

Summary
Human ficolin‐2 (FCN‐2) and mouse ficolin‐A (FCN‐A, a ficolin‐2‐like molecule in mouse) are activators of the lectin complement pathway, present in normal plasma and usually associated with infectious diseases, but little is known about the role of FCN‐A/2 in inflammatory bowel disease (IBD). In our present study, we found that patients with IBD exhibited much higher serum FCN‐2 levels than healthy controls. In the dextran sulphate sodium‐induced acute colitis mouse model, FCN‐A knockout mice showed much milder disease symptoms with less histological damage, lower expression levels of pro‐inflammatory cytokines [interleukin‐6 (IL‐6), IL‐1β and tumour necrosis factor‐α (TNF‐α)], chemokines (CXCL1/2/10 and CCL4) and higher levels of the anti‐inflammatory cytokine IL‐10 compared with wild‐type mice. We demonstrated that FCN‐A/2 exacerbated the inflammatory pathogenesis of IBD by stimulating M1 polarization through the TLR4/MyD88/MAPK/NF‐κB signalling pathway in macrophages. Hence, our data suggest that FCN‐A/2 may be used as a novel therapeutic target for IBD.
Keywords: ficolin‐2, ficolin‐A, inflammatory bowel disease, macrophage, Toll‐like receptor 4
Abbreviations
- BMDM
bone‐marrow‐derived macrophages
- CD
Crohn's disease
- CRP
C‐reactive protein
- DAI
Disease Activity Index
- DSS
dextran sulphate sodium
- FBS
fetal bovine serum
- FCN‐2
ficolin‐2
- FCN‐A
ficolin‐A
- IBD
inflammatory bowel disease
- IL
interleukin
- IFN
interferon
- IHC
immunohistochemistry
- KO
knockout
- LPMCs
lamina propria mononuclear cells
- LV
liposome vehicle
- mAb
monoclonal antibody
- pc
pcDNA3.1
- pV
pVAX‐1
- TLR
Toll‐like receptor
- TNF
tumour necrosis factor
- UC
ulcerative colitis
- WT
wild‐type
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn's disease (CD), is an intestinal inflammation disorder characterized by weight loss, abdominal pain, diarrhoea and haematochezia.1, 2 It is currently recognized as a complex multifactorial disease that may be regulated by the interplay among immunity, environmental factors, genetic susceptibility and enteric luminal contents.3, 4, 5 However, the precise mechanisms of IBD are still not fully understood.
Macrophages, as the most abundant mononuclear phagocytes in the healthy intestinal lamina propria, are heterogeneous and versatile cells that could undergo phenotypically or functionally dynamic switch in response to microenvironment signals.6, 7 Two major macrophage subpopulations with different functions, including classically activated/inflammatory (M1) and alternatively activated/anti‐inflammatory (M2) macrophages, have long been recognized.8 M1 polarization can be induced by Toll‐like receptor (TLR) ligands, interferon‐γ (IFN‐γ), and results in the increased production of pro‐inflammatory cytokines. These mediators promote the differentiation and activation of T helper type 1 and type 17 cells and further aggravate inflammation and tissue damage. M2 polarization is stimulated by interleukin‐4 (IL‐4) and IL‐13 and leads to low levels of pro‐inflammatory cytokines and higher expression of IL‐10, the mannose receptor (CD206), and arginase I (Arg‐1).9, 10, 11 Previous studies have shown that macrophage M1 polarization plays an important role in the onset, progression and prognosis of inflammation and the imbalance of M1 and M2 macrophages is implicated in the development of experimental IBD.12, 13, 14, 15
Ficolins (FCNs) belong to family of collectins that activate the lectin pathway of complement.16, 17 In humans, three FCNs have been characterized: FCN‐1 (M‐FCN), FCN‐2 (L‐FCN) and FCN‐3 (H‐FCN). In mice, two types of FCNs, FCN‐A and FCN‐B, have been identified. Based on the structural and functional properties and phylogenetic analysis, mouse FCN‐A resembles human FCN‐2. FCN‐A/2 is mainly synthesized in the liver and present in the circulation, and has a lectin‐like activity in the recognition of N‐acetyl glucosamine, lipopolysaccharides, β‐1,3‐d‐glucan, lipoteichoic acid and various acetylated compounds.18, 19, 20
Recently, a number of reports have shown that the dysfunction or abnormal expression of FCN‐A/2 may play crucial roles in immune responses, inflammatory processes, viral and bacterial diseases.21, 22, 23, 24, 25, 26 However, little is known regarding the roles and mechanisms of FCN‐A/2 in intestinal inflammation. As the precise mechanisms of IBD are not yet completely understood, it is, therefore, necessary to further elucidate the immune roles and mechanisms of FCN‐A/2 in IBD. In the present study, we demonstrate that FCN‐A/2 effectively aggravated inflammation and pathological damage in dextran sulphate sodium (DSS) ‐induced mouse colitis by promoting the M1 polarization of macrophages, and FCN‐A/2 might be a promising therapeutic target for the treatment of IBD and its complications.
Materials and methods
Clinical specimens
This study was approved by the Ethics Committees of the Wuhan University School of Medicine. Written informed consent was obtained from all participants. From 2011 to 2013, a total of 48 patients with active UC and 51 patients with active CD were enrolled from the Department of Gastroenterology of Zhongnan Hospital of Wuhan University. The diagnosis of IBD was based on standard clinical, endoscopic and histological criteria. Disease activities were assessed using the Truelove and Witts' criteria for patients with UC and CD Activity Index for patients with CD.27, 28 All patients underwent laboratory investigations including determination of the serum C‐reactive protein (CRP) level. Healthy donors (HDs) were recruited from the Medical Examination Centre of Zhongnan Hospital based on clinical and laboratory findings with no signs or symptoms of UC and CD. Serum samples were collected and stored at −80° until analysis.
Measurement of serum FCN‐2 and FCN‐A concentrations
The sandwich ELISA method was used to measure the concentrations of serum FCN‐2 as previously described.23 First, 96‐well plates were coated with rabbit anti‐FCN‐2 (prepared in our laboratory) (1 : 100 dilution) in carbonate–bicarbonate buffer (pH 9·6) overnight at 4°. The plates were blocked for 1 hr at room temperature withPBS plus 0·5% BSA. Each serum sample (100 μl) was added and incubated at 37° for 2 hr. Then, the plates were coated with monoclonal anti‐human FCN‐2 GN5 (HyCult Biotechnology b.v., Uden, the Netherlands) (1 : 1000 dilution) and incubated at 37° for 1 hr to detect FCN‐2. After washing, the plates were incubated with 100 μl of horseradish peroxidase‐conjugated goat anti‐mouse IgG (1 : 1000) in PBST‐BSA. After washing, the reaction was developed with 100 μl/well tetramethylbenzidine substrate (Sigma‐Aldrich, St Louis, MO) for 15 min in the dark. The colour‐development reaction was stopped with 100 μl of 0·5 m H2SO4, and the absorption was read at 450 nm.
An indirect ELISA was used to measure the concentrations of mouse serum FCN‐A. Briefly, 96‐well ELISA plates were coated with 100 μl of each serum sample and incubated at 37° for 2 hr. The plates were washed three times and blocked with 5% BSA overnight. Subsequently, a rabbit anti‐FCN‐A polyclonal antibody (prepared in our laboratory) (1 : 1000 dilution) was added to each well and incubated at 37° for 1 hr. The following method is described above.
Construction of eukaryotic expression plasmids
Full‐length FCN‐2 (GenBank accession no. NM004108) and FCN‐A (GenBank accession no. NM007995) were amplified and cloned in‐frame into the eukaryotic expression vectors pcDNA3.1(‐)Myc‐His (pc) or pVAX‐1 (pV) (Invitrogen, Carlsbad, CA) to generate the plasmids pcDNA3.1‐FCN‐2 (pc‐FCN‐2) and pVAX1‐FCN‐A (pV‐FCN‐A), respectively.29 The constructs were confirmed by restriction enzyme digestion along with DNA sequencing analysis. All the DNA preparations were produced using endotoxin‐free purification columns (Qiagen, Valencia, CA).
Animals
The animal experimental protocols were performed in compliance with all guidelines and approved by the Institutional Animal Care and Use Committee of Wuhan University. Wild‐type C57BL/6, mice were purchased from the Centre of Animal Experiments of Wuhan University. TLR4 knockout (KO) (C57BL/6) mice were kindly provided by Professor Hongliang Li. MyD88 KO (C57BL/6) mice were kindly provided by Dr Zhinan Yin. A targeting construct was produced to disrupt the FCN‐A of C57BL/6J mice by homologous recombination according to a previous report.30 The FCN‐A KO mice were maintained by backcrossing to C57BL/6 mice, and the FCN‐A KO mice were further screened and determined as FCN‐A−/− homozygote by PCR using primers (forward: 5′‐TTGGGT GGAGAGG CTATT‐3′, reverse: 5′‐TTTCCATGGGT CACGA CGAGATC‐3′). FCN‐A−/− used in this study belonged to the third filial generation.
Female FCN‐A KO mice (8–10 weeks old) were randomly separated into four groups and immunized with each plasmid using in vivo electroporation according to a previously described protocol.31, 32 Three days before immunization, 0·5% procaine was injected for local anaesthesia into the posterior tibialis muscle. Three days later, plasmids pV‐FCN‐A, pc‐FCN‐2 or pV/pc empty vectors (20 μg DNA/each mouse) were introduced into the mice, respectively, by injecting into the same position on the posterior tibialis muscle using in vivo electroporation by intramuscular injection with an electric square porter (Shanghai TERESA Healthcare sci‐Tech Co., Ltd, Shanghai, China). Electroporation was administered with six 1 Hz pulses of 60 V/cm, and 50 milliseconds in duration and 1 second apart.33 Exogenous FCN‐A/2 protein expressions in the mouse muscle/liver tissues and sera were detected by Western blot and ELISA, respectively.
Cell culture and cell lines
The murine macrophage cell line RAW 264.7 from the Wuhan University Centre for Type Culture Collection (Wuhan, China) was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml of penicillin‐streptomycin in a 5% CO2 humidified incubator at 37°.
Human monocytic cell line (THP‐1) was cultured with RPMI‐1640 supplemented with 10% FBS under a humidified 5% CO2 incubator at 37°. The monocytes were stimulated with PMA (100 ng/ml, Sigma, St Louis, MO) for 48 hr to induce differentiation into M0 macrophage.34
Bone‐marrow‐derived macrophages preparation
Bone marrow cells isolated from the leg bones of wild‐type (WT) (TLR4+/+/MyD88+/+), TLR4−/− and MyD88−/− mice were treated with erythrocyte‐lysing buffer and cultured in Dulbecco's modified Eagle's medium (Gibco Ltd, Paisley, UK) supplemented with 10% FBS (Gibco), 1% antibiotic/antimycotic and 50 ng/ml macrophage colony‐stimulating factor (Peprotech, Rocky Hill, NJ) for 6 days to induce differentiation into M0 macrophages.35 The purity of the F4/80+ bone‐marrow‐derived macrophages (BMDM) was assessed by flow cytometric analysis.
Induction of experimental mouse colitis
Specific pathogen‐free male mice (weighing 20–25 g and 8–10 weeks old) were acclimated for 1 week in the animal housing conditions. Then, acute colitis was induced by administering 4% (weight/volumr) DSS (36 000–50 000 MW; MP Biomedicals, Santa Ana, CA) dissolved in normal drinking water and refreshed every day for 7 days. The mice were allowed free access to water and food during the experiments. Body weight and faecal scores (consistency and faecal bleeding) were determined daily after DSS challenge in mice. The change in body weight of animals from different genotypes or treatments was expressed as the percentage of body weight loss in comparison with those at day 0. The disease activity index (DAI) consisted of the following parameters: body weight loss (0 points, < 5% weight loss; 1 point, 5–10% weight loss; 2 points, 10–15% weight loss; 3 points, 15–20% weight loss; and 4 points, > 20% weight loss), stool consistency (0 points, formed pellets; 2 points, pasty/semi‐formed stool; and 4 points, liquid stool) and faecal bleeding (0 points, no rectal bleeding; 2 points, haemoccult‐positive; and 4 points, visible gross bleeding). The scores of the parameters were added, and their mean value was calculated. The mouse colons were collected on the 7th day after DSS challenge, and the lengths of the colons were measured.
Histopathological assessment, immunohistochemistry and fluorescence imaging
Each mouse colon was harvested. The colon tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned (5 mm) for haematoxylin & eosin (HE) staining and immunohistochemistry (IHC). The histopathology was scored by an independent pathologist as previously described. Two independent parameters were measured: the extent of inflammation (0, none; 1, slight; 2, moderate; 3, severe; 4, massive) and the extent of crypt damage (0, none; 1, the basal one‐third portion damaged; 2, the basal two‐thirds portion damaged; 3, the entire crypt damaged but the surface epithelium intact; 4, the entire crypt and epithelium lost).
For IHC, tissue sections were deparaffinized using xylene and dehydrated in a alcohol gradient. The sections were treated with 1 × citrate buffer for antigen retrieval and washed with 1 × PBS, pH 7·5. The endogenous peroxidase activity was quenched with methanol and 3% H2O2, and then the sections were blocked with normal goat serum for 30 min. The IHC was performed using an ABC kit (Vector Laboratories, Burlingame, CA). The tissues were first incubated with primary anti‐IL‐6 or anti‐CD68 antibodies (Abcam, Cambridge, MA) overnight at 4° followed by a biotinylated secondary IgG and then incubated with streptavidin–peroxidase for 1 hr. The colour of the final product was developed using 3,3‐diaminobenzidine (DAKO, Carpinteria, CA). The sections were counterstained with Mayer's haematoxylin. All histological assessments were performed by two independent blinded observers.
For immunofluorescence staining, colon tissue sections (5 μm) were incubated in 10% normal goat serum for 1 hr and stained with rabbit anti‐inducible nitric oxide synthase (iNOS), rabbit anti‐IL‐6 or rat anti‐CD68 antibodies (Abcam) overnight at 4°. The tissue sections were washed with PBS and incubated with either Alexa Fluor 555®‐labelled goat anti‐rabbit IgG or Alexa Fluor 488‐labelled goat anti‐rat IgG. Fluorescence images were collected using a DP80 Microscope (Olympus Corp., Tokyo, Japan).
Microarray analysis
For differential gene expression profiling between WT and FCN‐A KO (C57BL/6J background) mice, total RNA was extracted from livers of WT and FCN‐A KO mice, respectively. The RNA integrity was assessed using an Agilent Bioanalyzer 2100 (the Agilent Mouse, Design ID: 049801; Agilent Technologies, Santa Clara, CA). The sample labelling, microarray hybridization, washing and scanning were performed based on the manufacturer's standard protocols (Oebiotech, Shanghai, China). Briefly, total RNAs were transcribed to double‐stranded cDNA, then synthesized into cRNA and labelled with Cyanine‐3‐CTP. The labelled cRNAs were hybridized onto the Agilent Mouse Gene Expression v1 microarray (Agilent). After washing, the arrays were scanned by the Agilent Scanner G2505C. feature extraction software (version 10.7.1.1; Agilent Technologies) was used to analyse the array images to obtain raw data. The raw data were normalized with the quantile algorithm. Differentially expressed genes were then identified through fold change. Hierarchical clustering and heat maps were generated using the r statistical system (http://cran.us.r-project.org/).
Reverse transcription‐quantitative real‐time PCR
The total RNA was extracted from macrophages using Trizol reagent (Invitrogen) and reverse transcribed using a cDNA reverse transcription kit (Toyobo, Osaka, Japan). The reverse‐transcribed cDNA (11 μl) was used as a template in quantitative PCR containing SYBR Green Real‐time PCR Master Mix (BioRad, Hercules, CA) and 0·4 μm forward and reverse primers. The primers used are listed in Table 1.
Table 1.
Primers used for quantitative PCR
| mRNA | Forward primer (5′‐3′) | Reverse primer (5′‐3′) |
|---|---|---|
| Mouse | ||
| TNF‐α | ATTCGAGTGACAAGCCTGTAGCCCA | CTGGGAGTAGACAAGGTACAACCCA |
| IL‐6 | TGTCTATACCACTTCACAAGTCGGAG | GCACAACTCTTTTCTCATTTCCAC |
| IL‐1β | GCCCATCCTCTGTGACTCA | AGGCCACAGGTATTTTGTC |
| IFN‐γ | ACTGACTTGAATGTCCAACGCA | ATCTGACTCCTTT TTCGCTTCC |
| IL‐10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| IL‐12 | CGCAGCACTTCAGAATCACA | TCTCCCACAGGAGGTTTCTG |
| iNOS | CAGCTGGGCTGTACAAACCTT | CATTGGAAGTGAAGCGTTT CG |
| Arg‐1 | GGAATCTGCATGGGCAACCTGTGT | AG GGTCTACGTCTCGCAAGCCA |
| CXCL1 | CCCAAACCGAAGTCATAGCCACAC | TTGTCAGAAGCCAGCGTTCACCAG |
| CXCL2 | GGCTGTTGTGGCCAGTGAA | TGTTCAGTATCTTTTGGATGATTTTCTG |
| CXCL10 | GCCGTCATTTTCTGCCTCAT | GCTTCCCTATGGCCCTCATT |
| CCL4 | GCCCTCTCTCTCCTCTTGCT | GAGGGTCAGAGCCCATTG |
| GADPH | CCTCGTCCCGTAGAC AAAATC | TGAAGGGGTCGTTGATGGC |
| Human | ||
| TNF‐α | CCCCAGGGCTCCAGAAGGT | TGGGGCAGAGGGTTGATTAGTTG |
| iNOS | AGCTGAACTTGAGCGAGGAG | GGAAAAGACTGCACCGAAGA |
| Arg‐1 | CAGATATGCAGGGAGTCACC | CAGAAGAATGGAAGAGTCAG |
| CD80 | GGGAAAGTGTACGCCCTGTA | GCTACTTCTGTGCCCACCAT |
| CD206 | GAGGGAATCTGGTCTCCATACAA | AAGTGGAGTCCTTCATGTGATAGGT |
| IL‐10 | AACAAGAGCAAGGCCGTGG | GAAGATGTCAAACTCACTCATGGC |
| GADPH | GCACCGTCAAGGCTGAGAAC | TGGTGAAGACGCCAGTGGA |
Purification of recombinant proteins
The recombinant GST‐FCN‐A, and GST proteins were purified as previously described.23 Briefly, the prokaryotic expression plasmids pGEX‐KG‐FCN‐A, pGEX‐KG‐FCN‐2 and pGEX‐KG were transformed into Escherichia coli BL21 (DE3) [pLysS] cells (Invitrogen), the expressions of the recombinant proteins were induced by isopropyl β‐d‐1‐thiogalactopyranoside (IPTG), and the proteins were purified using glutathione Sepharose 4B (GE Healthcare, Piscataway, NJ). The purified GST‐FCN‐A, GST‐FCN‐2 and GST proteins were further treated with Detoxi‐Gel endotoxin removing gel (Pierce, Pierce, Rockford, IL), and then identified by SDS–PAGE. Endotoxin levels in the purified proteins were measured using an EndoLISA kit (Hyglos GmbH, Bernried, Germany).
Enzyme‐linked immunosorbent assay
RAW264.7 cells or M0 BMDMs were stimulated with the recombinant proteins FCN‐A‐GST and GST for different time‐points. PMA‐stimulated THP‐1 cells were treated with different concentrations of GST or GST‐FCN‐2 proteins for 24 hr. The levels of IL‐1β, IL‐6, TNF‐α and IL‐10 in the cell supernatants or cell lysates were detected by ELISA (eBioscience, San Diego, CA).
The quantification of tissue cytokines was also determined by ELISA. Frozen colon tissue samples were homogenized in ice‐cold radio‐immunoprecipitation assay (Thermo Fisher Scientific, Waltham, MA) lysis buffer. The concentrations of IL‐1β, IL‐6 and TNF‐α were measured in the whole‐tissue extracts by ELISA following the manufacturer's instructions (eBioscience) and expressed as pg per mg of total proteins.
Western blot analysis
Macrophages (2 × 105 cells/ml) were stimulated with the FCN‐A/2 protein (10 μg/ml) for different time‐points. The treated cells were washed twice with PBS and then lysed for 30 min on ice in radio‐immunoprecipitation assay solution containing a protease inhibitor cocktail and phosphatase inhibitors (Sigma‐Aldrich). The expressions of proteins in the cell lysates were examined using anti‐nuclear factor‐κB (NF‐κB) p‐p65, anti‐p‐c‐Jun N‐terminal kinase (JNK), anti‐p‐IL‐1 receptor‐associated kinase 1 (IRAK1) and anti‐p‐extracellular signal regulated kinase 1/2 (ERK1/2) antibodies (Affinity Biosciences, Cincinnati, OH). Anti‐β‐actin (Affinity Biosciences) was used as an internal control. RAW264.7 cells (2 × 105 cells/ml) were pretreated or not with one of the following inhibitors: BAY11‐7085 (NF‐κB inhibitor, 10 μm; Sigma), SP600125 (JNK inhibitor, 10 μm; Sigma) and PD98059 (ERK inhibitor, 10 μm; Sigma).
Co‐immunoprecipitation and immuno‐blotting
For co‐immunoprecipitation, RAW264.7 cell membrane proteins were extracted using a membrane protein extraction kit (Novagen; Merck KgaA, Darmstadt, Germany). Extracted membrane proteins were pre‐cleared with 20 μl of protein A/G Sepharose (Santa Cruz Biotechnology, Dallas, TX) for 1 hr, and then incubated with the purified GST‐FCN‐A or GST proteins at 4° for 2 hr; then anti‐TLR4 (Cell Signaling Technology, Danvers, MA) or rabbit normal IgG overnight at 4°. The immune complexes were incubated with 50 μl of protein A/G Sepharose for 4 hr at 4° and were washed three times. Immune complexes were eluted by boiling for 5 min at 95° in 2 × SDS loading buffer, followed by immunoblotting and detected with rabbit polyclonal antibody FCN‐A and monoclonal antibody (mAb) anti‐TLR4.
Determination of the myeloperoxidase activity
Briefly, colon tissues were weighed, and the myeloperoxidase (MPO) activity was measured in homogenized intestinal tissue samples according to Ren et al.36 Before homogenization, 200 μl of lysis buffer was added to each 10‐mg piece of tissue. The samples were centrifuged twice (1500 g at 4° for 15 min) and stored at −80° until the MPO activity assay. A mouse MPO ELISA kit (Abcam, Waltham, MA) was used to determine the MPO levels in the mouse colon lysates. The sample absorbance was measured using a spectrophotometer at 450 nm following the instructions provided by the instrument manufacturer.
Isolation of lamina propria mononuclear cells and flow cytometry analysis
The isolation of lamina propria mononuclear cells (LPMCs) was performed as previously described.37 Briefly, colons were opened longitudinally and washed twice to remove faecal content by shaking in calcium/magnesium‐free Hanks' buffered salt solution containing 5 mm EDTA for 20 min at 37°. The supernatants were removed, and the remaining tissues were cut into 1‐mm pieces and incubated with Hanks' buffered salt solution containing 4% FBS, 1 mg/ml type IV collagenase and 40 μg/ml DNase I (Sigma) for 50 min at 37° in a shaking water bath. After filtration, the cells were washed, suspended in PBS, and stained with the following fluorescent mAbs: phycoerythrin‐anti‐CD45, FITC‐anti‐CD11b, allophycocyanin‐anti‐F4/80, allophycocyanin‐anti‐CD11C and phycoerythrin‐Cy5‐anti‐Gr1 (BioLegend, San Diego, CA) for 30 min at 4°. All samples were processed on an Accuri C6 flow cytometer and results were analysed using the Accuri C6 Flow software (BD Bioscience, Franklin Lakes, NJ).
In vivo model of macrophage and neutrophil depletion in mice
To deplete macrophages, each mouse was injected intraperitoneally with 100 μl/10 g body weight of clodronate liposome (F70101C‐N; FormuMax Scientific Inc, Palo Alto, CA) 2 days before DSS administration.38 Liposome vehicle (LV) (F70101‐N; FormuMax Scientific Inc., Sunnyvale, CA) was used as a negative control. The efficiencies of mouse macrophage depletion were determined by flow cytometry and immunohistochemical staining analysis, as indicated by significant decreases of CD11b+ F4/80+ cells in splenocytes and CD68+ macrophages in colon tissues.39
To deplete neutrophils, each mouse was intravenously injected with anti‐Gr1 mAb (BioLegend, 400 μg/mouse) 2 days before DSS administration, rat IgG2b mAb (BioLegend) was used as an isotype control. The depletion of mouse neutrophils was determined by flow cytometry, as indicated by a loss of CD11b+ Gr1+ cells from peripheral blood.40
Adoptive transfer of BMDMs
For adoptive transfer experiments, an FCN‐A KO mouse was intravenously injected with clodronate liposome to deplete macrophages, and then injected with plasmids pV‐FCN‐A, pc‐FCN‐2 or pV/pc empty vectors (20 μg DNA/each mouse) once, respectively, through intramuscular injection 2 days before 4% DSS administration. M0 BMDMs (1 × 106) were injected intravenously into each mouse on days 0, 3 and 5. Each recipient mouse was induced to develop colitis by drinking 4% DSS every day.41
Statistical analysis
All data are presented as the mean ± SEM. The differences between groups were estimated using t‐test, Mann–Whitney U‐test, or one‐way analysis of variance followed by the Bonferroni post‐test analysis. Significant differences were accepted when the P‐value was < 0·05. Spearman's correlation and rank correlation were used for correlation analysis. The statistical analyses were calculated and plotted using graphpad prism version 5.0 (GraphPad Software, San Diego, CA).
Results
Patients with IBD and mice with acute colitis exhibit much higher serum FCN‐2/A levels than healthy donors
Human serum FCN‐2 concentrations were determined by ELISA using samples from 48 patients with UC, 51 patients with CD and 41 HDs. The clinical characteristics of the study population are shown in Table 2. The patients with UC and CD had significantly higher serum FCN‐2 levels compared with healthy controls (Fig. 1a). The mean concentrations of FCN‐2 were 5·78 μg/ml and 6·20 μg/ml in patients with UC and patients with CD, respectively, and 3·70 μg/ml in HDs. There were no statistically significant differences in age and gender between the HD group and the IBD patient groups (Table 2). We further examined the correlation between serum FCN‐2 levels and disease activities. FCN‐2 levels were positively correlated with the degrees of disease severity in patients with UC (r = 0·634, P < 0·0001) (Fig. 1b) and patients with CD (r = 0·596, P < 0·0001) (Fig. 1c). CRP is a common biochemical activity marker of inflammation. Our analysis also showed a positive correlation between serum FCN‐2 levels and serum CRP levels of patients with UC (r = 0·294, P = 0·031) (Fig. 1d) and with CD (r = 0·354, P = 0·008) (Fig. 1e). Together, these data suggest that FCN‐2 may be involved in the pathogenesis of inflammatory bowel disease.
Table 2.
Characteristics of clinical samples
| Characteristics | Ulcerative colitis | Crohn's disease | Healthy donors |
|---|---|---|---|
| Numbers | 48 | 51 | 41 |
| Male/Female | 26/22 | 37/14 | 20/21 |
| Age (Mean ± SD) | 43·81 ± 16·71 | 37·44 ± 13·92 | 40·41 ± 19·62 |
| Diarrhoea (n/%) | 37 (77·1%) | 30 (58·8%) | – |
| Abdominal pain (n/%) | 26 (54·2%) | 34 (66·6%) | – |
| Haematochezia (n/%) | 45 (93·8%) | 10 (19·6%) | – |
| Fever (n/%) | 3 (6·3%) | 6 (11·7%) | – |
| Perianal fistulae (n/%) | 1 (2·1%) | 4 (7·8%) | – |
| Extraintestinal symptom (n/%) | 3 (6·3%) | 5 (9·8%) | – |
| Operation history (n/%) | 4 (8·3%) | 10 (19·6%) | – |
| Disease activity | – | – | – |
| Mild (n/%) | 13 (27·1%) | – | – |
| Moderate (n/%) | 18 (37·5%) | 24 (47%) | – |
| Severe (n/%) | 17 (35·4%) | 27 (52·9%) | – |
| CRP (mg/l) | 7·14 ± 1·26 | 10·15 ± 2·00 |
N, numbers of individuals; %, positive percentages; CRP, C‐reactive protein.
Figure 1.
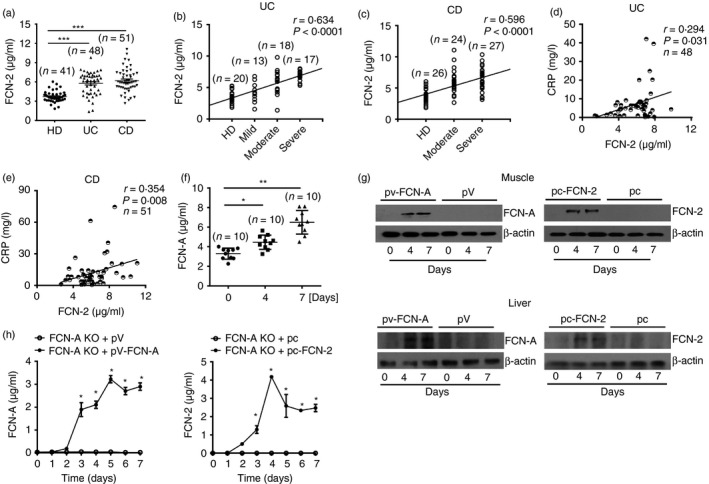
Patients with inflammatory bowel disease (IBD) and mice with acute colitis exhibited much higher serum ficolin‐2/A (FCN‐2/A) levels than healthy donors (HDs). (a) FCN‐2 concentrations were detected by ELISA in serum samples from 48 patients with ulcerative colitis (UC), 51 patients with Crohn's disease (CD) patients and 41 HDs. (b, c) Spearman's Rank correlation analysis was performed to analyse the correlation between serum FCN‐2 concentrations and degrees of disease activity of patients with UC (b) and CD (c). (d, e) Spearman's correlation analysis was performed to analyse the correlation between serum FCN‐2 levels and serum C‐reactive protein (CRP) levels in UC (d) and CD (e) patients. (f) Mouse sera were harvested at Day 0, 4 and 7 in dextran sulphate sodium (DSS) ‐treated wild‐type (WT) mice. Serum FCN‐A concentrations were detected by ELISA (n = 10 per group). (g) Exogenous FCN‐A and FCN‐2 expressions in mouse muscle and liver tissues from FCN‐A knockout (KO) mice were detected by Western blotting. (h) Exogenous FCN‐A and FCN‐2 expressions in the sera from FCN‐A KO mice were detected by ELISA (n = 6 per group). Values are mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01 and ***P < 0·001.
We also measured serum FCN‐A levels after DSS‐induced mouse colitis. Compared with no DSS‐treated mice (at Day 0), elevated serum FCN‐A levels were observed at Days 4 and 7 in mice after DSS‐induced mouse colitis (Fig. 1f). To further investigate the expressions of FCN‐A/2 in DSS‐induced colitis in mice, WT mice, FCN‐A KO mice and FCN‐A KO mice injected with pV‐FCN‐A, pc‐FCN‐2, or empty vectors through intramuscular electroporation were analysed for research. As shown in Fig. 1(g), 4 and 7 days after plasmid injection, FCN‐A/2 expressions were detected in both muscle and liver tissues of FCN‐A KO mice compared with empty vector (pV or pc) groups (Fig. 1g). We also determined that the FCN‐A/2 molecules were present in the sera of FCN‐A KO mice after the injection of the FCN‐A/2‐encoding plasmids on different days (Fig. 1h). Serum FCN‐A/2 was observed to be significantly elevated from 3 to 7 days after plasmid injection (Fig. 1h). These data suggest that injection of the FCN‐A/2‐encoding plasmids induced FCN‐A/2 expressions in vivo and that the FCN‐A/2 molecules were released into the circulation and tissues.
FCN‐A/2 can aggravate DSS‐induced colitis in mice
The severity of 4% DSS‐induced colitis in mice was further determined. We found that FCN‐A KO mice showed much milder disease symptoms compared with WT mice, as indicated by less weight loss (Fig. 2a), lower DAI scores (Fig. 2d) and a longer colon (Fig. 2e,f). Compared with the FCN‐A KO mice injected with empty vector, mice injected with pV‐FCN‐A or pc‐FCN‐2 had more weight loss (Fig. 2b,c), higher DAI scores (Fig. 2d), and shorter colon lengths (Fig. 2e,f). Haematoxylin & eosin staining showed aggravated colonic epithelium loss and inflammation in both WT mice and FCN‐A KO mice administered with exogenous FCN‐A/2, whereas FCN‐A KO mice had minimal colonic epithelial inflammation (Fig. 2g,h). These findings provide strong evidence that FCN‐A/2 can aggravate the development of DSS‐induced colitis in vivo.
Figure 2.
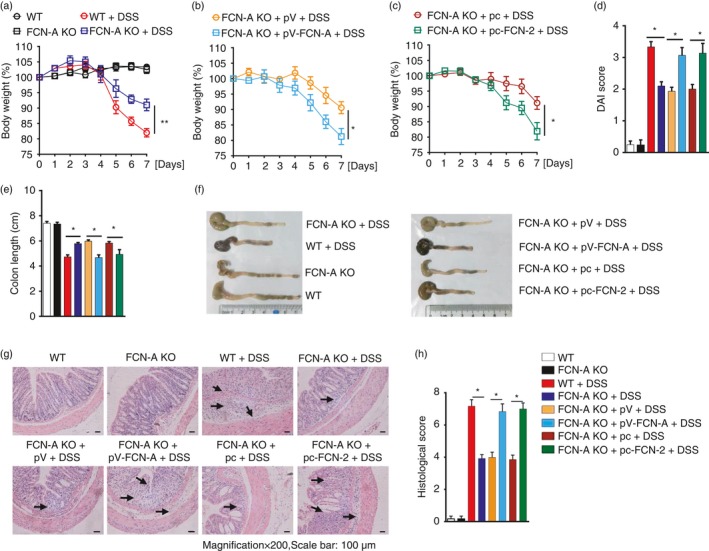
Ficolin‐A/2 (FCN‐A/2) exacerbated the dextran sulphate sodium (DSS) ‐induced acute colitis in mice. (a–c) Comparison of body weights between each group of mice as indicated. (d) Comparison of Disease Activity Index (DAI) scores and (e) colon length between each group of mice as indicated. (f) Representative data of the colon length analysis for each group of mice. (g) haematoxylin & eosin staining and (h) histopathological score of colon tissues from each group of mice. Eight mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01. pV, pVAX1; pV‐FCN‐A, pVAX1‐FCN‐A; pc, pcDNA3.1; pc‐FCN‐2, pc‐DNA3.1‐FCN‐A.
FCN‐A/2 enhances the production of pro‐inflammatory cytokines and chemokines in colon tissue of DSS‐treated mice
To identify the potential role of FCN‐A in physiological and pathological processes, we used the liver tissues from untreated WT and FCN‐A KO mice to conduct a gene expression microarray analysis. The results of the microarray analysis showed that the mRNA expression of several cytokines and receptors, including CXCL1/10, IFN‐α, TNF‐α, was down‐regulated in FCN‐A KO mice compared with WT mice (Fig. 3a). Consistent with these results, the mRNA expression levels of colonic pro‐inflammatory cytokines (TNF‐α, IL‐1β, IL‐12, IFN‐γ and IL‐6) and chemokines (CXCL1, CXCL2, CXCL10 and CCL4) were lower, as determined by reverse transcription‐quantitative real‐time PCR (RT‐qPCR) analysis, in DSS‐treated FCN‐A KO mice compared with WT mice and FCN‐A KO mice given exogenous FCN‐A/2, particularly for IL‐6 (Fig. 3b). IHC analysis also indicated much lower IL‐6 expression in DSS‐treated FCN‐A KO mice compared with WT mice, and the administration of exogenous FCN‐A/2 increased IL‐6 expression in colonic tissues of FCN‐A KO mice (see Supplementary material, Fig. S1c). Conversely, mRNA expression levels of anti‐inflammatory cytokine IL‐10 were higher in DSS‐treated FCN‐A KO mouse colon tissues compared with WT and FCN‐A KO mice given exogenous FCN‐A/2 (Fig. 3b). These data suggest that FCN‐A/2 promotes pro‐inflammatory cytokines (TNF‐α, IL‐1β, IL‐12, IFN‐γ and IL‐6) and chemokines (CXCL1, CXCL2, CXCL10 and CCL4) and inhibits anti‐inflammatory cytokine IL‐10.
Figure 3.
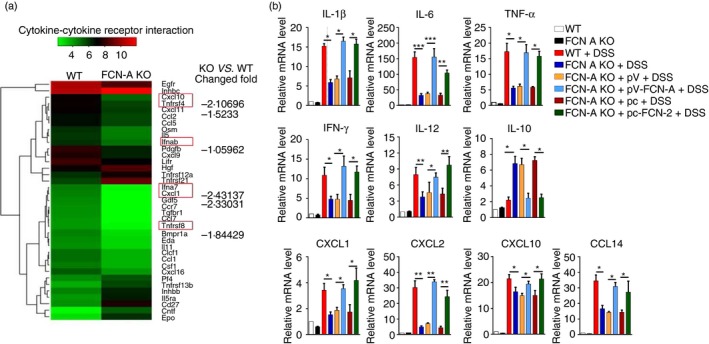
Ficolin‐A/2 (FCN‐A/2) enhanced the production of pro‐inflammatory cytokines and chemokines in colon tissue of dextran sulphate sodium (DSS) ‐treated mice. (a) According to the gene expression microarray data from mouse liver tissues, the mRNA expression ratios of cytokine‐related genes between untreated FCN knockout (KO) and wild‐type (WT) mice were analysed by heatmap. (b) The mRNA expression levels of colonic inflammatory cytokines and chemokines were detected using RT‐qPCR. Eight mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01 and ***P < 0·001.
FCN‐A KO mice exhibit significantly less leucocyte infiltration in colon tissues than WT mice
We next investigated whether FCN‐A/2 affected the intestinal infiltration of leucocytes (including CD45+ leucocytes, CD11b+ Gr‐1+ neutrophils, CD11b+ F4/80+ macrophages and CD11b+ CD11c+ dendritic cells) by flow cytometric analysis. By determining the percentages of the different cell types in the LPMCs isolated from colon tissues following DSS challenge, we found much lower percentages of infiltrating CD45+ leucocytes, macrophages and neutrophils in the colon tissues of FCN‐A KO mice compared with WT mice (Fig. 4a–c and see Supplementary material, Fig. S2a,b). However, no difference in the DC percentage between the groups was observed (Fig. 4d and see Supplementary material, Fig. S2a,b). We also found that the intensity of CD68+ macrophages was more prominent in both WT and FCN‐A KO mice injected with plasmid FCN‐2 or FCN‐A than in FCN‐A KO mice (Fig. 4e,f).
Figure 4.

The inflammatory cell infiltration was significantly decreased in the colon tissues of ficolin‐A (FCN‐A) knockout (KO) mice. Flow cytometry analysis of the percentages of (a) CD45+ leucocytes, (b) CD11b+ F4/80+ macrophages, (c) CD11b+ Gr1+ neutrophils and (d) CD11b+ CD11c+ dendritic cells in the colon tissues of each group of mice. (e) IHC analysis of the colon tissues with an anti‐CD68 for determining the presence of macrophages (arrows, brown). (f) Quantitative analysis of CD68 staining by image pro plus 6·0 software. Six mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05, ns: not significant.
FCN‐A/2‐mediated exacerbation of DSS‐induced colitis is dependent on macrophages
To further determine which types of immune cells participate in the FCN‐A/2‐stimulated intestinal inflammation, mouse macrophages and neutrophils were depleted using clodronate liposomes or an anti‐Gr1 antibody, respectively. The depletion efficiencies were confirmed by flow cytometry (see Supplementary material, Fig. S2c,e). Immunohistochemical analysis of CD68+ macrophages in the colon tissues from DSS‐treated WT mice with either clodronate or liposome vehicle was conducted to detect the depletion efficiencies of clodronate liposomes during the progression of DSS‐induced colitis (see Supplementary material, Fig. S2d). When macrophages were depleted with clodronate liposomes, mice exhibited no differences of the body weights (Fig. 5b), DAI scores (Fig. 5c), colon length (Fig. 5d) and histological pathological damage score (Fig. 5e) compared with those of LV‐treated, non‐depleted mice (Fig.5a,c–e) between DSS‐induced WT and FCN‐A KO mice.
Figure 5.
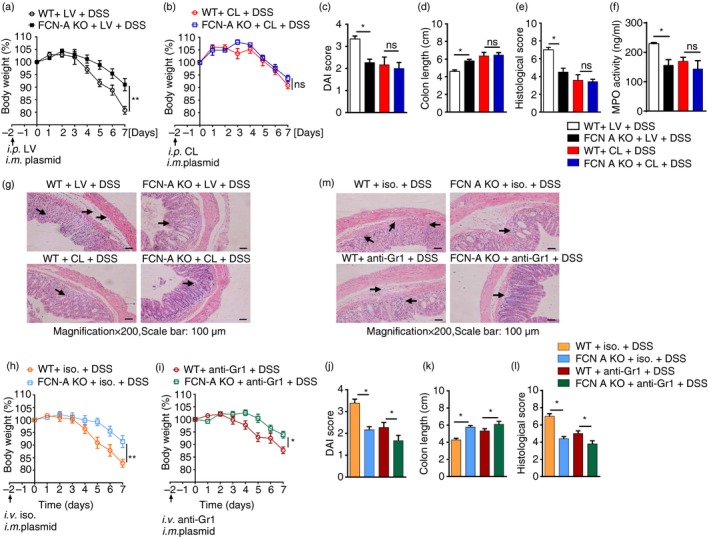
Effects of macrophage and neutrophil depletion on colitis in dextran sulphate sodium (DSS) ‐treated wild‐type (WT) mice and ficolin‐A knockout (FCN‐A KO) mice. (a–g) The severity of DSS‐induced colitis in WT mice and FCN‐A KO mice was compared after macrophage depletion. (a, b) Body weight, (c) Disease Activity Index (DAI) score, (d) colon length, and (f) myeloperoxidase (MPO) activity. (e) Statistical analysis of histopathological score, and (g) representative data of haematoxylin & eosin (H&E) staining. (h–m) The severity of DSS‐induced colitis in WT mice and FCN‐A KO mice was compared after neutrophil depletion. (h, i) Body weight, (j) DAI score, and (k) colon length. (l) Statistical analysis of histopathological score, and (m) representative data of H&E staining. Six mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01, ns: not significant. CL: Clodronate liposomes. LV: liposome vehicle. ios.: isotype control.
However, when we depleted neutrophils, a significant decrease in body weight was still observed in WT mice compared with FCN‐A KO mice (Fig. 5h,i). Similarly, we also observed significantly increased DAI scores (Fig. 5j), shorter colon length (Fig. 5k) and increased histological scores (Fig. 5l,m) in WT mice compared with FCN‐A KO mice. The infiltration of neutrophils into colon tissues, as indicated by MPO activity, was compared between DSS‐treated WT and FCN‐A KO mice after macrophage depletion. However, no significant differences in neutrophil infiltration were observed (Fig. 5f). As in our previous experiments, we observed similar results that FCN‐2 enhanced acute colitis (see Supplementary material, Fig. S3a–f). These data suggest that the FCN‐A/2‐mediated promotion of DSS‐induced colitis is mainly dependent on macrophages but not on neutrophils.
FCN‐A stimulates M1 macrophage polarization and pro‐inflammatory cytokine production by activating the MAPK/NF‐κB signalling pathway
As the exacerbation of colitis by FCN‐A is mainly dependent on macrophages, we further determined the effects of FCN‐A on macrophages in vitro. The contaminant endotoxin in the purified proteins was strictly removed (see Supplementary material, Fig. S4a). RAW 264.7 cells were treated with either GST‐FCN‐A or GST, and the expressions of the M1 macrophage marker iNOS and the M2 macrophage marker Arg‐1 were examined by Western blot analysis. We observed that FCN‐A induced iNOS expression and decreased Arg‐1 expression and these effects occurred in a dose‐dependent manner, whereas the control protein GST did not have any effect (Fig. 6a). The levels of pro‐inflammatory cytokines in the cell supernatants were examined by ELISA, and the secretions of IL‐1β, IL‐6 and TNF‐α from RAW264.7 cells were significantly increased after stimulation with FCN‐A for different time‐points, whereas the control protein GST had no effect (Fig. 6b). We also examined the effect of FCN‐A on the IL‐10 expression level. We found that FCN‐A could attenuate the IL‐10 secretion in IL‐4‐induced M2 macrophages (see Supplementary material, Fig. S4b).
Figure 6.
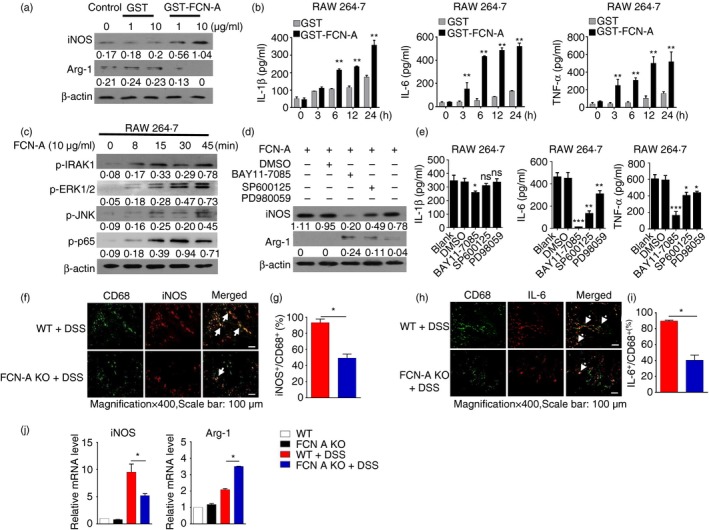
Ficolin‐A/2 (FCN‐A/2) promoted M1 polarization and pro‐inflammatory cytokine production by activating the mitogen‐activated protein kinase/ nuclear factor‐κB (MAPK/NF‐κB) signalling pathway. In vitro, RAW264.7 cells were stimulated with GST‐FCN‐A/GST proteins for 24 hr; GST was used as a control. (a) The expressions of inducible nitric oxide synthase (iNOS) and Arg‐1 were examined by Western blot analysis. (b) The pro‐inflammatory cytokines interleukin‐1β (IL‐1β), IL‐6 and tumour necrosis factor‐α (TNF‐α) in cell supernatants were detected by ELISA. (c) Immunoblot analysis of p‐IRAK1, p‐p65, p‐ERK1/2, and p‐JNK in the lysates of RAW246.7 cells after stimulation with FCN‐A for 0–45 min. (d) The following signalling inhibitors, NF‐κB inhibitor (BAY‐117082, 10 μm), JNK inhibitor (SP600125, 10 μm) and ERK inhibitor (PD98059, 10 μm), were added to cells and incubated for 1 hr before GST‐FCN‐A (10 μg/ml) treatment for 24 hr. Protein expression levels of iNOS and Arg‐1 in cell lysates were examined by Western blot analysis. (e) The levels of IL‐1β, IL‐6 and TNF‐α in the cell supernatants were measured by ELISA. (f, g) In vivo, representative immunofluorescence (f) and statistical analysis (g) of the iNOS + CD68+ colonic macrophages in dextran sulphate sodium (DSS) treated‐mice (in f, solid arrow, the green fluorescence‐labelled CD68 merged with the red fluorescence‐labelled iNOS appeared as yellow). (h, i) In vivo, representative immunofluorescence (h) and statistical analysis (i) of the IL‐6+ CD68+ colonic macrophages in DSS‐treated‐mice (in h, solid arrow, the green fluorescence‐labelled CD68 merged with the red fluorescence‐labelled IL‐6 to appear as yellow). (j) The mRNA levels of iNOS and Arg‐1 in the colonic tissues of DSS‐treated mice were detected by RT‐qPCR. Six mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001, ns: not significant.
We further examined the effect of FCN‐A on the activation of signalling pathways in macrophages. We found that FCN‐A caused the increased expression of p‐IRAK1, p‐ERK1/2, p‐JNK and p‐p65 in RAW264.7 cells (Fig. 6c). To clarify whether the promotion of M1 macrophage polarization by FCN‐A was dependent on the activation of the mitogen‐activated protein kinase (MAPK)/NF‐κB signalling pathway, we further examined iNOS expression in the presence of the inhibitors of the key signalling pathways. Western blot analysis revealed that the expression of iNOS in FCN‐A‐stimulated RAW264.7 cells was significantly inhibited in the presence of the NF‐κB inhibitor BAY11‐7082 and the JNK inhibitor SP600125 but not the ERK inhibitor PD98059 (Fig. 6d). The NF‐κB inhibitor also significantly inhibited IL‐6 and TNF‐α secretion from FCN‐A‐stimulated RAW 264.7 cells and slightly inhibited the secretion of IL‐1β (Fig. 6e). The JNK inhibitor had a significant inhibitory effect on the secretion of IL‐6, a minor inhibitory effect on the secretion of TNF‐α, and no effect on the secretion of IL‐1β (Fig. 6e). The ERK inhibitor only had minor inhibitory effects on the secretion of IL‐6 and TNF‐α and no effect on IL‐1β (Fig. 6e). These results suggest that the increased activation of MAPK/NF‐κB signalling by FCN‐A is necessary for the M1 macrophage polarization and pro‐inflammatory cytokine production in RAW264.7 cells. The effects of FCN‐2 on PMA‐stimulated THP‐1 were also evaluated. In comparison with GST, GST‐FCN‐2‐treated THP‐1 exhibited higher mRNA levels of M1 phenotype markers such as iNOS, TNF‐α and IL‐6, whereas a set of M2 phenotype markers like IL‐10, Arg‐1 and CD206 were down‐regulated (see Supplementary material, Fig. S4c). GST‐FCN‐2 stimulated THP‐1 secreted higher levels of the pro‐inflammatory cytokines, IL‐1β, IL‐6 and TNF‐α with activation of MAPK/NF‐κB signalling (see Supplementary material, Fig. S4d–g).
We also compared the in vivo phenotype and infiltration of macrophages in the colon tissues of DSS‐treated WT and FCN‐A KO mice. By immunofluorescence staining, we observed that fewer double‐positive iNOS+ CD68+ (Fig. 6f,g and see Supplementary material, S5a,c), and IL‐6+ CD68+ (Fig. 6h,i and see Supplementary material, S5b,d) macrophages infiltrated the colon tissues of FCN‐A KO mice compared with the WT mice. The RT‐qPCR results showed significantly higher expression of iNOS in the colonic tissues of DSS‐treated WT mice compared with FCN‐A KO mice (Fig. 6j), and the administration of exogenous FCN‐A or FCN‐2 induced increased iNOS and decreased Arg‐1 expression (see Supplementary material, Fig. S5e). These data suggest that FCN‐A/2 results in more M1‐directed macrophage polarization and pro‐inflammatory cytokine production in the colon tissues.
FCN‐A activates M1 macrophages and promotes pro‐inflammatory cytokine production via the TLR4/MyD88/MAPK/NF‐κB pathway
Toll‐like receptor 4 is an important pattern recognition receptor on macrophages. To investigate the role of FCN‐A in TLR4 signalling of macrophage, we examined the interaction between FCN‐A and TLR4 on macrophage membrane by using Co‐immunoprecipitation. The GST‐FCN‐A/GST proteins were purified and identified by SDS–PAGE (Fig. 7a). The complex of purified GST‐FCN‐A/GST proteins with cellular membrane proteins of macrophages were used to conduct Co‐immunoprecipitation with anti‐TLR4 or rabbit IgG control. Co‐immunoprecipitation analysis of the interaction between GST‐FCN‐A and TLR4 was detected by immunoblotting (Fig. 7b). However, we did not observe any interaction between GST and TLR4 (Fig. 7b). The activation of TLR4 and its adaptor protein MyD88 causes the phosphorylation of MAPK/NF‐κB and the production of pro‐inflammatory cytokines. To explore whether TLR4 and MyD88 are involved in the FCN‐A‐induced activation of the MAPK/NF‐κB pathway, we measured the expression of iNOS and pro‐inflammatory cytokines in FCN‐A‐stimulated mouse BMDMs. We prepared BMDMs as described in the Material and methods section, and the purity of the BMDMs was assessed by flow cytometric analysis (see Supplementary material, Fig. S2f). Compared with TLR4+/+ MyD88+/+ BMDMs, much lower iNOS and higher Arg‐1 expression levels were observed in TLR4−/− BMDMs (Fig. 7c) and MyD88−/− BMDMs (Fig. 7e), whereas the expression of the housekeeping gene β‐actin was not changed. The expressions of the pro‐inflammatory cytokines IL‐1β in the cell lysates and secreted IL‐6 and TNF‐α were significantly decreased in TLR4−/− BMDMs (Fig. 7d) and MyD88−/− BMDMs (Fig. 7f) compared with the control groups. The expression levels of p‐IRAK1, p‐ERK, p‐JNK and p‐p65 were substantially lower in both TLR4−/− and MyD88−/− BMDMs compared with the TLR4+/+ MyD88+/+ BMDMs after FCN‐A stimulation for different time‐points (Fig. 7g,h). These results suggest that FCN‐A can activate M1 macrophages and promote pro‐inflammatory cytokine production in vitro via the TLR4/MyD88/MAPK/NF‐κB pathway.
Figure 7.
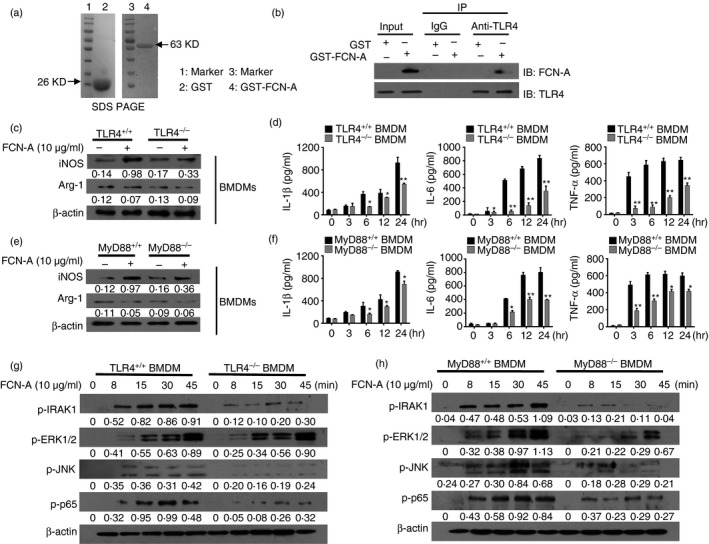
Ficolin‐A (FCN‐A) promoted the M1 polarization of bone‐marrow‐derived macrophages (BMDMs) through a Toll‐like receptor 4 (TLR4)/MyD88‐dependent pathway in vitro. (a) The protein expressions of the purified GST‐FCN‐A and GST were determined by SDS–PAGE. (b) The extracted membrane proteins from RAW264.7 cells were incubated with the purified GST‐FCN‐A or GST proteins. Co‐immunoprecipitation analysis of the interaction between TLR4 of macrophage and GST‐FCN‐A was performed by using anti‐TLR4. Rabbit IgG was used as a negative control in co‐immunoprecipitation. (c, e) BMDMs, isolated from wild‐type (WT), TLR4−/− or MyD88−/− mice, were stimulated with FCN‐A (10 μg/ml) for 24 hr, then the expressions of iNOS and Arg‐1 from BMDMs were examined by Western blot analysis. (d, f) The levels of pro‐inflammatory cytokines IL‐1β in cell lysates, and secreted IL‐6, tumour necrosis factor‐α (TNF‐α) were detected by ELISA. (g, h) Western blot analysis of p‐IRAK1, p‐p65, p‐ERK1/2 and p‐JNK in the BMDM lysates of TLR4−/−, MyD88−/− or WT after stimulation with FCN‐A for 0–45 min. In d and f, values are mean ± SEM from three independent experiments. *P < 0·05, **P < 0·01.
Adoptive transfer of TLR4−/−/MyD88−/− macrophages attenuates the FCN‐A‐mediated exacerbation of DSS‐induced colitis
To explore whether TLR4/MyD88 signalling in macrophages is responsible for the FCN‐A‐dependent increase in the susceptibility of mice to DSS‐induced colitis, adoptive transfer experiments were conducted (see Supplementary material, Fig. S6). After macrophage depletion, pV or pV‐FCN‐A‐administered FCN‐A KO mice were adoptively transferred with TLR+/+ and TLR4−/− BMDMs, respectively. Compared with mice adoptively transferred with TLR4−/− BMDMs, mice given pV‐FCN‐A adoptively transferred with TLR4+/+ BMDMs were more susceptible to DSS‐induced colitis, showing remarkably more severe colitis symptoms, including lower body weight (Fig. 8a), higher DAI score (Fig. 8b), shorter colon length (Fig. 8c), and higher histological pathological damage score (Fig. 8d,e), as well as higher colonic levels of the pro‐inflammatory cytokines IL‐6, TNF‐α and IL‐1β (Fig. 8f–h). We observed similar effects between FCN‐A KO mice that were adoptively transferred with MyD88+/+ and MyD88−/− BMDMs (Fig.9a–h). The effects of TLR4/MyD88 signalling in macrophages were also involved in the FCN‐2‐exacerbated colitis (see Supplementary material, Fig. S7). Together, these results suggest that TLR4/MyD88 signalling in macrophages is required for the exacerbation of colitis by FCN‐A/2.
Figure 8.
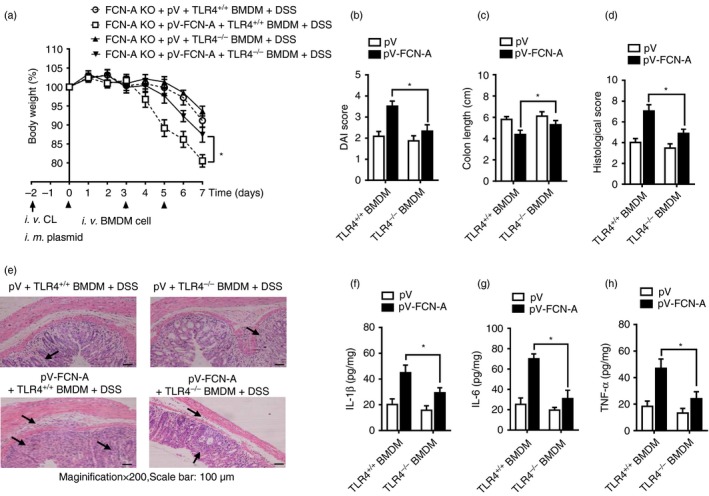
Adoptive transfer of TLR4−/− macrophages attenuated the exacerbation of dextran sulphate sodium (DSS) ‐induced colitis by ficolin‐A (FCN‐A). After macrophage depletion, mice were adoptively transferred with bone‐marrow‐derived macrophages (BMDMs) as described in the Supplementary material (Fig. S6). (a) Body weight, (b) Disease Activity Index (DAI) score, (c) colon length, (d) statistical analysis of histopathological score, and (e) representative data of haematoxylin & eosin (H&E) staining. (f–h) The levels of the interleukin‐1β (IL‐1β), IL‐6 and tumour necrosis factor‐α (TNF‐α) in the homogenate from colonic tissues were detected by ELISA. Six mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05.
Figure 9.
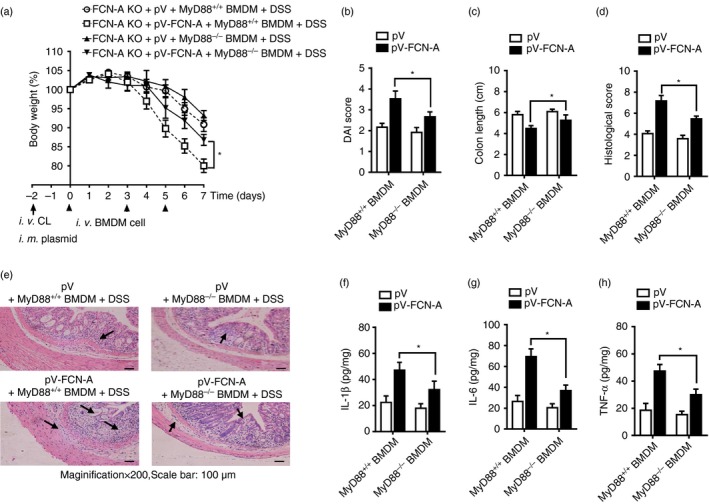
Adoptive transfer of MyD88−/− macrophages attenuated the exacerbation of dextran sulphate sodium (DSS)‐induced colitis by ficolin‐A (FCN‐A). After macrophage depletion, mice were adoptively transferred with bone‐marrow‐depleted macrophages (BMDMs) as described in the Supplementary material (Fig. S6). (a) Body weight, (b) Disease Activity Index (DAI) score, (c) colon length. (d) statistical analysis of histopathological score and (e) representative data of haematoxylin & eosin (H&E) staining. (f–h) The levels of interleukin‐1β (IL‐1β), IL‐6 and tumour necrosis factor‐α (TNF‐α) in the homogenates from colonic tissues were detected by ELISA. Six mice were analysed in each group. Values are mean ± SEM from three independent experiments. *P < 0·05.
Discussion
Common treatments for IBD, such as corticosteroids and immunosuppressive drugs, have undesirable adverse effects that could counteract their therapeutic benefits in the long term. In recent years, novel biological therapies designed against a specific inflammatory mediator have emerged and found some of their first real clinical applications in treating IBD.42, 43
The contribution of innate immunity to IBD remains an area of intense interest. Several studies have suggested a pathogenic role for innate immunity in the onset and severity of IBD. FCNs are innate immune members of the lectin family, which act as pattern recognition receptors. They are soluble oligomeric defence proteins with lectin‐like activity and are able to recognize pathogen‐associated molecular patterns, which are carbohydrate molecules on the surface of pathogens and apoptotic, necrotic, or malignant cells.44 Upon binding to their specific pathogen‐associated molecular patterns, FCNs may trigger the activation of the immune system either (i) by initiating the activation of complement via the lectin pathway, (ii) through a primitive type of opsonophagocytosis, or (iii) by stimulating the secretion of inflammatory cytokines by macrophages, so limiting the infection and concurrently orchestrating the subsequent adaptive immune response. Our and others' previous studies have indicated that FCN‐A/2 was an anti‐bacterial and anti‐viral innate immune molecule.21, 22, 23, 24, 25 In our present study, FCN‐A/2 played important pro‐inflammatory roles in the induction phase of acute colitis. In Fig. 3(a), analysis of the gene expression array showed the fold changes of the inflammatory cytokines in livers between WT and FCN‐A KO mice. Recently, in the same way, Pedrotti et al.45, 46 demonstrated that systemic cytokines could impair DSS colitis by priming lymphocytes and macrophages. Our work showed that FCN‐A/2 increased the population of M1‐type macrophages and neutrophil infiltration and aggravated the inflammation and tissue damage in the colon by inducing higher levels of pro‐inflammatory cytokines (IL‐1β, IL‐6, and TNF‐α. especially IL‐6), chemokines (CXCL1/2/10 and CCL4) and lower levels of anti‐inflammatory cytokine IL‐10. Because of the presence of resident macrophages and other immune cells in the liver, so cytokines and FCN‐A/2 secreted from the liver could affect other macrophages through paracrine and endocrine routes, or secreted into blood, which might affect macrophages in the colonic tissue and inflammatory colitis. FCN‐A/2, as a systemic signal, could also greatly affect local inflammatory responses.
Intestinal macrophages are important tissue immune cells that are ideally situated at the interface with the enteric luminal environment to respond to microbes and other potential stimuli.47, 48 Macrophages play a crucial role in the pathogenesis of IBD. Modulating the M1/M2 polarization status of macrophages can alter the course of the local inflammatory reaction. We found that the in vivo depletion of macrophages in mice completely diminished the differences of DSS‐colitis induced by FCN‐A/2, further indicating that the modulation of DSS‐colitis by FCN‐A/2 is dependent on macrophages. In vitro data demonstrated that FCN‐A could induce the M1 polarization of macrophages and the production of pro‐inflammatory cytokines by promoting the activation of the MyD88/MAPK/NF‐κB signalling pathway. In accordance with these results, in vivo, FCN‐A and FCN‐2 can induce the M1 macrophage phenotype in the colon and the production of pro‐inflammatory cytokines and chemokines. These findings support the role of M1 macrophage polarization in the effects of FCN‐A/2 on the development of inflammatory diseases. Together, these data demonstrate that FCN‐A/2 can act as a new regulator of macrophage polarization mediating the inflammatory response in experimental mouse colitis.
Accumulating evidence demonstrates that IL‐6 selectively regulates the migration and resistance against apoptosis of all CD4+‐expressing T cells regardless of their activation state, which is a key factor in IBD pathogenesis.49, 50 In this study, we found a significantly increased level of IL‐6 secreted from FCN‐A/2‐stimulated macrophages in vivo and in vitro. Besides IL‐6, as our results indicated, other cytokines such as TNF‐α, IFN‐γ, CXC chemokines (CXCL1/2, CXCL10) and CCL4, were also up‐regulated after FCN‐A/2 stimulation, which also play important roles in IBD pathogenesis. As a neutrophil‐associated chemokine, CXCXL1/2, which was mainly induced in macrophages, can specifically mediate neutrophil infiltration by binding to the common receptor CXCR2.51 Our results showed that when mice were depleted of macrophages in vivo, the degree of neutrophil infiltration was diminished. We hold the opinion that the neutrophil infiltration was influenced by the chemokines secreted by macrophages, such as CXCL1/2. Our present study has shown that the levels of the CXCR3 ligand CXCL10 increased and that the infiltration of CD45+ leucocytes was enhanced in FCN‐A‐stimulated experimental colitis when compared with control. Our data support the above view on the importance of these cytokines and chemokines in the progression of the symptoms of IBD.
Here we provide the first evidence for cross‐talk between FCN‐A/2 and TLR4 signalling in macrophages. We demonstrated that in vivo adoptive transfer of TLR4−/− and MyD88−/− macrophages attenuated the exacerbation of DSS‐induced colitis by FCN‐A, which suggests that FCN‐A/2 exacerbated the inflammatory pathogenesis of IBD by stimulating M1 polarization through the TLR4/MyD88 signalling pathway in macrophages. We propose that FCN‐A/2 might bind to the TLR4 receptor of macrophages and then stimulate M1 polarization and pro‐inflammatory cytokine release. Our present results showed that FCN‐A promotes M1 macrophage activation and pro‐inflammatory cytokine release through the regulation of the TLR4/MyD88/MAPK/NF‐κB signalling pathway (Figs 6, 7, 8, 9). The detailed mechanism of the interaction between FCN‐A/2 and macrophage receptors is currently being investigated in our laboratory.
The complement system plays a pivotal role in immune responses and inflammatory processes.52, 53 In early clinical studies, complement activation was shown to be associated with the progression of UC and CD. The synthesis of complement components in the intestine of UC patients might be part of the local inflammatory amplification process.54 In our study, we found that serum FCN‐2/A increased in both patients with IBD and in mice with acute colitis. In Fig. 1(a), it seems that two populations can be found in the UC and CD groups, one population with levels similar to the HDs and one population with elevated levels. We speculate that this might be due to different disease severities or to the treatment that these patients received. As the results show in Fig. 1(b,c), FCN‐2 levels were positively correlated with the degrees of disease severity in patients with UC and CD, and acute inflammatory protein CRP levels (Fig. 1e), as well. Sandahl et al.55 reported that the lectin pathway of the complement system was down‐regulated in patients with CD who respond to anti‐TNF‐α therapy. Consistent with other group's work, our present data also suggest that the lectin pathway proteins may exacerbate inflammation in IBD and changes in lectin pathway protein levels may be associated with clinical outcome. Roles of FCNs in rodent experimental colitis models require further study. C3, fB and C5a are the key components of the complement cascade, and their deficiencies have been reported to be beneficial against a DSS‐induced model due to the reduction of intestinal inflammation in C3−/− and fB−/− mice.56, 57 However, the exacerbated disease observed in C1q/MBL−/− mice appears to be due, at least in part, to commensal microorganisms gaining access to the systemic circulation as a result of impaired host defence, leading to a breakdown of tolerance.56
In conclusion, our present data suggest that targeted ficolin‐2/A inhibitors may be of therapeutic benefit in the acute phases of IBD. Nevertheless, DSS‐induced colitis is only one of several models of IBD, and further studies are also needed to address the role of FCN‐A/2 in chronic disease to better understand the potential benefits of ficolin‐2/A‐based complement inhibition for the treatment of this condition and to delineate new insights into the immune interplay underlying IBD.
Disclosures
None declared.
Supporting information
Figure S1. Analysis of interleukin‐6 expressions in mouse colonic tissues.
Figure S2. Flow cytometry analysis of immune cells. Flow cytometry analysis of infiltrated leucocytes in mouse colonic tissues.
Figure S3. Macrophage and neutrophil depletion experiments.
Figure S4. The effects of ficolin‐2/A on the polarization of macrophages.
Figure S5. The effects of exogenous ficolin‐A/2 on M1 polarization and interleukin‐6 expressions of colonic CD68+ macrophages.
Figure S6. The procedure of adoptive transfer experiments.
Figure S7. The effects of adoptive transfer of TLR4−/−/MyD88−/− macrophages on the exacerbation of dextran sulphate sodium‐induced colitis induced by ficolin‐2.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31221061, 31270176 and 31370197), National Outstanding Youth Foundation of China (81025008), the Hubei Province's Outstanding Medical Academic Leader Programme (523‐276003), Hubei Province Major Knowledge Innovation Project (Natural Science Foundation) (2016CFA062), Hubei Province Technological Innovation Special Foundation Major Project (2016ACA150), and the Science and Technology Program of Wuhan (201150530141).
Contributor Information
Yi‐Fei Yang, Email: yangyifeinihao163@163.com.
Xiao‐Lian Zhang, Email: zhangxiaolian@whu.edu.cn.
References
- 1. Podolsky DK. Inflammatory bowel disease. N Engl J Med 2002; 347:417–29. [DOI] [PubMed] [Google Scholar]
- 2. Ko JK, Auyeung KK. Inflammatory bowel disease: etiology, pathogenesis and current therapy. Curr Pharm Des 2014; 20:1082–96. [DOI] [PubMed] [Google Scholar]
- 3. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev 2014; 13:3–10. [DOI] [PubMed] [Google Scholar]
- 4. Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EJ, Tysk C et al Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013; 62:630–49. [DOI] [PubMed] [Google Scholar]
- 5. Latiano A, Palmieri O, Pastorelli L, Vecchi M, Pizarro TT, Bossa F et al Associations between genetic polymorphisms in IL‐33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE 2013; 8:e62144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuhl AA, Erben U, Kredel LI, Siegmund B. Diversity of intestinal macrophages in inflammatory bowel diseases. Front Immunol 2015; 6:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zigmond E, Jung S. Intestinal macrophages: well educated exceptions from the rule. Trends Immunol 2013; 34:162–8. [DOI] [PubMed] [Google Scholar]
- 8. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C et al Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal 2014; 26:192–7. [DOI] [PubMed] [Google Scholar]
- 9. Steinbach EC, Plevy SE. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm Bowel Dis 2014; 20:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010; 32:593–604. [DOI] [PubMed] [Google Scholar]
- 11. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8:958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hunter MM, Wang A, Parhar KS, Johnston MJ, Van Rooijen N, Beck PL et al In vitro‐derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 2010; 138:1395–405. [DOI] [PubMed] [Google Scholar]
- 13. Weisser SB, Brugger HK, Voglmaier NS, McLarren KW, van Rooijen N, Sly LM. SHIP‐deficient, alternatively activated macrophages protect mice during DSS‐induced colitis. J Leukoc Biol 2011; 90:483–92. [DOI] [PubMed] [Google Scholar]
- 14. You Y, Zhou C, Li D, Cao ZL, Shen W, Li WZ et al Sorting nexin 10 acting as a novel regulator of macrophage polarization mediates inflammatory response in experimental mouse colitis. Sci Rep 2016; 6:20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang X, Yu J, Shi Q, Xiao Y, Wang W, Chen G et al Tim‐3 promotes intestinal homeostasis in DSS colitis by inhibiting M1 polarization of macrophages. Clin Immunol 2015; 160:328–35. [DOI] [PubMed] [Google Scholar]
- 16. Fujimori Y, Harumiya S, Fukumoto Y, Miura Y, Yagasaki K, Tachikawa H et al Molecular cloning and characterization of mouse ficolin‐A. Biochem Biophys Res Commun 1998; 244:796–800. [DOI] [PubMed] [Google Scholar]
- 17. Hein E, Nielsen LA, Nielsen CT, Munthe‐Fog L, Skjoedt MO, Jacobsen S et al Ficolins and the lectin pathway of complement in patients with systemic lupus erythematosus. Mol Immunol 2015; 63:209–14. [DOI] [PubMed] [Google Scholar]
- 18. Le Y, Lee SH, Kon OL, Lu J. Human L‐ficolin: plasma levels, sugar specificity, and assignment of its lectin activity to the fibrinogen‐like (FBG) domain. FEBS Lett 1998; 425:367–70. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I et al Human M‐ficolin is a secretory protein that activates the lectin complement pathway. J Immunol 2005; 175:3150–6. [DOI] [PubMed] [Google Scholar]
- 20. Kwon S, Kim MS, Kim D, Lee KW, Choi SY, Park J et al Identification of a functionally relevant signal peptide of mouse ficolin A. J Biochem Mol Biol 2007; 40:532–8. [DOI] [PubMed] [Google Scholar]
- 21. Zhao Y, Ren Y, Zhang X, Zhao P, Tao W, Zhong J et al Ficolin‐2 inhibits hepatitis C virus infection, whereas apolipoprotein E3 mediates viral immune escape. J Immunol 2014; 193:783–96. [DOI] [PubMed] [Google Scholar]
- 22. Hu YL, Luo FL, Fu JL, Chen TL, Wu SM, Zhou YD et al Early increased ficolin‐2 concentrations are associated with severity of liver inflammation and efficacy of anti‐viral therapy in chronic hepatitis C patients. Scand J Immunol 2013; 77:144–50. [DOI] [PubMed] [Google Scholar]
- 23. Luo F, Chen T, Liu J, Shen X, Zhao Y, Yang R et al Ficolin‐2 binds to HIV‐1 gp120 and blocks viral infection. Virol Sin 2016; 31:406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo F, Sun X, Wang Y, Wang Q, Wu Y, Pan Q et al Ficolin‐2 defends against virulent Mycobacteria tuberculosis infection in vivo, and its insufficiency is associated with infection in humans. PLoS ONE 2013; 8:e73859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen T, Hu Y, Ding Q, Yu J, Wang F, Luo F et al Serum ficolin‐2 concentrations are significantly changed in patients with hepatitis B virus infection and liver diseases. Virol Sin 2015; 30:249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang G, Liang Y, Zheng T, Song R, Wang J, Shi H et al FCN2 inhibits epithelial‐mesenchymal transition‐induced metastasis of hepatocellular carcinoma via TGF‐β/Smad signaling. Cancer Lett 2016; 378:80–6. [DOI] [PubMed] [Google Scholar]
- 27. Truelove SC, Witts LJ. Cortisone in ulcerative colitis; final report on a therapeutic trial. Br Med J 1955; 2:1041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harvey RF, Bradshaw JM. A simple index of Crohn's‐disease activity. Lancet 1980; 1:514. [DOI] [PubMed] [Google Scholar]
- 29. Endo Y, Sato Y, Matsushita M, Fujita T. Cloning and characterization of the human lectin P35 gene and its related gene. Genomics 1996; 36:515–21. [DOI] [PubMed] [Google Scholar]
- 30. Pan Q, Chen H, Wang F, Jeza VT, Hou W, Zhao Y et al L‐ficolin binds to the glycoproteins hemagglutinin and neuraminidase and inhibits influenza A virus infection both in vitro and in vivo . J Innate Immun 2012; 4:312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li P, Wan Q, Feng Y, Liu M, Wu J, Chen X et al Engineering of N‐glycosylation of hepatitis C virus envelope protein E2 enhances T cell responses for DNA immunization. Vaccine 2007; 25:1544–51. [DOI] [PubMed] [Google Scholar]
- 32. Liu M, Chen H, Luo F, Li P, Pan Q, Xia B et al Deletion of N‐glycosylation sites of hepatitis C virus envelope protein E1 enhances specific cellular and humoral immune responses. Vaccine 2007; 25:6572–80. [DOI] [PubMed] [Google Scholar]
- 33. Ren Y, Min YQ, Liu M, Chi L, Zhao P, Zhang XL. N‐glycosylation‐mutated HCV envelope glycoprotein complex enhances antigen‐presenting activity and cellular and neutralizing antibody responses. Biochim Biophys Acta 2016; 8:1764–75. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y, Shi L, Mei H, Zhang J, Zhu Y, Han X et al Inflamed macrophage microvesicles induce insulin resistance in human adipocytes. Nutr Metab (Lond) 2015; 12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun X, Pan Q, Yuan C, Wang Q, Tang XL, Ding K et al A single ssDNA aptamer binding to mannose‐capped lipoarabinomannan of Bacillus Calmette–Guérin enhances immunoprotective effect against tuberculosis. J Am Chem Soc 2016; 138:11680–9. [DOI] [PubMed] [Google Scholar]
- 36. Ren T, Grants I, Alhaj M, McKiernan M, Jacobson M, Hassanain HH et al Impact of disrupting adenosine A(3) receptors (A(3)(‐)/(‐) AR) on colonic motility or progression of colitis in the mouse. Inflamm Bowel Dis 2011; 17:1698–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc 2007; 2:2307–11. [DOI] [PubMed] [Google Scholar]
- 38. Weisser SB, van Rooijen N, Sly LM. Depletion and reconstitution of macrophages in mice. J Vis Exp 2012; 66:e4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N et al Critical roles of macrophages in the formation of intracranial aneurysm. Stroke 2011; 42:173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma Y, Chen H, Wang Q, Luo F, Yan J, Zhang XL. IL‐24 protects against Salmonella typhimurium infection by stimulating early neutrophil Th1 cytokine production, which in turn activates CD8+ T cells. Eur J Immunol 2009; 12:3357–68. [DOI] [PubMed] [Google Scholar]
- 41. Ji J, Shu D, Zheng M, Wang J, Luo C, Wang Y et al Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci Rep 2016; 6:24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kniazev OV, Parfenov AI, Ruchkina IN, Lazebnik LB. Sagynbaeva VE. Ter Arkh 2013; 85:55–9. [PubMed] [Google Scholar]
- 43. Xiao YT, Yan WH, Cao Y, Yan JK, Cai W. Neutralization of IL‐6 and TNF‐α ameliorates intestinal permeability in DSS‐induced colitis. Cytokine 2016; 83:189–92. [DOI] [PubMed] [Google Scholar]
- 44. Ren Y, Ding Q, Zhang X. Ficolins and infectious diseases. Virol Sin 2014; 29:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pedrotti LP, Sena AA, Rodriguez GM, Cejas H, Correa SG. Intestinal mononuclear cells primed by systemic interleukin‐12 display long‐term ability to aggravate colitis in mice. Immunology 2017; 150:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pedrotti LP, Barrios BE, Maccio‐Maretto L, Bento AF, Sena AA, Rodriguez‐Galan MC et al Systemic IL‐12 burst expands intestinal T‐lymphocyte subsets bearing the α 4 αβ 7 integrin in mice. Eur J Immunol 2016; 46:70–80. [DOI] [PubMed] [Google Scholar]
- 47. Isidro RA, Appleyard CB. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol 2016; 311:G59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Serini S, Ottes VR, Fasano E, Calviello G. Epigenetic regulation of gene expression and M2 macrophage polarization as new potential omega‐3 polyunsaturated fatty acid targets in colon inflammation and cancer. Expert Opin Ther Targets 2016; 20:843–58. [DOI] [PubMed] [Google Scholar]
- 49. Zhang SJ, Wang L, Ming L, Guo XB, Wang HM, Li XW et al Blockade of IL‐6 signal exacerbates acute inflammatory bowel disease via inhibiting IL‐17 producing in activated CD4+ Th17 population. Eur Rev Med Pharmacol Sci 2013; 17:3291–5. [PubMed] [Google Scholar]
- 50. Mitsuyama K, Matsumoto S, Masuda J, Yamasakii H, Kuwaki K, Takedatsu H et al Therapeutic strategies for targeting the IL‐6/STAT3 cytokine signaling pathway in inflammatory bowel disease. Anticancer Res 2007; 27:3749–56. [PubMed] [Google Scholar]
- 51. Spehlmann ME, Dann SM, Hruz P, Hanson E, McCole DF, Eckmann L. CXCR2‐dependent mucosal neutrophil influx protects against colitis‐associated diarrhea caused by an attaching/effacing lesion‐forming bacterial pathogen. J Immunol 2009; 183:3332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laufer J, Katz Y, Passwell JH. Extrahepatic synthesis of complement proteins in inflammation. Mol Immunol 2001; 38:221–9. [DOI] [PubMed] [Google Scholar]
- 53. Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement‐related diseases. EMBO J 2015; 34:2735–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pagoldh M, Lange S, Jennische E, Almer S, Bostrom EA, Eriksson A. Faecal analysis and plasma complement factor 3c levels at admission for an acute attack of ulcerative colitis are predictive of the need for colectomy. Eur J Gastroenterol Hepatol 2014; 26:295–300. [DOI] [PubMed] [Google Scholar]
- 55. Sandahl TD, Kelsen J, Dige A, Dahlerup JF, Agnholt J, Hvas CL et al The lectin pathway of the complement system is downregulated in Crohn's disease patients who respond to anti‐TNF‐α therapy. J Crohns Colitis 2014; 8:521–8. [DOI] [PubMed] [Google Scholar]
- 56. Schepp‐Berglind J, Atkinson C, Elvington M, Qiao F, Mannon P, Tomlinson S. Complement‐dependent injury and protection in a murine model of acute dextran sulfate sodium‐induced colitis. J Immunol 2012; 188:6309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Woodruff TM, Arumugam TV, Shiels IA, Reid RC, Fairlie DP, Taylor SM. A potent human C5a receptor antagonist protects against disease pathology in a rat model of inflammatory bowel disease. J Immunol 2003; 171:5514–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of interleukin‐6 expressions in mouse colonic tissues.
Figure S2. Flow cytometry analysis of immune cells. Flow cytometry analysis of infiltrated leucocytes in mouse colonic tissues.
Figure S3. Macrophage and neutrophil depletion experiments.
Figure S4. The effects of ficolin‐2/A on the polarization of macrophages.
Figure S5. The effects of exogenous ficolin‐A/2 on M1 polarization and interleukin‐6 expressions of colonic CD68+ macrophages.
Figure S6. The procedure of adoptive transfer experiments.
Figure S7. The effects of adoptive transfer of TLR4−/−/MyD88−/− macrophages on the exacerbation of dextran sulphate sodium‐induced colitis induced by ficolin‐2.