

Abstract
Fetal exposure to drugs cannot be readily estimated from single time point cord blood sampling at the time of delivery. Therefore, we developed a physiologically based pharmacokinetic (PBPK) model to estimate fetal drug exposure throughout pregnancy. In this study, we report verification of this novel maternal-fetal PBPK (m-f-PBPK) model for drugs that passively diffuse across the placenta and are not metabolized/transported there. Our recently built m-f-PBPK model was populated with gestational age-dependent changes in maternal drug disposition and maternal-fetal physiology. Using midazolam as an in vivo calibrator, the transplacental passive diffusion clearance of theophylline and zidovudine was first estimated. Then, for verification, the predicted maternal plasma (MP) and umbilical venous (UV) plasma drug concentrations by our m-f-PBPK were compared against those observed at term. Overall, our m-f-PBPK model well predicted the maternal and fetal exposure to the two verification drugs, theophylline and zidovudine, at term, across a range of dosing regimens, with nearly all observed MP and UV plasma drug concentrations falling within the 90% prediction interval [i.e., 5th–95th percentile range of a virtual pregnant population (n = 100)]. Prediction precision and bias of theophylline MP and UV were 14.5% and 12.4%, and 9.4% and 7.5%, respectively. Furthermore, for zidovudine, after the exclusion of one unexpectedly low MP concentration, prediction precision and bias for MP and UV were 50.3% and 30.2, and 28.3% and 15.0%, respectively. This m-f-PBPK should be useful to predict fetal exposure to drugs, throughout pregnancy, for drugs that passively diffuse across the placenta.
Introduction
Medication use during pregnancy is remarkably common. The average number of over-the-counter and prescription drugs used by women during pregnancy increased from 2.5 in 1976–1978 to 4.2 in 2006–2008 in the United States alone (Mitchell et al., 2011). The same study also revealed that, over the same period, use of such drugs during the first trimester increased from 1.6 to 2.6. The above statistics are not surprising as pregnant women need to be treated for many medical conditions, either pre-existing [e.g., epilepsy, asthma, human immunodeficiency virus (HIV) infection] or pregnancy-induced (e.g., gestational hypertension, diabetes, pre-eclampsia). In addition, the fetus is sometimes the therapeutic target, for example, to prevent vertical transmission of HIV (e.g., zidovudine, lamivudine), to treat fetal supraventricular tachycardia (e.g., digoxin), or to prevent fetal respiratory distress syndrome (e.g., dexamethasone) (Evans et al., 1993).
Regardless of whether the fetus is the intended target of pharmacotherapy during pregnancy, the fetus is de facto exposed to all drugs taken by the mother. Clearly, quantitative assessment of fetal exposure to drugs, especially during early gestation when the fetus is more prone to teratogenic effects (http://www.columbia.edu/itc/hs/medical/humandev/2004/Chpt23-Teratogens.pdf), is important from both efficacy and toxicity standpoint. However, fetal drug exposure cannot be ethically studied before the time of delivery, when a single time cord blood sample [usually umbilical venous (UV) plasma] can be safely obtained. As we have shown before (submitted manuscript), this UV plasma concentration, except at steady state after maternal drug infusion, does not provide information on fetal drug exposure, a determinant of drug efficacy and/or toxicity. Furthermore, these term or near-term data cannot be readily extrapolated to early gestation. Therefore, there is a pressing need to develop other in silico methods to predict fetal drug exposure, such as physiologically based pharmacokinetic (PBPK) modeling and simulation approaches.
Recently, we expanded our previously verified maternal PBPK model to include the fetus [maternal-fetal (m-f)-PBPK model] (submitted manuscript). The placenta, amniotic fluid, and fetal organs important for drug disposition, as well as the gestational age-dependent fetal physiologic changes (when available), were included in this m-f-PBPK model. Once developed, all models need to be verified. However, in this case, the only data available for verification are the UV and maternal plasma (MP) drug concentrations, often obtained simultaneously, at the time of birth. Therefore, as a first step, the primary objective of this study was to verify our novel m-f-PBPK model for drugs that cross the placenta predominantly by passive diffusion and for which UV and MP concentrations at the time of delivery are available in the literature (i.e., theophylline and zidovudine).
Materials and Methods
The General Maternal-Fetal PBPK Model Structure and Key Assumptions.
The general m-f-PBPK structure (Supplemental Fig. 1b) and the key assumptions made in constructing the model have been described in detail (submitted manuscript). In brief, a fetal PBPK model was developed to replace the lumped, noneliminating placental-fetal unit in our verified maternal PBPK model (Ke et al., 2012, 2013, 2014). The resulting m-f-PBPK contained organs that are relevant to fetal disposition of pharmaceutical agents, including those involved in drug disposition (e.g., fetal liver, fetal kidneys) and in drug efficacy/toxicity (e.g., fetal brain). The model also contained compartments representing the placenta and the amniotic fluid. The ordinary differential equations defining mass balance in the maternal PBPK have been described previously (Gaohua et al., 2012; Ke et al., 2012), whereas those describing the fetal PBPK are provided in Supplemental Material. Briefly, all tissues except the placenta were regarded as well-stirred tissues; that is, the unbound tissue drug concentration is in instant equilibrium with the unbound drug concentration in the emergent venous blood. The placenta was modeled as a permeability-limited tissue, and therefore it was subdivided into maternal-placental blood, placenta tissue, and fetal-placental blood compartments. Only the unbound, unionized fraction of drug can passively diffuse across the placenta. The bidirectional unbound maternal-placental and fetal-placental transplacental passive diffusion clearances (CLPD,u) across the placenta were assumed to be equal (Tuntland et al., 1999). Our model has the capability of including placental transport and fetoplacental metabolism of drugs when quantitative data on transporters and metabolic enzymes become available. Because this fetal PBPK does not contain an umbilical vein compartment per se, the predicted fetal plasma drug concentration in the central venous blood compartment was assumed to represent that in umbilical vein.
Known gestational age-dependent changes in maternal and fetal physiology (e.g., blood flows, organ volumes, etc.) from recent literature meta-analyses were incorporated into the m-f-PBPK (Abduljalil et al., 2012) (submitted manuscript). The change in drug unbound fraction in plasma (fu,p) was assumed to result from altered serum albumin concentration during pregnancy and was accounted for, as described previously (Ke et al., 2012). Maternal hepatic 3A and 1A2 activity was assumed to increase by 99% [as measured by 1′-hydroxymidazolam formation clearance (Hebert et al., 2008)] and decrease by 65% [as indicated by salivary caffeine clearance (Tracy et al., 2005)] during the third trimester (T3), respectively. Current clinical data suggest that maternal UDP-glucuronosyltransferase (UGT)2B7 activity is unchanged during pregnancy (Anderson, 2005; Tasnif et al., 2016). Therefore, we assumed that maternal hepatic or extrahepatic UGT2B7 activity does not alter during pregnancy. Maternal renal glomerular filtration rate was assumed to increase by 33% at term [gestational week (GW) 40] (Abduljalil et al., 2012). For zidovudine, its renal net secretion clearance in the mother was assumed to be unaltered during pregnancy.
General Workflow of PBPK Model Development and Model Verification Criterion.
Drug-specific parameters of midazolam and theophylline in nonpregnant subjects (Table 1) were obtained from our previous publications to populate their respective m-f-PBPK models (Ke et al., 2012, 2013), whereas those of zidovudine were first refined based on the Simcyp Simulator Version 14 (Simcyp, A Certara Company, Sheffield, UK) compound library using our previously published approach (Ke et al., 2014). Briefly, refinements of zidovudine drug-specific parameters were made if the predicted zidovudine population mean plasma concentration-time (C-T) curve in nonpregnant population using our MATLAB 13-compartment PBPK model (Supplemental Fig. 1a) significantly deviated from that observed. The refined zidovudine pharmacokinetics (PK) parameters were subsequently used to populate the zidovudine m-f-PBPK model. In addition, the available interindividual variability in physiologic parameters and in drug-specific absorption, distribution, metabolism, and elimination processes in healthy volunteers (for zidovudine) or in the pregnant women (for midazolam, theophylline, and zidovudine at GW 0) was predicted within the Simcyp Simulator. In brief, a compound profile was first constructed for each of these drugs within the Simcyp Simulator using their respective drug-specific parameters. Interindividual variabilities associated with these parameters were those specified by the Simcyp Simulator. Then, the above predicted absorption, distribution, metabolism, and elimination characteristics for each virtual individual were used for the simulations conducted by the MATLAB 13-compartment PBPK model (for zidovudine only) or by the m-f-PBPK model (for midazolam, theophylline, and zidovudine). For the pregnant population, the reported gestational age-dependent changes in maternal and fetal physiology (Abduljalil et al., 2012) (submitted manuscript) and the changes in maternal hepatic enzyme activity based on phenotyping studies conducted in pregnant women were taken into account, as described above.
TABLE 1.
Summary of midazolam, theophylline, and zidovudine drug-dependent parameters
| Parameter | Midazolam Value | Methods/ Reference | Theophylline Value | Methods/ Reference | Zidovudine Value | Methods/Reference |
|---|---|---|---|---|---|---|
| Molecular weight | 325.8 | Librarya | 180.2 | Librarya | 267.2 | Librarya |
| Log Po:w | 3.13 | Optimizedb | −0.02 | Librarya | 0.05 | Librarya |
| pKa | 10.95,6.2 | Librarya | 8.8,0.99 | Librarya | 9.70 | Librarya |
| B/P ratio | 0.66 | Librarya | 0.82 | Librarya | 0.91 | Librarya |
| fu,p* | 0.032 | Librarya | 0.58 | Librarya | 0.80 | Librarya |
| Fa | 0.88 | Librarya | 0.97 | Librarya | 0.83 | Predicted by ADAM model |
| ka (h−1) | 4.0 | Optimizedc | 1.0 | Reportede | 4.05 | Reportedf |
| Fg | 0.58 | Librarya | 1.0 | Reportedd | 1 | Assumedg |
| Vss (L/Kg) | 1.1 | Reportedd | 0.39 | Reportedd | 1.10 | Optimizedh |
| CLiv (L/h) | 23.0 | Librarya | 3.0 | Reportedd | 91.0 | Reportedi |
| CLr (L/h) | 0.085 | Librarya | 0.45 | Reportedd | 15.5 | Libraryj |
| CLhep,int,u (L/h) | 1672.3 | Librarya | 4.60 | Reportedd | 30.9 | Calculatedk |
| fm and fe | fm,3A = 92%, | Reportedd | fm,1A2 = 68%, | Reportedd | fm,UGT2B7 = 67%, | Reportedl |
| fe ≈ 0% | fm,3A = 7%, | fe = 17% | ||||
| fm,2E1 = 10%, | ||||||
| fe = 15% |
Midazolam is a weak base (Andersin, 1991), whereas zidovudine is a weak acid (Gallicano, 2000). In contrast, theophylline is neutral (Hardman, 1962).
Refers to the SimCYP Simulator compound library (version 14).
Previously optimized and validated to match the predicted volume of distribution at steady state (Vss) to the reported Vss value of 1.10 L/kg in the literature (Ke et al., 2012).
Optimized based on sensitivity analysis to match reported absorption in pregnant subjects (Kanto et al., 1983).
Validated literature value used in our pregnancy PBPK model (Ke et al., 2012, 2013).
Phoenix estimate from reported oral absorption data in healthy volunteers (Aslaksen et al., 1981).
Phoenix estimate from reported oral absorption data in nonpregnant subjects (Klecker et al., 1987).
No report on zidovudine Fg is available. Zidovudine Fg was assumed 1 because human intestinal microsomes showed negligible UGT2B7 activity measured by two UGT2B7 probes (diclofenac and gemfibrozil) (Furukawa et al., 2014).
The reported average zidovudine Vss is 1.4 ± 0.4 L/kg (Collins and Unadkat, 1989). This Vss was optimized through manual sensitivity analysis in the range of 0.8–1.6 L/kg to match the predicted peak plasma concentration (Cmax) to the reported Cmax following i.v. infusion in nonpregnant population (Klecker et al., 1987; Cload, 1989).
Calculated based on the reported average i.v. clearance of 1.3 L/h/kg assuming 70 kg body weight.
Reported zidovudine fraction excreted in the urine (fe) ranges from 14% to 20%. The SimCYP Simulator compound library value of 15.5L/h (fe = 17%) was used.
Back calculation from well-stirred liver model using hepatic blood flow of 90 L/h assuming hepatic clearance of 20.7 L/h.
Estimated from urinary data (Cload, 1989; Stagg et al., 1992).
For each of the above test compounds, MP and UV drug C-T profiles were simulated in a virtual population consisting of 100 pregnant women at GW 40. The model was deemed to have met our verification criterion if the observed individual UV and MP drug concentrations (except midazolam MP concentrations, in which the mean values from 8 subjects were used) at the time of delivery (extracted or digitized from literature using MATLAB Grabit m.file; available free online at http://www.mathworks.com/matlabcentral) fell within the 90% prediction interval (5th– 95th percentile range of the virtual population) calculated based on the interindividual variability in the maternal PK. Additionally, model prediction precision and prediction bias were evaluated by calculating the mean absolute prediction error and the mean prediction error, where Pred and Obs denote the predicted and observed values, respectively (Sheiner and Beal, 1985).
Estimation of In Vivo Transplacental Passive Diffusion Clearance of Drugs.
Because data on the in vivo transplacental passive diffusion clearance (CLPD) of drugs are not available in the literature, we chose midazolam as an in vivo calibrator to estimate CLPD of theophylline and zidovudine (the same approach can be used for any drug). First, we optimized the in vivo CLPD,u of midazolam (see below). Then, the CLPD,u of drug X (theophylline or zidovudine) was estimated by scaling CLPD,u of midazolam using eq. 1.
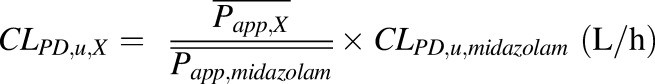 |
(1) |
where CLPD,u,midazolam is the optimized in vivo CLPD,u (L/h) of midazolam (see below), and  and
and 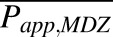 are the mean apparent membrane permeability (Papp) values (nm × s−1) of drug X and midazolam, respectively. Reported Papp values of model drugs were collected from multiple sources in the literature (Table 2). The average Papp values of midazolam, theophylline, and zidovudine used were obtained by computing the mean values of Papp reported by five, five, and seven independent studies, respectively. For physiologic relevance and to avoid the confounding factor of binding, we only included studies conducted in established epithelial cell lines that form tight junctions between cells in monolayer cultures (i.e., Madin-Darby canine kidney and Caco-2) in the absence of serum/binding proteins.
are the mean apparent membrane permeability (Papp) values (nm × s−1) of drug X and midazolam, respectively. Reported Papp values of model drugs were collected from multiple sources in the literature (Table 2). The average Papp values of midazolam, theophylline, and zidovudine used were obtained by computing the mean values of Papp reported by five, five, and seven independent studies, respectively. For physiologic relevance and to avoid the confounding factor of binding, we only included studies conducted in established epithelial cell lines that form tight junctions between cells in monolayer cultures (i.e., Madin-Darby canine kidney and Caco-2) in the absence of serum/binding proteins.
TABLE 2.
Papp values from in vitro studies and the estimated CLPD,u
The predicted theophylline maternal fu,p is 0.66 at term. The predicted zidovudine maternal fu,p is ∼0.80 at term. The estimated CLPD,u values were based on mean Papp from in vitro reports.
| Drug | Papp Value (nm × s−1) | CLPD,u (L/h) | Resources | |||
|---|---|---|---|---|---|---|
| Mean | S.D. | Median | Range | |||
| Midazolam | 489.9 | 158.7 | 490.0 | 320–699 | 500.0 | (Yamashita et al., 2000; Mahar Doan et al., 2002; Taub et al., 2002; Tolle-Sander et al., 2003; Gertz et al., 2010) |
| Theophylline | 335.5 | 162.2 | 260.0 | 231–620 | 342.4 | (Yamashita et al., 2000; Masungi et al., 2004; Collett et al., 2008; Teksin et al., 2010; Di et al., 2011) |
| Zidovudine | 212.4 | 217.2 | 69.3 | 31.8–543.3 | 216.8 | (Yazdanian et al., 1998; Irvine et al., 1999; de Souza et al., 2009) |
Midazolam m-f-PBPK Model.
First, the predicted midazolam plasma C-T profile, following a single 15 mg oral dose in 13 pregnant women (GW = 40) proceeding elective cesarean section surgery, was compared against the observed data (Kanto et al., 1983). Midazolam drug-specific parameters, previously validated, are outlined in Table 1 (Ke et al., 2012). CYP3A metabolism occurs in maternal gut, liver, and fetal liver. The observed 99% increase in CYP3A activity was confined to maternal liver, as we have previously shown that only maternal hepatic CYP3A activity appears to be induced during pregnancy (Zhang et al., 2008; Ke et al., 2012). To match the observed midazolam maternal absorption profile, a lag time of 0.1 hour was introduced and a first-order absorption rate constant of 4.0 hour−1 was chosen through sensitivity analysis (data not shown). Because fetal liver predominantly expresses CYP3A7 (Shuster et al., 2014), fetal hepatic intrinsic clearance of midazolam (CLf,hep,int) was estimated using eq. 2.
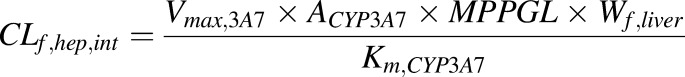 |
(2) |
Where Vmax,3A7 and Km,CYP3A7 are the maximal velocity and Michaelis–Menten constant determined in recombinant CYP3A7, ACYP3A7 is CYP3A7 abundance per mg microsomal protein, MPPGL is the fetal hepatic microsomal protein concentration per gram fetal liver, and Wf,liver is fetal liver weight at term. Reported in vitro Vmax/Km value of midazolam hydroxylation in recombinant CYP3A7 is 2.1 mL/h/nmol P450 (Williams et al., 2002), and it is estimated that fetal liver microsomes contain 0.3 nmol P450 proteins per mg protein (Barter et al., 2007). Fetal MMPGL is 26 mg/g liver (Barter et al., 2008), and the average fetal liver at term weighs ∼130 g (Abduljalil et al., 2012). Therefore, fetal hepatic CLf,hep,int mediated by CYP3A7 was estimated as 2.13 L/h. The CLPD,u value of midazolam was optimized through sensitivity analysis. Briefly, the magnitude of midazolam CLPD,u was varied to reduce the residual sum of squares between the predicated and observed fetal C-T profile [residual sum of squares; calculated as 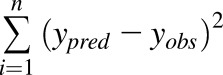 , where ypred and yobs refer to the predicted and observed fetal plasma midazolam concentrations, respectively]. The observed fetal C-T profile was created by pooling the reported midazolam UV plasma concentrations from seven newborns in the same study (Kanto et al., 1983).
, where ypred and yobs refer to the predicted and observed fetal plasma midazolam concentrations, respectively]. The observed fetal C-T profile was created by pooling the reported midazolam UV plasma concentrations from seven newborns in the same study (Kanto et al., 1983).
Theophylline m-f-PBPK Model.
Maternal theophylline drug-specific parameters were those previously published (Table 1) (Ke et al., 2013). Because the major placental CYP1A isoform, CYP1A1, has negligible contribution to theophylline metabolism (Ha et al., 1995) and no report on CYP1A2 expression in the term placenta or fetal liver is available, the placenta and fetal liver were considered as nonmetabolizing organs for theophylline. Using the estimated CLPD,u,theophylline (eq. 1), maternal and fetal theophylline C-T profiles following multiple oral doses of theophylline were simulated and compared with the observed data using the verification criterion described above. The observed theophylline data were from a study in which 10 asthmatic women with normal pregnancies were administered 160 mg theophylline (in the form of aminophylline) every 6 hours for 30 hours prior to delivery (Ron et al., 1984). The observed maternal and fetal C-T profiles were created by pooling single time point MP and UV plasma theophylline concentrations from 10 maternal-fetal pairs.
Zidovudine m-f-PBPK Model.
In nonpregnant adults following an i.v. dose, only ∼17% of zidovudine is excreted unchanged in the urine, whereas 67% of the i.v. dose is recovered in the urine as 5′-O-glucuronide via UGT2B7-mediated glucuronidation (Cload, 1989; Stagg et al., 1992). Other identified metabolites include its active triphosphate metabolite formed via sequential intracellular phosphorylation (Veal et al., 1994), 3′-amino-3′-deoxythymidine, and 3′-amino-3′-deoxythymidine glucuronide (Stagg et al., 1992). Average zidovudine i.v. plasma clearance is 91 L/h normalized to 70 kg body weight (Collins and Unadkat, 1989). Thus, plasma zidovudine glucuronidation clearance is estimated as 61 L/h. These data, in conjunction with the observed zidovudine absolute bioavailability (F) of ∼63% (Klecker et al., 1987), suggest considerable extrahepatic zidovudine metabolism. However, although UGT2B7 is expressed in the gut, liver, and kidneys (Ohno and Nakajin, 2009), investigations on the extrahepatic metabolism of zidovudine revealed that zidovudine is not glucuronidated by gut microsomes and that renal glucuronidation is minimal (Cretton et al., 1991; Howe et al., 1992; Knights et al., 2016). Moreover, published in vitro-to-in vivo approach based on human liver microsomal data substantially underpredicted zidovudine glucuronidation clearance by a factor of 30.5 (Kilford et al., 2009). Based on these analyses, we speculate that there are unidentified extrahepatic, nonrenal pathways responsible for zidovudine metabolism (glucuronidation and nonglucuronidation) in vivo. Therefore, to recapitulate zidovudine i.v. clearance, zidovudine hepatic unbound intrinsic clearance (CLhep,int,u) was calculated using the well-stirred liver model
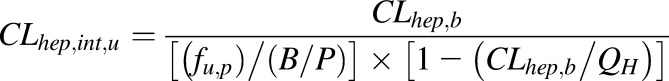 |
(3) |
Where fup is the fraction unbound in the plasma, CLhep,b is hepatic blood clearance 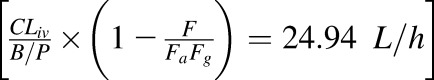 , B/P is the blood to plasma concentration ratio, and QH is the hepatic blood flow (90 L/h). A systemic plasma metabolic clearance of 52.8 L/h (
, B/P is the blood to plasma concentration ratio, and QH is the hepatic blood flow (90 L/h). A systemic plasma metabolic clearance of 52.8 L/h ( was assigned to the unidentified extrahepatic zidovudine metabolic pathways. Using the above detailed PK parameters (Table 1), the predicted zidovudine plasma C-T curves using the 13-compartment PBPK model were compared with those observed following 1-hour i.v. infusion of 2.5 mg/kg (Figs. 1, 2) or two different single oral doses (200 and 300 mg) in nonpregnant asymptomatic HIV-infected male patients with normal hepatic and renal functions (Cload, 1989; Gallicano et al., 1993; Anderson et al., 2000).
was assigned to the unidentified extrahepatic zidovudine metabolic pathways. Using the above detailed PK parameters (Table 1), the predicted zidovudine plasma C-T curves using the 13-compartment PBPK model were compared with those observed following 1-hour i.v. infusion of 2.5 mg/kg (Figs. 1, 2) or two different single oral doses (200 and 300 mg) in nonpregnant asymptomatic HIV-infected male patients with normal hepatic and renal functions (Cload, 1989; Gallicano et al., 1993; Anderson et al., 2000).
Fig. 1.

Predicted population mean (black solid lines) and observed (open circles) MP (A) and UV plasma (B) midazolam C-T profiles following a single 15 mg oral dose at term (GW 40) (Kanto et al., 1983). In (A), the predicted MP C-T profile is overlaid with the observed maternal plasma data (mean ± S.D.; n = 8). In (B), each observed UV data point was derived from a single maternal-fetal pair from another subset of subjects from the same study (n = 7). The observed mean midazolam MP concentrations at various time points fell within the 90% prediction interval (5th–95th percentile boundaries; lower and upper gray dashed lines, respectively) for a virtual population consisting of 100 maternal–fetal pairs (A). Consequently, all of the observed individual UV plasma concentrations were within the 90% prediction interval (B). Insets are the same data plotted to highlight early time points.
Fig. 2.

Predicted population mean (black solid lines) and observed (open circles) MP (A) and UV plasma (B) theophylline C-T profiles following five doses of 160 mg oral theophylline every 6 hours at term (GW 40) (Ron et al., 1984). Each pair of observed data (MP and UV) was derived from a single maternal-fetal pair (n = 10). All observed MP drug concentrations, except one data point, and subsequently all observed UV data points, fell within the 90% prediction interval (5th–95th percentile boundaries; lower and upper gray dashed lines, respectively), for a virtual population consisting of 100 maternal-fetal pairs.
Term human placenta perfusion studies have demonstrated that placental glucuronidation of zidovudine is nonexistent (Liebes et al., 1990; Schenker et al., 1990; Bawdon et al., 1992) or negligible (Collier et al., 2002). The information on fetal glucuronidation of zidovudine is currently unavailable, but is likely to be negligible given the size of the fetal liver (∼130 g at GW 40 versus 1.5 kg in adults). Therefore, no irreversible loss of zidovudine was assumed to be present in the fetoplacental unit. Using the estimated zidovudine CLPD,u, the predicted MP and UV C-T profiles of zidovudine using the m-f-PBPK model were compared against the observed zidovudine plasma concentrations obtained at labor (O'Sullivan et al., 1993) following a single 200 mg oral dose (n = 8) or multiple 200 mg oral dose preceding a 1-hour i.v. infusion (140 mg/h) (n = 7). In the latter study, the MP and UV plasma concentrations (observed data) were simultaneously obtained from seven maternal-fetal pairs at the time of birth.
Results
Estimated In Vivo Transplacental Passive Diffusion Clearance of Midazolam.
Incorporation of a lag time and optimization of first-order absorption rate constant resulted in excellent agreement between the predicted and the observed maternal C-T profiles (Fig. 1A). Subsequent sensitivity analysis on midazolam CLPD,u demonstrated that although fetal exposure to midazolam was relatively insensitive to changes in CLPD,u (Supplemental Fig. 2), a value of 500 L/h (term fu,p = 0.045; CLPD of 22.7 L/h) best described the fetal exposure to midazolam (minimum residual sum of squares) (Supplemental Table 1). The resulting UV plasma concentrations, except for one data point, were in close agreement with the observed UV plasma concentrations, falling within the 90% prediction interval (Fig. 1B). Additionally, using midazolam as the calibrator, the resultant CLPD,u for theophylline and zidovudine were 342.4 L/h and 216.8 L/h, respectively (Table 2).
Theophylline.
Using the estimated theophylline CLPD,u of 342.4 L/h (term fu,p = 0.66 ; CLPD = 226.4L/h), the predicted MP (Fig. 2A) and UV (Fig. 2B) drug concentrations were in good agreement with the observed data. All predictions (except a single MP concentration) met our verification criterion (i.e., observed plasma concentration falls within the 90% prediction interval). Model prediction precision and bias for MP and UV concentrations were 14.5% and 12.4% and 9.4% and 7.8%, respectively.
Zidovudine.
First, the predicted zidovudine mean plasma C-T profile in nonpregnant population (n = 100) was compared against the observed zidovudine mean plasma C-T profiles following various dosing regimens (Fig. 3). Predicted population mean data matched the observed data with precision of <40% and bias ranging from −30% to 9%. After model verification in nonpregnant population, zidovudine drug-specific parameters were incorporated into our zidovudine m-f-PBPK model. Using the estimated CLPD,u of 216.8 L/h (term fu,p = 0.8; CLPD = 172.6 L/h; Table 2), the predicted MP zidovudine plasma before the onset of labor passed our prediction criterion (Fig. 4A), whereas the majority of maternal-fetal zidovudine plasma concentrations obtained during delivery fell within the 90% prediction interval. In the latter scenario, one of seven MP (Fig. 4B) and two of seven UV (Fig. 4C) plasma concentrations fell outside this 90% prediction interval. Prediction precision and bias for MP and UV plasma drug concentrations were 135.2% and 118.4% and 121.4% and 110.3%, respectively. However, these larger precision and bias data were largely due to the unexpected low MP concentration of 0.17 μg/mL at 43.3 hours (0.88 hour post the initiation of 1-hour i.v. infusion to the mother) and consequently low UV concentration. When this point was excluded, the resultant prediction precision and bias for MP and UV drug concentrations reduced considerably to 50.3% and 30.2%, and 28.3% and 15.0%, respectively.
Fig. 3.

Predicted population mean (black solid lines) and observed (open circles) plasma zidovudine C-T profiles following a 1-hour 2.5 mg/kg i.v. infusion dose (A) (Cload, 1989) (n = 8) or a single 200 mg oral dose (B) (Gallicano et al., 1993) (n = 10) or a single 300 mg oral dose (C) (Anderson et al., 2000) (n = 4) in nonpregnant population. All observed plasma drug concentrations (mean ± S.D.), except one terminal-phase plasma concentration (A), in HIV-infected asymptomatic male volunteers fell within the 90% prediction interval (5th–95th percentile boundaries; lower and upper gray dashed lines, respectively) from a virtual population consisting of 100 male subjects.
Fig. 4.

Predicted population mean (black solid lines) and observed (open circles) MP (A and B) and UV plasma (C) zidovudine C-T profiles in HIV-infected, asymptomatic women with uncomplicated pregnancies (O’Sullivan et al., 1993). A 200 mg oral dose of zidovudine (5 times per day) was commenced prior to onset of labor (A) (GW 40, n = 8). Then, during labor, 4 hours after last dose, 140 mg zidovudine was i.v. infused over 1-hour period to the same volunteers (B and C) (n = 7). In (A), all of the observed mean maternal plasma zidovudine concentrations fell within the 90% prediction interval (5th and 95th percentile range; lower and upper gray dashed lines, respectively) for a virtual population consisting of 100 maternal-fetal pairs. In (B) and (C), each data point (open circles) represents an observed single time point zidovudine concentration from paired MP (B) or UV plasma (C) samples. Six of seven observed MP concentrations, and five of seven UV plasma concentrations, fell within the 90% prediction interval. Insets are the same data replotted with a different time scale to illustrate the dosing regimen.
Discussion
In the current work, we have verified a novel m-f-PBPK model using drugs that passively permeate the placenta and are not known or expected to be metabolized there. Our fetal PBPK model was constructed to be consistent with the distinctive fetal vascular physiology and to allow future incorporation of transport and metabolism within the placenta. The model contains a three-compartment placenta consisting of maternal-placental blood, placental tissue, and fetal-placental blood. The model also accounts for the unique fetal hepatic blood supply as the fetal liver is primarily perfused by UV blood flow (Supplemental Fig. 1b). However, a significant portion of the latter (∼30–70%) is shunted to the fetal systemic circulation via the ductus venosus. Although this is not the first report of a fetal PBPK model (Yoon et al., 2011; Loccisano et al., 2013; De Sousa Mendes et al., 2016), to our best knowledge it is the first full m-f-PBPK model that: 1) features fetal physiologic aspects that are relevant to pharmaceutical drugs; 2) systematically incorporates the gestational age-dependent changes in maternal drug disposition and maternal-fetal physiology; 3) accounts for the interindividual variability in maternal plasma C-T profile; and 4) well predicts the systemic exposure of test pharmaceutical drugs in maternal-fetal pairs at term.
Crucial for predicting the fetal exposure of passive diffusion drugs is the magnitude of in vivo passive diffusion clearance across the placenta. In our model, the mass transfer of a passive diffusion drug from the mother to her fetus is described by equal bidirectional maternal-placental and placental-fetal CLPD,u (Tuntland et al., 1999). In essence, the rate of drug transfer across the placenta is rate-determined by CLPD,u (after accounting for binding) or placental blood flow, whichever is lower. In theory, CLPD,u equals the intrinsic permeability–surface area product. At a given gestational age, the magnitude of this CLPD,u should be directly proportional to its intrinsic permeability (i.e., permeability after adjusting for plasma protein binding) across the syncytiotrophoblast that separates the maternal and fetal circulation. Of note, the differing longitudinal changes in plasma drug-binding protein concentrations across the placenta can have a significant impact on the transplacental passage of drugs (Hill and Abramson, 1988; McNamara and Alcorn, 2002) and have been accounted for in our m-f-PBPK model. Despite its importance, in vitro-to-in vivo of passive diffusion clearance across the placenta remains a challenge. Immortalized cell lines of human placenta origin, such as BeWo and Jar, cannot form tight junctions, and therefore are poorly suited to estimate placental drug diffusion (Kitano et al., 2004). Although ex vivo dually perfused human placentae may represent the most physiologically relevant system, only theophylline and zidovudine have been studied (Liebes et al., 1990; Schenker et al., 1990; Omarini et al., 1992; Dancis et al., 1993). Furthermore, none of these studies provide sufficient data for whole-organ scale-up of CLPD. Therefore, we hypothesized that for a passive diffusion drug, the in vivo CLPD,u of the drug can be predicted by calibrating its in vitro permeability against the positive control, midazolam. The magnitude of the in vivo CLPD,u of a new drug entity can then be calculated, assuming that it will be proportional to its passive diffusion permeability relative to that of midazolam in epithelial cell lines that form tight junctions (eq. 2).
Our in vivo calibrator midazolam [Biopharmaceutics Classification System class I (Benet, 2010)] crosses the placenta predominantly via passive diffusion. Due to its high passive membrane permeability, the contribution of P-glycoprotein toward midazolam tissue distribution is negligible (Tolle-Sander et al., 2003; Doran et al., 2005). Among the three test compounds, only midazolam maternal population average plasma concentrations (n = 8) have been reported at term. Therefore, midazolam was chosen as our in vivo calibrator to estimate the in vivo placental diffusion clearance of both theophylline and zidovudine (see next paragraph).
Consistent with our previous findings, a 99% induction in hepatic CYP3A alone was sufficient to explain the clinically observed changes in maternal midazolam disposition during T3 (Ke et al., 2012). As expected, midazolam readily crosses the placenta. The optimized CLPD resulted in all observed UV midazolam plasma concentrations falling within the 90% prediction interval, suggesting ∼50% extraction ratio by the placenta and a blood flow–limited extraction by the fetal placental flow (CLPD of 22.7 L/h versus term placental and UV blood flows of ∼50 L/h and ∼20 L/h, respectively) (Abduljalil et al., 2012). The first verification drug, theophylline, is also a Biopharmaceutics Classification System class I drug (Benet, 2010). To date, only OAT2 has been indicated in theophylline tissue uptake, but this transporter is absent in human placenta (Kobayashi et al., 2005; Mao et al., 2014). Theophylline is mainly cleared by CYP1A2 with minor contributions from CYP 3A and CYP2E1 as well as a small renal component (Ke et al., 2013). The incorporation of a 65% reduction in maternal hepatic CYP1A2 activity (Tracy et al., 2005) along with the 99% increase in CYP3A activity satisfactorily explained the observed maternal plasma concentrations (Fig. 2A). Using the predicted CLPD,u of 342.4 L/h, fetal exposure was well described (Fig. 2B). Of note, as a result of moderate protein binding (fu,p = 0.66 at term) and relatively high CLPD,u (342.4 L/h), the predicted transplacental passage of theophylline was blood flow limited.
The second verification drug, zidovudine, is a nucleoside reverse-transcriptase inhibitor and structure analog of thymidine. Although it has been shown to be transported in vitro by several transporters expressed in human placenta (e.g., P-glycoprotein, breast cancer resistance protein, multidrug resistance-associated protein 5, equilibratuve nucleoside transporter 2, and OAT4), several ex vivo placenta perfusion studies indicate that zidovudine crosses the placental via passive diffusion (Liebes et al., 1990; Schenker et al., 1990; Dancis et al., 1993). Zidovudine is mainly cleared by UGT2B7 in vivo with a renal clearance exceeding renal filtration. The activity of UGT2B7 is generally regarded not to be affected by pregnancy (Anderson, 2005) and is supported by our simulation. Several studies have shown that zidovudine is a substrate for human OATs (i.e., OAT1-4), all of which are expressed in the kidney (Takeda et al., 2002). Interestingly, although pregnancy may increase renal OAT1 activity [measured by the 55% increase in amoxicillin net renal secretion during T3, which may be attributed to enhanced renal OAT1 activity and/or reduced reabsorption (Andrew et al., 2007)], our simulations demonstrated that maternal physiologic changes along with the increased renal filtration clearance sufficiently described the maternal disposition of zidovudine (Fig. 4, A and B). Zidovudine demonstrates good permeability and was also estimated to have blood flow– limited distribution into the fetal compartment.
Overall, the m-f-PBPK model described well-predicted maternal and fetal exposure to the two verification drugs (zidovudine and theophylline) at term across a range of dosing regimens, with nearly all simulated plasma drug concentrations falling within the 90% prediction interval in both the mother and her fetus. Because these drugs passively diffuse across the placenta and are not significantly metabolized in the fetal liver or the placenta, the overall unbound fetal plasma area under the curve (AUC) is predicted to be equal to the unbound maternal plasma AUC. Due to the sparse fetal plasma drug concentration data available, the fetal AUC of the verification drugs could not be estimated. Therefore, our goal was to dynamically predict the fetal plasma drug concentrations available in the literature (i.e., predict the time-variant fetal plasma C-T curve). Such dynamic prediction is a true test of any m-f-PBPK model, including when the drugs are extensively transported or metabolized in the placenta. In the future, our aim is to incorporate these processes in our model when quantification data on expression of placental transporters and enzymes are available.
The application of PBPK models for predicting drug disposition in the coupled maternal-fetal pairs is still in its infancy. Therefore, as is the case with other PBPK models, our model has several limitations. First, although interindividual variability in maternal drug disposition, where available, was incorporated in our model, due to lack of data on variability of feto-placental parameters, such variability could not be incorporated in the model. This may underestimate the true variability in fetal exposure. Second, midazolam was not a sensitive calibrator and both theophylline and zidovudine demonstrated blood flow–limited passive placental diffusion clearance. Ideally, the passive diffusion clearance of test compounds should span a much wider range, including hydrophilic drugs that passively diffuse across the placenta with CLPD much lower than the placental blood flow. Unfortunately, coupled maternal-fetal PK data for such drugs are not available. Third, until placental transporters and metabolic enzyme expression are available, our model can be applied to only drugs that passively diffuse across the placenta and are not metabolized/transported there. To address this limitation, proteomics-based quantification of placental transporters and enzymes is currently underway in our laboratory. Fourth, although our model can predict fetal exposure to passive diffusion drugs across fetal developmental stages (GW 14-term), many fetal physiologic parameters are not available for the first half of pregnancy, and the fetal skin is not keratinized at <20 weeks of gestation (Polin et al., 2004), potentially enabling the bidirectional diffusion of drugs through the fetal skin to the amniotic fluid. Although the latter limitation can be overcome by including such a possibility in a future version of our model, the former issue reflects an inherent limitation of studies in this special population, that is, the difficulty of measuring physiologic parameters of the fetus at earlier gestational age. As a result, we have less confidence in predicting fetal drug exposure at <20 weeks of gestation.
The clinical implications of the current study relate to addressing an urgent need for an understudied and vulnerable population: quantitative assessment of fetal exposure to drugs in the maternal-fetal dyad. Drug use during pregnancy is a reality. When the perceived benefits outweigh risks, pharmacotherapy of pregnant women is initiated, in most cases, without prior knowledge on the maternal-fetal disposition of the drug. However, obtaining any fetal drug exposure data at term is fraught with logistical and ethical issues. As a result, there is a paucity of fetal exposure data in this population. As pointed out earlier, existing fetal drug exposure data are limited to term pregnancy. Furthermore, such data for early gestational age fetuses are virtually impossible to obtain, rendering fetuses orphan population with respect to drug exposure and drug efficacy/toxicity. Instead, the proposed PBPK model can provide information on fetal drug exposure based on sound physiologic data and modeling. Although data to verify our model for gestational ages other than term are not available, the term verification data presented above lend considerable confidence that our m-f-PBPK model can be used to a priori predict fetal exposure to drugs (that passively diffuse across the placenta) during pregnancy. When earlier gestational age data become available, our model can be verified for these gestational ages.
Acknowledgments
The authors thank Drs. Nina Isoherranen and Kazuya Ishida for insightful comments.
Abbreviations
- AUC
area under the curve
- C-T
concentration-time
- CLPD
transplacental passive diffusion clearance
- CLPD,u
unbound transplacental passive diffusion clearance
- fu,p
fraction unbound in plasma
- GW
gestational week
- HIV
human immunodeficiency virus
- m-f-PBPK
maternal-fetal PBPK
- MP
maternal plasma
- OAT
organic anion transporter
- Papp
apparent membrane permeability
- PBPK
physiologically based pharmacokinetic
- T3
third trimester
- UGT
UDP-glucuronosyltransferase
- UV
umbilical venous
Authorship Contributions
Participated in research design: Zhang, Unadkat.
Conducted experiments: Zhang.
Performed data analysis: Zhang.
Wrote or contributed to the writing of the manuscript: Zhang, Unadkat.
Footnotes
This work was supported by National Institutes of Health National Institute on Drug Abuse [Grant P01DA032507].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. (2012) Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet 51:365–396. [DOI] [PubMed] [Google Scholar]
- Andersin R. (1991) Solubility and acid-base behaviour of midazolam in media of different pH, studied by ultraviolet spectrophotometry with multicomponent software. J Pharm Biomed Anal 9:451–455. [DOI] [PubMed] [Google Scholar]
- Anderson GD. (2005) Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet 44:989–1008. [DOI] [PubMed] [Google Scholar]
- Anderson PL, Noormohamed SE, Henry K, Brundage RC, Balfour HH, Jr, Fletcher CV. (2000) Semen and serum pharmacokinetics of zidovudine and zidovudine-glucuronide in men with HIV-1 infection. Pharmacotherapy 20:917–922. [DOI] [PubMed] [Google Scholar]
- Andrew MA, Easterling TR, Carr DB, Shen D, Buchanan ML, Rutherford T, Bennett R, Vicini P, Hebert MF. (2007) Amoxicillin pharmacokinetics in pregnant women: modeling and simulations of dosage strategies. Clin Pharmacol Ther 81:547–556. [DOI] [PubMed] [Google Scholar]
- Aslaksen A, Bakke OM, Vigander T. (1981) Comparative pharmacokinetics of theophylline and aminophylline in man. Br J Clin Pharmacol 11:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter ZE, Bayliss MK, Beaune PH, Boobis AR, Carlile DJ, Edwards RJ, Houston JB, Lake BG, Lipscomb JC, Pelkonen OR, et al. (2007) Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr Drug Metab 8:33–45. [DOI] [PubMed] [Google Scholar]
- Barter ZE, Chowdry JE, Harlow JR, Snawder JE, Lipscomb JC, Rostami-Hodjegan A. (2008) Covariation of human microsomal protein per gram of liver with age: absence of influence of operator and sample storage may justify interlaboratory data pooling. Drug Metab Dispos 36:2405–2409. [DOI] [PubMed] [Google Scholar]
- Bawdon RE, Sobhi S, Dax J. (1992) The transfer of anti-human immunodeficiency virus nucleoside compounds by the term human placenta. Am J Obstet Gynecol 167:1570–1574. [DOI] [PubMed] [Google Scholar]
- Benet LZ. (2010) Predicting drug disposition via application of a Biopharmaceutics Drug Disposition Classification System. Basic Clin Pharmacol Toxicol 106:162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cload PA. (1989) A review of the pharmacokinetics of zidovudine in man. J Infect 18 (Suppl 1):15–21. [DOI] [PubMed] [Google Scholar]
- Collett A, Stephens RH, Harwood MD, Humphrey M, Dallman L, Bennett J, Davis J, Carlson GL, Warhurst G. (2008) Investigation of regional mechanisms responsible for poor oral absorption in humans of a modified release preparation of the alpha-adrenoreceptor antagonist, 4-amino-6,7-dimethoxy-2-(5-methanesulfonamido-1,2,3,4 tetrahydroisoquinol-2-yl)-5-(2-pyridyl)quinazoline (UK-338,003): the rational use of ex vivo intestine to predict in vivo absorption. Drug Metab Dispos 36:87–94. [DOI] [PubMed] [Google Scholar]
- Collier AC, Ganley NA, Tingle MD, Blumenstein M, Marvin KW, Paxton JW, Mitchell MD, Keelan JA. (2002) UDP-glucuronosyltransferase activity, expression and cellular localization in human placenta at term. Biochem Pharmacol 63:409–419. [DOI] [PubMed] [Google Scholar]
- Collins JM, Unadkat JD. (1989) Clinical pharmacokinetics of zidovudine: an overview of current data. Clin Pharmacokinet 17:1–9. [DOI] [PubMed] [Google Scholar]
- Cretton EM, Xie MY, Bevan RJ, Goudgaon NM, Schinazi RF, Sommadossi JP. (1991) Catabolism of 3′-azido-3′-deoxythymidine in hepatocytes and liver microsomes, with evidence of formation of 3′-amino-3′-deoxythymidine, a highly toxic catabolite for human bone marrow cells. Mol Pharmacol 39:258–266. [PubMed] [Google Scholar]
- Dancis J, Lee J, Mendoza S, Liebes L. (1993) Nucleoside transport by perfused human placenta. Placenta 14:547–554. [DOI] [PubMed] [Google Scholar]
- de Souza J, Benet LZ, Huang Y, Storpirtis S. (2009) Comparison of bidirectional lamivudine and zidovudine transport using MDCK, MDCK-MDR1, and Caco-2 cell monolayers. J Pharm Sci 98:4413–4419. [DOI] [PubMed] [Google Scholar]
- De Sousa Mendes M, Hirt D, Vinot C, Valade E, Lui G, Pressiat C, Bouazza N, Foissac F, Blanche S, Lê MP, et al. (2016) Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br J Clin Pharmacol 81:646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Whitney-Pickett C, Umland JP, Zhang H, Zhang X, Gebhard DF, Lai Y, Federico JJ, 3rd, Davidson RE, Smith R, et al. (2011) Development of a new permeability assay using low-efflux MDCKII cells. J Pharm Sci 100:4974–4985. [DOI] [PubMed] [Google Scholar]
- Doran A, Obach RS, Smith BJ, Hosea NA, Becker S, Callegari E, Chen C, Chen X, Choo E, Cianfrogna J, et al. (2005) The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos 33:165–174. [DOI] [PubMed] [Google Scholar]
- Evans MI, Pryde PG, Reichler A, Bardicef M, Johnson MP. (1993) Fetal drug therapy. West J Med 159:325–332. [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Naritomi Y, Tetsuka K, Nakamori F, Moriguchi H, Yamano K, Terashita S, Tabata K, Teramura T. (2014) Species differences in intestinal glucuronidation activities between humans, rats, dogs and monkeys. Xenobiotica 44:205–216. [DOI] [PubMed] [Google Scholar]
- Gallicano K. (2000) Antiretroviral-drug concentrations in semen. Antimicrob Agents Chemother 44:1117–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallicano K, Sahai J, Ormsby E, Cameron DW, Pakuts A, McGilveray I. (1993) Pharmacokinetics of zidovudine after the initial single dose and during chronic-dose therapy in HIV-infected patients. Br J Clin Pharmacol 36:128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. (2012) A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol 74:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Harrison A, Houston JB, Galetin A. (2010) Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos 38:1147–1158. [DOI] [PubMed] [Google Scholar]
- Ha HR, Chen J, Freiburghaus AU, Follath F. (1995) Metabolism of theophylline by cDNA-expressed human cytochromes P-450. Br J Clin Pharmacol 39:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman HF. (1962) Molecular form of theophylline responsible for positive inotropic activity. Circ Res 10:598–607. [DOI] [PubMed] [Google Scholar]
- Hebert MF, Easterling TR, Kirby B, Carr DB, Buchanan ML, Rutherford T, Thummel KE, Fishbein DP, Unadkat JD. (2008) Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin Pharmacol Ther 84:248–253. [DOI] [PubMed] [Google Scholar]
- Hill MD, Abramson FP. (1988) The significance of plasma protein binding on the fetal/maternal distribution of drugs at steady-state. Clin Pharmacokinet 14:156–170. [DOI] [PubMed] [Google Scholar]
- Howe JL, Back DJ, Colbert J. (1992) Extrahepatic metabolism of zidovudine. Br J Clin Pharmacol 33:190–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine JD, Takahashi L, Lockhart K, Cheong J, Tolan JW, Selick HE, Grove JR. (1999) MDCK (Madin-Darby canine kidney) cells: a tool for membrane permeability screening. J Pharm Sci 88:28–33. [DOI] [PubMed] [Google Scholar]
- Kanto J, Sjövall S, Erkkola R, Himberg JJ, Kangas L. (1983) Placental transfer and maternal midazolam kinetics. Clin Pharmacol Ther 33:786–791. [DOI] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. (2013) A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos 41:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. (2012) A PBPK model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT Pharmacometrics Syst Pharmacol 1:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. (2014) Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br J Clin Pharmacol. 77:554-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. (2009) Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP-glucuronosyltransferase cofactors in alamethicin-activated human liver microsomes. Drug Metab Dispos 37:82–89. [DOI] [PubMed] [Google Scholar]
- Kitano T, Iizasa H, Hwang I-W, Hirose Y, Morita T, Maeda T, Nakashima E. (2004) Conditionally immortalized syncytiotrophoblast cell lines as new tools for study of the blood-placenta barrier. Biol Pharm Bull 27:753–759. [DOI] [PubMed] [Google Scholar]
- Klecker RW, Jr, Collins JM, Yarchoan R, Thomas R, Jenkins JF, Broder S, Myers CE. (1987) Plasma and cerebrospinal fluid pharmacokinetics of 3′-azido-3′-deoxythymidine: a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin Pharmacol Ther 41:407–412. [DOI] [PubMed] [Google Scholar]
- Knights KM, Spencer SM, Fallon JK, Chau N, Smith PC, and Miners JO (2016) Scaling factors for the in vitro-in vivo extrapolation (IV-IVE) of renal drug and xenobiotic glucuronidation clearance. Br J Clin Pharmacol. 81:1153-1164. [DOI] [PMC free article] [PubMed]
- Kobayashi Y, Sakai R, Ohshiro N, Ohbayashi M, Kohyama N, Yamamoto T. (2005) Possible involvement of organic anion transporter 2 on the interaction of theophylline with erythromycin in the human liver. Drug Metab Dispos 33:619–622. [DOI] [PubMed] [Google Scholar]
- Liebes L, Mendoza S, Wilson D, Dancis J. (1990) Transfer of zidovudine (AZT) by human placenta. J Infect Dis 161:203–207. [DOI] [PubMed] [Google Scholar]
- Loccisano AE, Longnecker MP, Campbell JL, Jr, Andersen ME, Clewell HJ., 3rd (2013) Development of PBPK models for PFOA and PFOS for human pregnancy and lactation life stages. J Toxicol Environ Health A 76:25–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. (2002) Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther 303:1029–1037. [DOI] [PubMed] [Google Scholar]
- Mao Q, Ganapathy V, Unadkat JD. (2014) Drug transport in the placenta, in Drug Transporters pp 341–353, (You GaM, Marilyn E. eds) John Wiley & Sons. [Google Scholar]
- Masungi C, Borremans C, Willems B, Mensch J, Van Dijck A, Augustijns P, Brewster ME, Noppe M. (2004) Usefulness of a novel Caco-2 cell perfusion system. I. In vitro prediction of the absorption potential of passively diffused compounds. J Pharm Sci 93:2507–2521. [DOI] [PubMed] [Google Scholar]
- McNamara PJ, Alcorn J. (2002) Protein binding predictions in infants. AAPS PharmSci 4:E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernandez-Diaz S; National Birth Defects Prevention Study (2011) Medication use during pregnancy, with particular focus on prescription drugs: 1976-2008. Am J Obstet Gynecol 205:51.e1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan MJ, Boyer PJ, Scott GB, Parks WP, Weller S, Blum MR, Balsley J, Bryson YJ, Zidovudine Collaborative Working Group (1993) The pharmacokinetics and safety of zidovudine in the third trimester of pregnancy for women infected with human immunodeficiency virus and their infants: phase I acquired immunodeficiency syndrome clinical trials group study (protocol 082). Am J Obstet Gynecol 168:1510–1516. [DOI] [PubMed] [Google Scholar]
- Ohno S, Nakajin S. (2009) Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos 37:32–40. [DOI] [PubMed] [Google Scholar]
- Omarini D, Barzago MM, Aramayona J, Bortolotti A, Lucchini G, Bonati M. (1992) Theophylline transfer across human placental cotyledon during in vitro dual perfusion. J Med 23:101–116. [PubMed] [Google Scholar]
- Polin RA, Fox WW, Abman SH. (2004) Fetal and Neonatal Physiology, W.B. Saunders, Philadelphia. [Google Scholar]
- Ron M, Hochner-Celnikier D, Menczel J, Palti Z, Kidroni G. (1984) Maternal-fetal transfer of aminophylline. Acta Obstet Gynecol Scand 63:217–218. [DOI] [PubMed] [Google Scholar]
- Schenker S, Johnson RF, King TS, Schenken RS, Henderson GI. (1990) Azidothymidine (zidovudine) transport by the human placenta. Am J Med Sci 299:16–20. [DOI] [PubMed] [Google Scholar]
- Sheiner LB, Beal SL. (1985) Pharmacokinetic parameter estimates from several least squares procedures: superiority of extended least squares. J Pharmacokinet Biopharm 13:185–201. [DOI] [PubMed] [Google Scholar]
- Shuster DL, Risler LJ, Prasad B, Calamia JC, Voellinger JL, Kelly EJ, Unadkat JD, Hebert MF, Shen DD, Thummel KE, et al. (2014) Identification of CYP3A7 for glyburide metabolism in human fetal livers. Biochem Pharmacol 92:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg MP, Cretton EM, Kidd L, Diasio RB, Sommadossi JP. (1992) Clinical pharmacokinetics of 3′-azido-3′-deoxythymidine (zidovudine) and catabolites with formation of a toxic catabolite, 3′-amino-3′-deoxythymidine. Clin Pharmacol Ther 51:668–676. [DOI] [PubMed] [Google Scholar]
- Takeda M, Khamdang S, Narikawa S, Kimura H, Kobayashi Y, Yamamoto T, Cha SH, Sekine T, Endou H. (2002) Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp Ther 300:918–924. [DOI] [PubMed] [Google Scholar]
- Tasnif Y, Morado J, Hebert MF. (2016) Pregnancy-related pharmacokinetic changes. Clin Pharmacol Ther 100:53–62. [DOI] [PubMed] [Google Scholar]
- Taub ME, Kristensen L, Frokjaer S. (2002) Optimized conditions for MDCK permeability and turbidimetric solubility studies using compounds representative of BCS classes I-IV. Eur J Pharm Sci 15:331–340. [DOI] [PubMed] [Google Scholar]
- Teksin ZS, Seo PR, Polli JE. (2010) Comparison of drug permeabilities and BCS classification: three lipid-component PAMPA system method versus Caco-2 monolayers. AAPS J 12:238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolle-Sander S, Rautio J, Wring S, Polli JW, Polli JE. (2003) Midazolam exhibits characteristics of a highly permeable P-glycoprotein substrate. Pharm Res 20:757–764. [DOI] [PubMed] [Google Scholar]
- Tracy TS, Venkataramanan R, Glover DD, Caritis SN, National Institute for Child Health and Human Development Network of Maternal-Fetal-Medicine Units (2005) Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A activity) during pregnancy. Am J Obstet Gynecol 192:633–639. [DOI] [PubMed] [Google Scholar]
- Tuntland T, Odinecs A, Pereira CM, Nosbisch C, Unadkat JD. (1999) In vitro models to predict the in vivo mechanism, rate, and extent of placental transfer of dideoxynucleoside drugs against human immunodeficiency virus. Am J Obstet Gynecol 180:198–206. [DOI] [PubMed] [Google Scholar]
- Veal GJ, Wild MJ, Barry MG, Back DJ. (1994) Effects of dideoxyinosine and dideoxycytidine on the intracellular phosphorylation of zidovudine in human mononuclear cells. Br J Clin Pharmacol 38:323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Hamman MA, Hall SD, Wrighton SA. (2002) Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30:883–891. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Furubayashi T, Kataoka M, Sakane T, Sezaki H, Tokuda H. (2000) Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur J Pharm Sci 10:195–204. [DOI] [PubMed] [Google Scholar]
- Yazdanian M, Glynn SL, Wright JL, Hawi A. (1998) Correlating partitioning and caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm Res 15:1490–1494. [DOI] [PubMed] [Google Scholar]
- Yoon M, Schroeter JD, Nong A, Taylor MD, Dorman DC, Andersen ME, Clewell HJ., 3rd (2011) Physiologically based pharmacokinetic modeling of fetal and neonatal manganese exposure in humans: describing manganese homeostasis during development. Toxicol Sci 122:297–316. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wu X, Wang H, Mikheev AM, Mao Q, Unadkat JD. (2008) Effect of pregnancy on cytochrome P450 3a and P-glycoprotein expression and activity in the mouse: mechanisms, tissue specificity, and time course. Mol Pharmacol 74:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]