

Abstract
Purpose:
Using effect estimates from genome-wide association studies (GWAS), we identified a genetic risk score (GRS) that has the strongest association with type 2 diabetes (T2D) status in a population-based cohort and investigated its potential for prospective T2D risk assessment.
Methods:
By varying the number of single-nucleotide polymorphisms (SNPs) and their respective weights, alternative versions of GRS can be computed. They were tested in 1,181 T2D cases and 9,092 controls of the Estonian Biobank cohort. The best-fitting GRS was chosen for the subsequent analysis of incident T2D (386 cases).
Results:
The best fit was provided by a novel doubly weighted GRS that captures the effect of 1,000 SNPs. The hazard for incident T2D was 3.45 times (95% CI: 2.31–5.17) higher in the highest GRS quintile compared with the lowest quintile, after adjusting for body mass index and other known predictors. Adding GRS to the prediction model for 5-year T2D risk resulted in continuous net reclassification improvement of 0.324 (95% CI: 0.211–0.444). In addition, a significant effect of the GRS on all-cause and cardiovascular mortality was observed.
Conclusion:
The proposed GRS would improve the accuracy of T2D risk prediction when added to the currently used set of predictors.
Genet Med 19 3, 322–329.
Keywords: genetic risk score, genetic risk, precision medicine, risk prediction, type 2 diabetes
Introduction
The increasing prevalence of type 2 diabetes (T2D) is one of the greatest challenges in public health, in both developed and developing countries. In 2012, approximately 8.3% of the world’s adult population was living with diabetes.1 Diabetes is a leading cause of cardiovascular disease, renal disease, blindness, and limb amputation. T2D, accounting for 80–90% of all diabetes in Europe, decreases life expectancy by 5–10 years.2
Because the onset of T2D can be postponed or partially prevented by changes in the lifestyles of high-risk subjects,3 the cost-effectiveness of lifestyle (and other) interventions can be increased by improving the precision of risk prediction, thereby enabling targeting of the individuals at highest risk.
Although obesity is the strongest predictor of T2D, it is also known that heritability of T2D is 26–69%, depending on age of onset,4,5 thus motivating the search for genetic predictors for T2D. However, despite the large number of published genome-wide association studies (GWAS) of T2D, there is still some skepticism regarding the practical value of identified single-nucleotide polymorphisms (SNPs) in personalized risk prediction for the disease. The main reason is that the effect of individual SNPs on complex common disease phenotypes is relatively weak and/or adds little to predictions based on lifestyle, demographic, and clinical factors.6,7
In GWAS, SNPs need to meet the stringent genome-wide threshold, usually set to P < 5 × 10−8, to be significantly associated with the trait. Even though the sample sizes in GWAS have been increasing steadily over the years, they are still insufficient for SNPs with small effects to pass that threshold.8 This could explain why it has been shown that all common variants across the genome actually explain a much higher proportion of heritability (50% or more) in many complex traits than one could see based on a small subset of significant SNPs only.9,10
To explain a meaningful proportion of variability in a complex trait and, more importantly, to use this knowledge in risk assessment at an individual level, one needs to construct a numeric summary measure of genetic risk—a genetic (polygenic) risk score (GRS) based on a large number of genotyped variants. Our aim is to develop a GRS with the best possible predictive power and investigate its potential to improve T2D risk stratification in the general population. For this purpose, two sources of data will be used: (i) results of large-scale meta-analyses of GWAS to obtain effect estimates for individual SNPs11 and (ii) individual-level data of a relatively large population-based cohort from the Estonian Biobank to compare versions of the GRS and decide on applicability of the best GRS in practical risk prediction. The best-fitting GRS for prevalent cases is then further validated in the analysis of incident T2D (data obtained by linking the Estonian Biobank cohort database to electronic health records of the participants).
Materials and Methods
Estonian Biobank cohort and genotyping
The Estonian Biobank (Estonian Genome Center, University of Tartu) was established with the long-term purpose of implementation of research results in public health and medicine in Estonia. Between 2002 and 2011, the Estonian Biobank recruited a cohort of 51,380 participants, including adults from all counties in Estonia and accounting for approximately 5% of the Estonian adult population during the recruitment period. Broad informed consent signed by participants enabled use of the data for various health research purposes as well as linkage of the data with other health-related databases and registries. An extensive phenotype questionnaire and measurement panel, together with follow-up data from linkage with national health-related registries and electronic health records (the Estonian Health Insurance database), allows assessment of the effects of classic epidemiological risk factors on the incidence of common complex diseases, such as T2D. The research project at the Estonian Genome Centre was approved by the Ethics Committee of Human Studies, University of Tartu, Estonia.
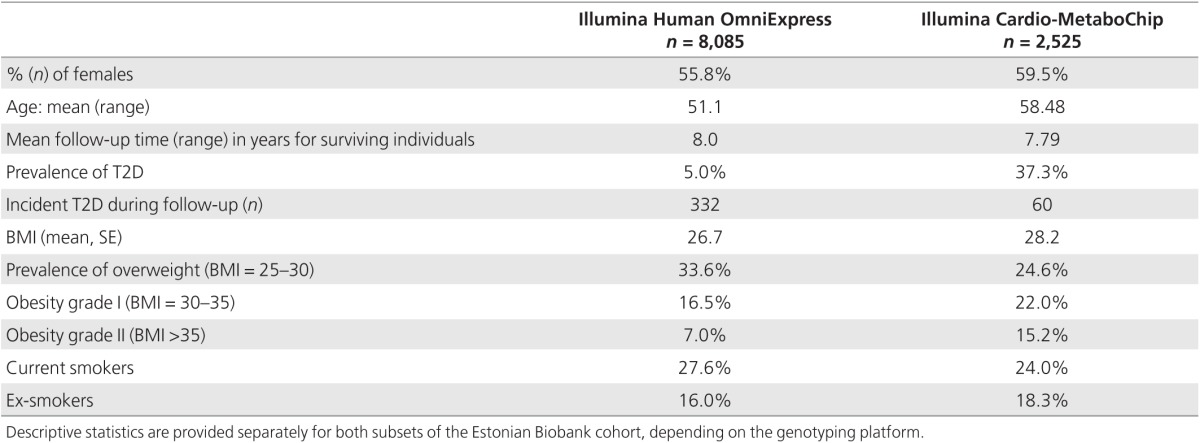
In the present study, a genotyped subset of 10,273 individuals (including 1,181 prevalent T2D cases) from the cohort was analyzed. The DNA samples of this subset were genotyped using either Illumina Human OmniExpress (a sample of 8,085 individuals) or Illumina Cardio-MetaboChip (a case–control sample of 942 T2D cases, 680 cases of coronary artery disease, and 903 random controls) genome-wide arrays. For 337 individuals (including 169 T2D cases) genotyped by both arrays, genotype data from the Cardio-MetaboChip array were used.
During the average follow-up time of 5.63 years, 386 incident T2D cases were observed (in 9,092 individuals free of T2D at recruitment) by 1 April 2014. A total of 1,994 individuals had died by 1 September 2015 (including 1,069 deaths due to cardiovascular causes).
The baseline phenotype data (Table 1) used for this study consist of age, gender, BMI and prevalent T2D status, history of hypertension or high blood glucose level, physical activity level (active versus inactive), and consumption of fresh fruit. For a subset (n = 6,064), data on plasma glucose level and lipoprotein profiles (low- and high-density lipoprotein (LDL and HDL) cholesterol, triglycerides, and total cholesterol) obtained by nuclear magnetic resonance (NMR) profiling were available (nonfasting measurements, with information on the time of last meal available for adjustment).
Table 1. Baseline characteristics of the two genotyped subsets of the Estonian Biobank cohort.
The genetic data used in this study were selected as follows. First, the Estonian Cardio-Metabochip sample was used in the large-scale GWAS meta-analysis for T2D susceptibility from the DIAGRAM Consortium.11 We therefore reran the meta-analysis to remove the effects of the Estonian sample because we intended to use the Cardio-Metabochip for developing the optimal GRS score. Second, only SNPs with P < 0.5 for association with T2D in the meta-analysis were chosen for further analysis. A set of independent SNPs with r2 ≤ 0.05 was then obtained via the LD-based clumping procedure of PLINK.12 Finally, clumped SNPs were retrieved from the Estonian Biobank database and filtered for genotyping and imputation quality and minor allele frequency, resulting in a set of 7,502 SNPs for further analysis and GRS construction (Supplementary Table S1 online). Full details on selection and weighting of SNPs can be found in the Supplementary Methods and Figures online.
Statistical analysis
Statistical analysis was performed using R version 3.1.0.13
The doubly weighted GRS
Suppose there are K independent markers with allele dosages 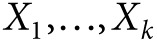 available for a study,
with estimated (linear or logistic) regression parameters from a GWAS
meta-analysis
available for a study,
with estimated (linear or logistic) regression parameters from a GWAS
meta-analysis 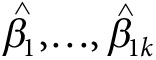 and
corresponding P values
and
corresponding P values . We define the general GRS as
. We define the general GRS as
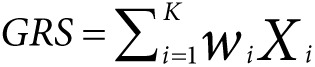 |
and different versions of GRS can be defined by varying the choice of weights
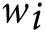 .
.
The conventionally used GRS (also referred to as the single-weighted GRS) is
defined by choosing 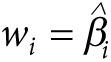 for all markers with
for all markers with 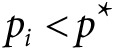 and
and 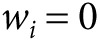 otherwise, where
otherwise, where 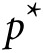 is a P value threshold (often
5 × 10–8 or
5 × 10–6). Let GRSk denote the
GRS where
is a P value threshold (often
5 × 10–8 or
5 × 10–6). Let GRSk denote the
GRS where 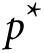 is
chosen so that exactly k (k < K) markers with the
smallest P values have nonzero weight in the score. For any choice of
k, GRSk suffers from a phenomenon called
“winner’s curse”—by selecting only SNPs with estimated
P values below a certain threshold, one systematically selects SNPs
with effect overestimated by chance. To correct for the resulting bias, we propose
a doubly weighted GRS, denoted by dGRSk, defined by selecting
is
chosen so that exactly k (k < K) markers with the
smallest P values have nonzero weight in the score. For any choice of
k, GRSk suffers from a phenomenon called
“winner’s curse”—by selecting only SNPs with estimated
P values below a certain threshold, one systematically selects SNPs
with effect overestimated by chance. To correct for the resulting bias, we propose
a doubly weighted GRS, denoted by dGRSk, defined by selecting
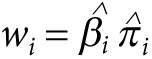 with
with
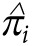 defined as
an estimated probability that the i-th marker belongs to the set of top k
SNPs with the strongest effect on the phenotype (“strongest” defined
as having the smallest P value on average of all possible studies). We
estimate such probability by simulating new values of potential parameter
estimates based on the observed estimates and their standard errors (more details
are provided in the Supplementary Methods and Figures online). We have
conducted simulation studies (not presented in this article) that demonstrated
that dGRSk indeed decreases the bias caused by the
“winner’s curse” in the single-weighted GRSk.
Although in practice the algorithm requires a large number of SNPs, choosing a
small value of k will result in near-zero weights for most of the SNPs
used.
defined as
an estimated probability that the i-th marker belongs to the set of top k
SNPs with the strongest effect on the phenotype (“strongest” defined
as having the smallest P value on average of all possible studies). We
estimate such probability by simulating new values of potential parameter
estimates based on the observed estimates and their standard errors (more details
are provided in the Supplementary Methods and Figures online). We have
conducted simulation studies (not presented in this article) that demonstrated
that dGRSk indeed decreases the bias caused by the
“winner’s curse” in the single-weighted GRSk.
Although in practice the algorithm requires a large number of SNPs, choosing a
small value of k will result in near-zero weights for most of the SNPs
used.
Comparison of different versions of the GRS in the association with prevalent T2D status
Using the data of the genotyped subsets of the Estonian Biobank cohort, we calculated both GRSk and dGRSk by varying k from 1 to all 7,502 of the initially selected SNPs. The effect of each GRS was assessed using age-, sex-, and genotype platform–adjusted logistic regression models for prevalent T2D status. Both BMI-adjusted and unadjusted models were fitted. The fit of (nonnested) models using a different version of the GRS as a covariate was compared using the Cox likelihood ratio test.14 The GRS producing the highest log-likelihood for both BMI-adjusted and unadjusted models was selected for further validation.
Because the GRS is a continuous measure, one also needs to specify a set of threshold values to classify individuals as being at “high,” “average,” or “low” genetic risk, for instance, to simplify the risk assessment in clinical practice. We investigated whether the quintiles of GRS can be used for that purpose, by using bar charts to compare the observed T2D prevalence in individuals aged 40–79 years across quintiles of the GRS and BMI category (<25, 25–30, 30–35, >35). In addition, bar charts were produced to study the distribution of individuals across GRS quintiles within the subset with prevalent T2D and in the subset of obese (BMI >35) T2D-free individuals aged 60 and older.
Validation of the GRS in the analysis of incident conditions
The GRS was further assessed for its effect on incident T2D in individuals without prevalent T2D at baseline and all-cause and cardiovascular mortality (in all individuals) using Cox proportional hazards modeling with age as the time scale. All models are adjusted for BMI category, smoking level (although smoking is not considered a standard predictor, we included it because of the significant effects of smoking level on T2D risk in our cohort), waist-to-hip ratio (WHR), waist circumference, physical activity level, history of high blood glucose, history of hypertension, fruit and vegetable consumption, and sex.
The analysis was restricted to the subset of 6,280 individuals aged 35–79 at recruitment (302 incident T2D cases) while censoring all T2D diagnoses beyond age 80 because diagnoses in the elderly are often related to significant risk-altering comorbidities (cancer or cardiovascular diseases). In addition, the Cox proportional hazards model was fitted in the subset with available glucose and lipid measurements (3,776 individuals, including 158 incident T2D cases) by adjusting for glucose, triglyceride, and HDL-cholesterol levels in addition to the covariates mentioned previously. The Kaplan-Meier graph of cumulative incidence of T2D was obtained for the subset with BMI >23.
Association of the GRS with other known T2D risk factors
The effects of GRS on BMI, WHR, plasma glucose, total cholesterol, HDL- and LDL-cholesterol, and triglyceride levels were estimated using age- and sex-adjusted linear regression analysis in individuals without prevalent T2D diagnosis. The effects of GRS on BMI and WHR were similarly assessed in individuals with prevalent T2D diagnosis.
Analysis of incremental value of GRS
For prevalent T2D, the area under the receiver operating characteristic (ROC) curve (AUC) was obtained from logistic regression fitted for individuals in the 40–79 age group who were genotyped on the OmniExpress platform. For incident T2D, Harrell’s c-statistic (concordance index) from the Cox proportional hazards models for individuals aged 35–79 with no prevalent T2D diagnosis was obtained.
To study reclassification and 5-year T2D risk predictions, Cox proportional hazards models with and without accounting for GRS were fitted. Improvement in the predictions was assessed using continuous net reclassification improvement (NRI) and integrated discrimination improvement (IDI).15 Confidence intervals for reclassification indices and c-statistics were estimated with bootstrapping.
Results
Comparison of different versions of the GRS
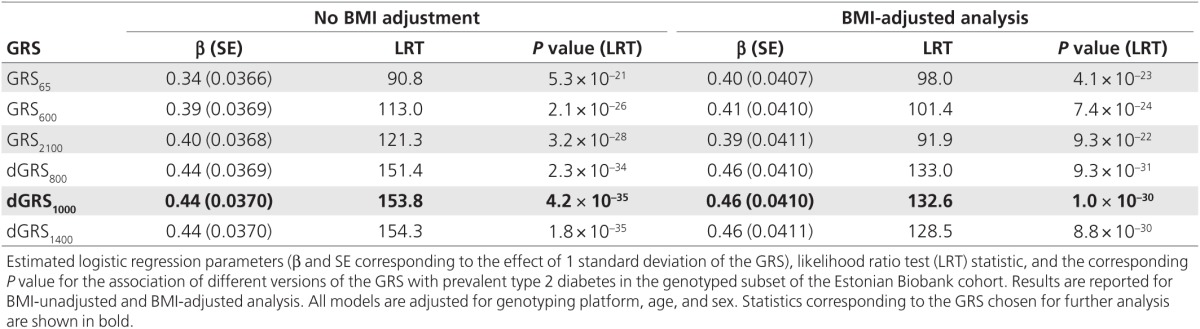
Results of the model fit for prevalent T2D status with GRSk and dGRSk for selected values of k are shown in Table 2. (More detailed results for values of k varied between 1 and 7,502 are provided in Supplementary Table S2 online and a corresponding plot of likelihood ratio statistics is shown in Supplementary Figure S1 online) Compared with the GRS65 (similar to ref. 16), the fit was considerably improved using a GRSk with a larger number of markers: the highest log-likelihood was achieved with GRS2100 (BMI-unadjusted models) and GRS600 (BMI-adjusted models). However, when dGRSk was used instead, with k = 300 or larger (up to 3,500), the fit became significantly better than that with any GRSk. The highest log-likelihood was achieved with dGRS1400 (BMI-unadjusted models) and dGRS800 (BMI-adjusted models); regardless of whether the analysis was adjusted for BMI, dGRS1000 provided a fit that was not significantly different from the best-fitting GRS (Cox test P > 0.05). Therefore, we used dGRS1000 in all subsequent analyses (weights shown in Supplementary Figure S2 online).
Table 2. Summary statistics for analyses performed with different GRS versions.
Association of dGRS1000 with prevalent T2D
The estimated odds ratio (OR) corresponding to 1 standard deviation (SD) difference in dGRS1000 was 1.56 (95% CI: 1.45–1.68) in the BMI-unadjusted model and 1.59 (95% CI: 1.46–1.72) in the BMI-adjusted model. The prevalence of T2D by BMI category and quintiles of dGRS1000 in the subset of the cohort with an age range of 45–79 is shown in Figure 1a (see Supplementary Figure S3 online for a more detailed plot). Although there was no significant interaction (P = 0.359) between BMI and dGRS1000, the association was strongest in overweight or moderately obese individuals (25 < BMI < 35), where the number of T2D cases in the highest GRS quintile was approximately comparable to the total number of cases in the three lowest quintiles.
Figure 1.

Bar charts illustrating the association between the dGRS1000 quintile, BMI category and prevalent T2D status. (a) T2D prevalence in genotyped individuals aged 45–79 in the Estonian Biobank cohort by dGRS1000 quintile and BMI category. The y-axis is scaled to match the average T2D prevalence by BMI category in the entire Estonian Biobank cohort (23,538 individuals aged 45–79, including 1,936 cases of prevalent T2D). The total number of T2D cases in each BMI-dGRS1000 category is shown on the top of each bar. (b) Distribution of dGRS1000 in all 1,181 genotyped individuals with prevalent T2D. Distribution of dGRS1000 is shown among all prevalent T2D cases, with dark blue color indicating individuals with BMI >35. (c) Distribution of dGRS1000 in severely obese T2D-free individuals aged 60 and older. Distribution of dGRS1000 is shown among T2D-free individuals who have BMI >35 and are older than 60.
Figure 1b indicates that approximately one-third of all prevalent T2D cases belong to the highest GRS quintile, but the differences in the observed frequencies of GRS quintiles were greatest in those with BMI <35. However, as indicated by Figure 1c, the majority of severely obese (BMI >35) but T2D-free individuals aged 60 and older belonged to the two lowest GRS quintiles.
Validation of the GRS in the analysis of incident conditions
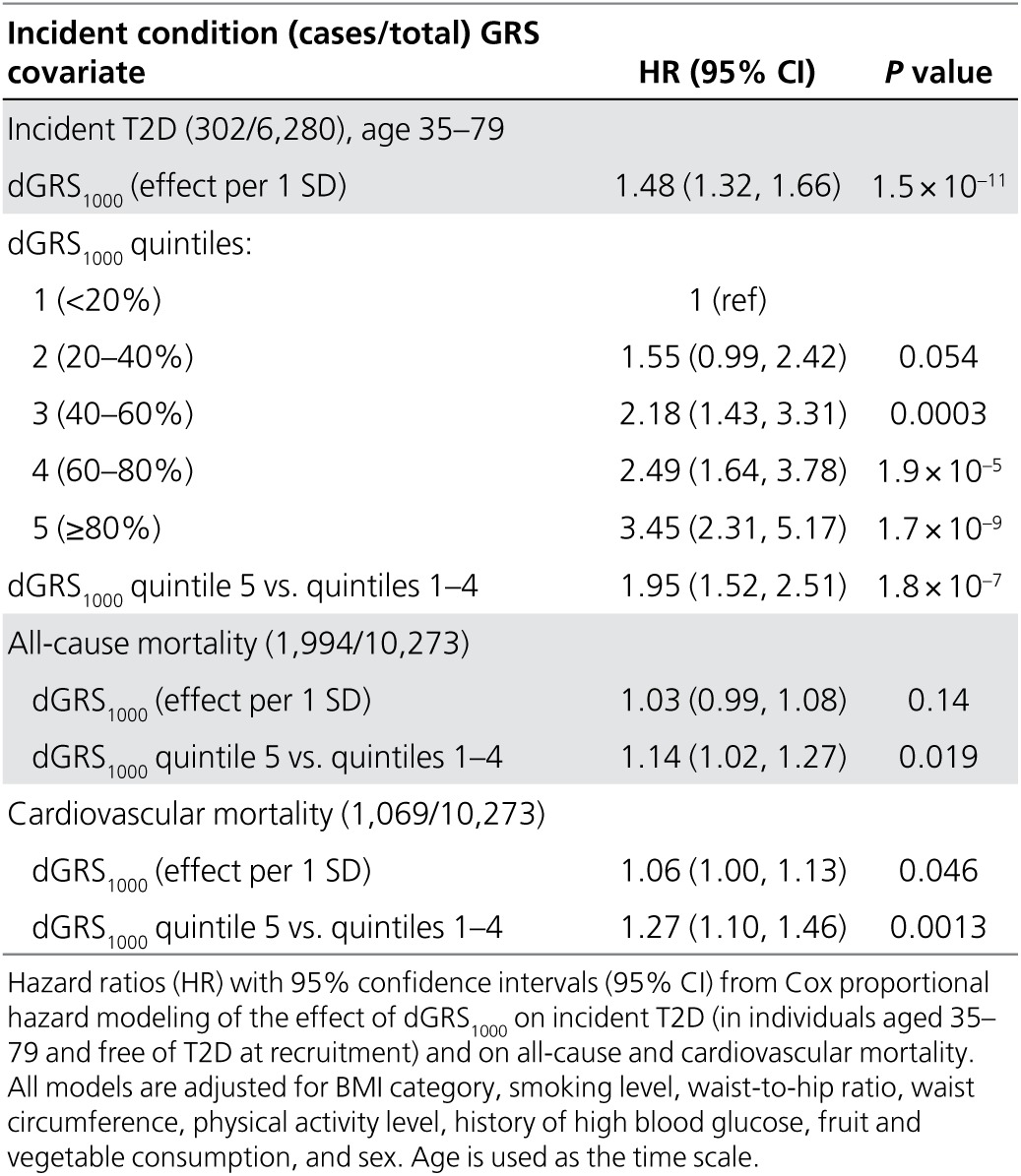
As shown in Table 3, dGRS1000 had a highly significant effect (HR = 1.48, 95% CI: 1.32–1.66 per 1 SD; P = 1.5 × 10−11) on the risk of developing T2D during follow-up and accounted for a large set of known covariates. According to the likelihood ratio test, the P value corresponding to the combined effect of the obesity-related parameters in this fitted model for incident T2D is 2.0 × 10–21, followed by the effect of dGRS1000. The difference in risks between lowest and highest GRS quintiles was more than threefold (HR = 3.45, 95% CI: 2.31–5.17). It is also important to note that the risk in the highest dGRS1000 quintile was approximately twice that in the rest of the sample (HR = 1.95, 95% CI: 1.52–2.51), indicating that this subset could be targeted for risk-reducing interventions. This is additionally supported by the fact that the highest dGRS1000 quintile was associated with 14% higher risk for all-cause mortality (P = 0.019) and 27% higher risk for cardiovascular mortality (P = 0.001).
Table 3. Analysis of the effect of dGRS1000 on incident T2D and on all-cause and cardiovascular mortality.
The differences in cumulative T2D incidence across dGRS1000 quintiles can also be seen in Figure 2, which shows that the cumulative risk differences are already obvious by the end of the first year of follow-up.
Figure 2.

Cumulative incidence of type 2 diabetes in 4,881 genotyped individuals free of T2D aged 35–79 and with BMI >23 at baseline. In the figure, 6.25-year follow-up is presented because only 25% of individuals were followed for more than 6.25 years. Cumulative incidence in presented separately in three dGRS1000 categories.
In the analysis of the effect of dGRS1000 on incident T2D while additionally adjusting for glucose, triglycerides, and HDL cholesterol, the HR corresponding to 1 SD difference in dGRS1000 was estimated to be 1.49 (95% CI: 1.27–1.76; P = 1.7 × 10−6), indicating that dGRS1000 has a significant effect on T2D risk that is independent of all available well-known risk factors.
The coefficients of all parameters in the model for incident T2D in the entire cohort of 30,094 initially T2D-free individuals (without genetic effects) and in the genotyped subset of 6,280 individuals (including the effect of dGRS1000) are shown in Supplementary Tables S3 and S4 online.
Association of dGRS1000 with known T2D risk factors
The estimated regression coefficients (β, SE, P value) from the analysis of the effect of dGRS1000 on BMI, WHR, plasma glucose, total cholesterol, triglycerides, HDL- and LDL-cholesterol levels are presented in Supplementary Table S5 online.
In individuals without prevalent T2D, a significant positive association of dGRS1000 with plasma glucose (P = 4.7 × 10−8) and triglyceride levels (P = 8.8 × 10−4) and a negative association with HDL-cholesterol level (P = 8.1 × 10−5) were found, suggesting that some individuals at high genetic risk may already have a clearly increased disease risk according to other criteria (or even prediabetes or undiagnosed T2D). No significant association of dGRS1000 with BMI was found in T2D-free individuals (P = 0.42), and there was only a weak positive association with WHR (P = 0.012). Thus, it is unlikely that dGRS1000 affected T2D risk through its possible effect on obesity.
The association between BMI and dGRS1000 in individuals with existing T2D diagnosis was found to be negative (P = 0.0039), suggesting that individuals at high genetic risk are more likely to develop T2D at a BMI lower than that of those with low genetic risk, as supported by the models for both prevalent and incident T2D, where the effects of BMI and GRS appeared to be additive. However, a cautious interpretation of the estimated effect size is needed because the T2D treatment may have affected the BMI of diseased individuals.
Analysis of incremental predictive ability of the dGRS1000
Incremental predictive ability of dGRS1000 was studied for both prevalent and incident T2D models. Including dGRS1000 in the logistic regression model for prevalent T2D improved AUC, irrespective of BMI adjustment (Supplementary Figure S4 online). Harrell’s c-statistic increased by 0.012 (95% CI: 0.004–0.023) after dGRS1000 was added to the model for incident T2D. More detailed results for c-statistics and likelihood ratio tests are shown in Supplementary Table S6 online.
Comparing 5-year predictions from models with and without dGRS1000 (Supplementary Figure S5 online) resulted in a continuous NRI of 0.324 (95% CI: 0.211 –0.444). Further investigation of components of the continuous NRI was undertaken as suggested;15 the continuous NRI for events was 0.115 (95% CI: 0.02–0.23) and for nonevents was 0.209 (95% CI: 0.182–0.23). The IDI was 0.013 (95% CI: 0.007–0.019).
Discussion
We have shown that the combined effect of a large number of genetic markers on T2D risk is of similar strength to (or even stronger than) that of some other known T2D risk factors. To use genetic predictors in practice, the effect of multiple markers needs to be summarized as a GRS. Our analysis shows that the strength of the GRS–disease association can vary greatly depending on how the GRS is calculated and how the SNPs (coded as effect allele count) are selected and weighted.
We propose a methodology called doubly weighting to increase the efficiency of the GRS and reduce the bias created by the “winner’s curse” (SNPs with an effect overestimated by chance in GWAS are oversampled in a list of top hits). A doubly weighted score, dGRSk, captures the effect of k “top” SNPs, but because it is uncertain whether a particular SNP belongs to the “top” set, probability weights for all available independent SNPs (7,502 in our case) are used to account for this uncertainty.
We found that when k ranges from 200 to 3,500 (5,200 in BMI-unadjusted models), the association between dGRSk and prevalent T2D in the Estonian Biobank cohort was clearly stronger than that of the best-fitting conventionally weighted GRS (Supplementary Table S2 online and Supplementary Figure S1 online). The best predictive accuracy was achieved with k = 1,000. This is an indicator that it is likely that the number of independent T2D-associated genomic loci is much larger than currently established by most recent GWAS studies.11,17 However, adding too many SNPs to the score will introduce too much random noise, diluting any possible signals. It is possible that an even larger-scale GWAS meta-analysis could reduce the amount of noise (smaller standard errors for parameter estimates); therefore, one could add more markers with weaker signals to the score, leading to further improvements in predictive accuracy.
Our analysis suggests that large-scale genome-wide studies have the potential to explain considerably more variability in the target phenotypes than currently estimated. We have shown that even a partial correction of some common biases in GRS (caused by the “winner’s curse”) may lead to a reduced amount of “missing heritability” (the proportion of genetic variability that is still unexplained by measurable genetic markers).
To demonstrate the potential of the GRS for practical use in personalized risk assessment, it is important to address the added value once the other known predictors are already accounted for. We assessed the effect of GRS when added to other predictors in a model for incident disease and found that the hazards of incident T2D in the top and bottom dGRS1000 quintiles differ by approximately 3.5 times. Although obesity is the strongest predictor for T2D, our data suggest that the effects of BMI and GRS on the T2D risk are additive; therefore, an individual with relatively high BMI but with low genetic risk can have the same overall T2D risk level as someone at high genetic risk but average BMI. Despite the observed significant association of the GRS with blood glucose, triglycerides, and HDL-cholesterol levels in T2D-free individuals, the effect of dGRS1000 on disease incidence remained practically unchanged after adjustment for these factors, thus suggesting the additive effect of genetic risk while accounting for these parameters.
In conclusion, our analysis suggests that the genetic risk factors for T2D that are captured in the dGRS1000 would improve the predictive accuracy when added to either a (nongenetic) T2D risk score (such as FINDRISC18) or a clinical risk assessment algorithm.
To quantify the potential improvement in risk assessment efficacy, ROC analysis (c-statistics) is an obvious methodological choice. Because age and BMI are already very strong predictors for T2D, the additional incremental value of GRS may seem small15 (as also shown in previous studies7). However, the clinically relevant quantity is not the relative contribution of GRS in comparison with age or BMI, for instance, but its ability to distinguish between different individual risk levels in subjects of the same age and BMI. Although the overall improvement in c-statistic for the incident T2D in individuals aged 35–79 is 1.2%, this increased to 2.1% in a subset with BMI of 25–35.
It has been debated whether genetic risk estimates based on DNA markers would add any meaningful information in cases where the family history of T2D is known, and it has been shown that complete family history provides better prediction than that achieved using 21 SNPs.19 However, the highly polygenic nature of T2D suggests that parent and offspring genetic risk may actually differ considerably because, on average, only half of the risk-affecting alleles are transmitted from each parent to the child. Moreover, in current family structures, it is increasingly difficult to obtain detailed information, if any, from both parents. Therefore, our study suggests that as the number of SNPs included in the GRS increases, the accuracy of GRS-based risk estimate improves in comparison to that based on family history.
One of the limitations of our study was that we have demonstrated the effect of GRS in one ethnically homogenous cohort only. We believe that the conclusions would be similar for other ethnicities as well, because trans-ethnic analyses of T2D have suggested that the effect sizes for common SNPs are relatively homogenous across ethnicities.17 However, one should be cautious about extrapolating our results to other populations without further validation.
We also see room for further improvements in the GRS methodology because our method offers only partial correction of the “winner’s curse” bias (SNPs with effects overestimated by chance still tend to obtain larger weights). One could also consider allowing for possible inclusion of correlated SNPs by taking the correlation structure (linkage disequilibrium between SNPs) into account.
In conclusion, we have shown that the proposed GRS is predictive for T2D risk in the long term (from birth on), as reflected by the sex- and age-adjusted comparisons of prevalent cases and controls. We have also demonstrated good predictive ability in the short term when other risk factors such as obesity, diet, physical activity, HDL cholesterol, and blood glucose level have already accounted for (analysis of T2D incidence). Although a longer follow-up time with a greater number of incident T2D cases would enable more precise effect estimation, our results indicate that a GRS with high accuracy, such as dGRS1000, would significantly improve the best existing risk assessment algorithms for T2D, encouraging its implementation in the practice of personalized medicine.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
EGCUT received financing from Estonian Research Council grants GP1GV9353 and IUT20-60, the Centre of Excellence for Genomics and Translational Medicine (GENTRANSMED), the University of Tartu (SP1GVARENG), the EU structural fund through the Archimedes Foundation, grant 3.2.1001.11-0033, and EU 2020 grant 692145 ePerMed.
Supplementary Material
References
- Novo Nordisk A/S. Changing diabetes barometer: diabetes—a chronic disease. http://www.changingdiabetesbarometer.com/about-diabetes.aspx. Accessed 1 January 2015.
- Mladovsky P, Allin S, Masseria C, et al. Health in the European Union: Trends and Analysis, 2009. http://www.euro.who.int/en/home. Accessed 10 January 2015.
- Tuomilehto J, Lindström J, Eriksson JG, et al.; Finnish Diabetes Prevention Study Group. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001;344:1343–1350. [DOI] [PubMed] [Google Scholar]
- Almgren P, Lehtovirta M, Isomaa B, et al.; Botnia Study Group. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia 2011;54:2811–2819. [DOI] [PubMed] [Google Scholar]
- Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance–a population-based twin study. Diabetologia 1999;42:139–145. [DOI] [PubMed] [Google Scholar]
- Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet 2013;14:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssenko V, Laakso M. Genetic screening for the risk of type 2 diabetes: worthless or valuable? Diabetes Care 2013;36 Suppl 2:S120–S126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng P, Zhao Y, Qian C, et al. Statistical analysis for genome-wide association study. J Biomed Res 2015;29:285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan D, Lander ES, Rosset S. Measuring missing heritability: inferring the contribution of common variants. Proc Natl Acad Sci USA 2014;111:E5272–E5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010;42:565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Voight BF, Teslovich TM, et al.; Wellcome Trust Case Control Consortium; Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing, 2008. http://www.r-project.org.Accessed 9 December, 2015.
- Cox DR. Tests of Separate Families of Hypotheses. Proc Fourth Berkeley Symp Math Stat Probab 1961;1:105–123. [Google Scholar]
- Pencina MJ, D’Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med 2011;30:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmud PJ, Cooper JA, Morris RW, et al.; UCLEB Consortium. Sixty-five common genetic variants and prediction of type 2 diabetes. Diabetes 2015;64:1830–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Go MJ, Zhang W, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 2014;46:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström J, Tuomilehto J. The diabetes risk score: a practical tool to predict type 2 diabetes risk. Diabetes Care 2003;26:725–731. [DOI] [PubMed] [Google Scholar]
- Do CB, Hinds DA, Francke U, Eriksson N. Comparison of family history and SNPs for predicting risk of complex disease. PLoS Genet 2012;8:e1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.