions of human γδ T cells. Immunobiology 2008; 213:173–82. [DOI] [PubMed] [Google Scholar]
- 12. Correia DV, Fogli M, Hudspeth K, da Silva MG, Mavilio D, Silva‐Santos B. Differentiation of human peripheral blood Vδ1 + T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 2011; 118:992–1001. [DOI] [PubMed] [Google Scholar]
- 13. Halary F, Peyrat MA, Champagne E, Lopez‐Botet M, Moretta A, Moretta L et al Control of self‐reactive cytotoxic T lymphocytes expressing γδ T cell receptors by natural killer inhibitory receptors. Eur J Immunol 1997; 27:2812–21. [DOI] [PubMed] [Google Scholar]
- 14. Poccia F, Cipriani B, Vendetti S, Colizzi V, Poquet Y, Battistini L et al CD94/NKG2 inhibitory receptor complex modulates both anti‐viral and antitumoral responses of polyclonal phosphoantigen‐reactive Vγ9Vδ2 T lymphocytes. J Immunol 1997; 159:6009–17. [PubMed] [Google Scholar]
- 15. Gougeon ML, Poccia F, Boullier S. Human γδ T lymphocytes in HIV disease: effector functions and control by natural killer cell receptors. Springer Semin Immunopathol 2000; 22:251–63. [DOI] [PubMed] [Google Scholar]
- 16. Tokuyama H, Hagi T, Mattarollo SR, Morley J, Wang Q, So HF et al Vγ9 Vδ2 T cell cytotoxicity against tumor cells is enhanced by monoclonal antibody drugs – rituximab and trastuzumab. Int J Cancer 2008; 122:2526–34. [DOI] [PubMed] [Google Scholar]
- 17. Gertner‐Dardenne J, Bonnafous C, Bezombes C, Capietto AH, Scaglione V, Ingoure S et al Bromohydrin pyrophosphate enhances antibody‐dependent cell mediated cytotoxicity induced by therapeutic antibodies. Blood 2009; 113:4875–84. [DOI] [PubMed] [Google Scholar]
- 18. Cairo C, Surendran N, Harris KM, Mazan‐Mamczarz K, Sakoda Y, Diaz‐Mendez F et al Vγ2Vδ2 T cell costimulation increases NK cell killing of monocyte‐derived dendritic cells. Immunology 2015; 144:422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maniar A, Zhang X, Lin W, Gastman BR, Pauza CD, Strome SE et al Human γδ T lymphocytes induce robust NK cell‐mediated antitumor cytotoxicity through CD137 engagement. Blood 2010; 116:1726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pauza CD, Riedel DJ, Gilliam BL, Redfield RR. Targeting γδ T cells for immunotherapy of HIV disease. Future Virol 2011; 6:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poccia F, Boullier S, Lecoeur H, Cochet M, Poquet Y, Colizzi V et al Peripheral Vγ9/Vδ2 T cell deletion and anergy to nonpeptidic mycobacterial antigens in asymptomatic HIV‐1‐infected persons. J Immunol 1996; 157:449–61. [PubMed] [Google Scholar]
- 22. Enders PJ, Yin C, Martini F, Evans PS, Propp N, Poccia F et al HIV‐mediated γδ T cell depletion is specific for Vy2+ cells expressing the Jy1.2 segment. AIDS Res Hum Retroviruses 2003; 19:21–9. [DOI] [PubMed] [Google Scholar]
- 23. Hebbeler AM, Propp N, Cairo C, Li H, Cummings JS, Jacobson LP et al Failure to restore the Vγ2‐Jγ1.2 repertoire in HIV‐infected men receiving highly active antiretroviral therapy (HAART). Clin Immunol 2008; 128:349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Peng H, Ma P, Ruan Y, Su B, Ding X et al Association between Vγ2Vδ2 T cells and disease progression after infection with closely related strains of HIV in China. Clin Infect Dis 2008; 46:1466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H, Pauza CD. HIV envelope‐mediated, CCR5/α4β7‐dependent killing of CD4‐negative γδ T cells which are lost during progression to AIDS. Blood 2011; 118:5824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wallace M, Scharko AM, Pauza CD, Fisch P, Imaoka K, Kawabata S et al Functional γδ T‐lymphocyte defect associated with human immunodeficiency virus infections. Mol Med 1997; 3:60–71. [PMC free article] [PubMed] [Google Scholar]
- 27. Riedel DJ, Sajadi MM, Armstrong CL, Cummings JS, Cairo C, Redfield RR et al Natural viral suppressors of HIV‐1 have a unique capacity to maintain γδ T cells. AIDS 2009; 23:1955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sajadi MM, Heredia A, Le N, Constantine NT, Redfield RR. HIV‐1 natural viral suppressors: control of viral replication in the absence of therapy. AIDS 2007; 21:517–9. [DOI] [PubMed] [Google Scholar]
- 29. Walker BD. Elite control of HIV Infection: implications for vaccines and treatment. Top HIV Med 2007; 15:134–6. [PubMed] [Google Scholar]
- 30. Bordon J, Evans PS, Propp N, Davis CE Jr, Redfield RR, Pauza CD. Association between longer duration of HIV‐suppressive therapy and partial recovery of the Vγ2 T cell receptor repertoire. J Infect Dis 2004; 189:1482–6. [DOI] [PubMed] [Google Scholar]
- 31. Chaudhry S, Cairo C, Venturi V, Pauza CD. The γδ T‐cell receptor repertoire is reconstituted in HIV patients after prolonged antiretroviral therapy. AIDS 2013; 27:1557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poonia B, Pauza CD. γδ T cells from HIV+ donors can be expanded in vitro by zoledronate/interleukin‐2 to become cytotoxic effectors for antibody dependent cellular cytotoxicity. Cytotherapy 2012; 14:173–81. [DOI] [PubMed] [Google Scholar]
- 33. Thompson K, Rojas‐Navea J, Rogers MJ. Alkylamines cause Vγ9Vδ2 T cell activation and proliferation by inhibiting the mevalonate pathway. Blood 2006; 107:651–4. [DOI] [PubMed] [Google Scholar]
- 34. Nussbaumer O, Gruenbacher G, Gander H, Thurnher M. DC‐like cell‐dependent activation of human natural killer cells by the bisphosphonate zoledronic acid is regulated by γδ T lymphocytes. Blood 2011; 118:2743–51. [DOI] [PubMed] [Google Scholar]
- 35. Nussbaumer O, Gruenbacher G, Gander H, Komuczki J, Rahm A, Thurnher M. Essential requirements of zoledronate‐induced cytokine and γδ T cell proliferative responses. J Immunol 2013; 191:1346–55. [DOI] [PubMed] [Google Scholar]
- 36. Di Virgilio F. The therapeutic potential of modifying inflammasomes and NOD‐like receptors. Pharmacol Rev 2013; 65:872–905. [DOI] [PubMed] [Google Scholar]
- 37. Tsai CY, Liong KH, Gunalan MG, Li N, Lim DS, Fisher DA et al Type I IFNs and IL‐18 regulate the antiviral response of primary human γδ T cells against dendritic cells infected with Dengue virus. J Immunol 2015; 194:3890–900. [DOI] [PubMed] [Google Scholar]
- 38. Cairo C, Propp N, Hebbeler AM, Colizzi V, Pauza CD. The Vc2/Vd2 T cell repertoire in Macaca fascicularis: functional responses to phosphoantigen stimulation by the Vc2/Jc1.2 subset. Immunology 2005; 115:197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cairo C, Sagnia B, Cappelli G, Colizzi V, Leke RG, Leke RJ et al Human cord blood γδ T cells expressing public Vγ2 chains dominate the response to bisphosphonate plus interleukin‐15. Immunology 2013; 138:346–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin‐18 and IL‐18 binding protein. Front Immunol 2013; 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urban EM, Li H, Armstrong C, Focaccetti C, Cairo C, Pauza CD. Control of CD56 expression and tumor cell cytotoxicity in human Vγ2Vδ2 T cells. BMC Immunol 2009; 10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noguchi A, Kaneko T, Kamigaki T, Fujimoto K, Ozawa M, Saito M et al Zoledronate‐activated Vγ9γδ T cell‐based immunotherapy is feasible and restores the impairment of γδ T cells in patients with solid tumors. Cytotherapy 2011; 13:92–7. [DOI] [PubMed] [Google Scholar]
- 43. Gioia C, Agrati C, Casetti R, Cairo C, Borsellino G, Battistini L et al Lack of CD27– CD45RA– Vγ9Vδ2+ T cell effectors in immunocompromised hosts and during active pulmonary tuberculosis. J Immunol 2002; 168:1484–9. [DOI] [PubMed] [Google Scholar]
- 44. Poccia F, Gioia C, Martini F, Sacchi A, Piacentini P, Tempestilli M et al Zoledronic acid and interleukin‐2 treatment improves immunocompetence in HIV infected persons by activating Vγ9Vδ2 T cells. AIDS 2009; 23:555–65. [DOI] [PubMed] [Google Scholar]
- 45. Angelini DF, Borsellino G, Poupot M, Diamantini A, Poupot R, Bernardi G et al FcγRIII discriminates between 2 subsets of Vγ9Vδ2 effector cells with different responses and activation pathways. Blood 2004; 104:1801–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Stimulators and inhibitors of interleukin‐18 production.