

Abstract
Accurately annotating biological functions of proteins is one of the key tasks in the postgenome era. Many machine learning based methods have been applied to predict functional annotations of proteins, but this task is rarely solved by deep learning techniques. Deep learning techniques recently have been successfully applied to a wide range of problems, such as video, images, and nature language processing. Inspired by these successful applications, we investigate deep restricted Boltzmann machines (DRBM), a representative deep learning technique, to predict the missing functional annotations of partially annotated proteins. Experimental results on Homo sapiens, Saccharomyces cerevisiae, Mus musculus, and Drosophila show that DRBM achieves better performance than other related methods across different evaluation metrics, and it also runs faster than these comparing methods.
1. Introduction
Proteins are the major components of living cells, they are the main material basis that form and maintain life activities. Proteins engage with various biological activities, such as catalysis of biochemical reactions and transport to signal transduction [1, 2]. High-throughput biotechniques produce explosive growth of biological data. Due to experimental techniques and the research bias in biology [3, 4], the gap between newly discovered genome sequences and functional annotations of these sequences is becoming larger and larger. The Human Proteome Project consortium recently claimed that we still have very little information about the cellular functions of approximately two-thirds of human proteins [5]. Wet-lab experiments can precisely verify functions of proteins, but it is time consuming and costly to do so. In practice, wet-lab techniques can only verify a portion of functions of proteins. In addition, it is difficult to efficiently verify functional annotations of massive proteins by wet-lab techniques. Therefore, it is important and necessary to develop computational models to make use of available functional annotations of proteins and a variety of types genomic and proteomic data, to automatically infer protein functions [2, 6].
Various computational methods have been proposed to predict functional annotations of proteins. These methods are often driven by data-intensive computational models. Data may come from amino acids sequences [7], protein-protein interactions [8], pathways [9], and multiple types of biological data fusion [10–12]. Gene Ontology (GO) is a major bioinformatics tool to unify gene products' attributes across all species, it uses GO terms to describe the gene products attributes [13], and these terms are structured in a directed acyclic graph (DAG). Each GO term in the graph can be viewed as a functional label and is associated with a distinct alphanumeric identifier, that is, GO:0008150 (biological process). GO is not static. Researchers and GO consortium contribute to updating GO as the revolved biological knowledge. Currently, most functional annotations of proteins are shallow and far from complete [3–5]. Given the true path rule of GO [13], if a protein is annotated with a GO term, then all the ancestor terms of that term are also annotated to the protein, but it is uncertain whether its descendant terms should be annotated to the protein or not. Therefore, it is more desirable to know the specific annotations of a protein, rather than the general ones, and the corresponding specific terms can provide more biological information than the shallow ones, which are ancestor terms of these specific terms. In this work, we investigate to predict deep (or specific) annotations of a protein based on the available annotations of proteins.
Functional associations between proteins and GO structure have been directly employed to predict protein functions [14–18]. Functional annotations of proteins can be encoded by a protein function association matrix, in which each row corresponds to a protein and each column represents a type of function. King et al. [14] directly used decision tree classifier (or Bayes classifier) on the pattern of annotations to infer additional annotations of proteins. But these two classifiers need sufficient annotations and they get rather poor performance on specific GO terms, which are annotated to fewer than 10 proteins. Khatri et al. [15] used truncated single value decomposition (tSVD) to replenish the missing functions of proteins based on protein function matrix. This approach is able to predict missing annotations in existing annotation databases and improve prediction accuracy. But this method does not take advantage of the hierarchical and flat relationships between GO terms. Previous researches have demonstrated that the ontology hierarchy plays important roles in predicting protein function [2, 16, 18]. Done et al. [16] used a vector space model and a number of weighting schemes, along with latent semantic indexing approach to extract implicit semantic relationships between proteins and those between functions to predict protein functions. This method is called NtN [16]. NtN takes into account GO hierarchical structure and can weigh different GO terms situated at different locations of GO DAG [19]. Tao et al. [17] proposed a method called information theory based semantic similarity (ITSS). ITSS first calculates the semantic similarity between pairwise GO terms in a hierarchy and then sums up these pairwise similarity for pairwise GO terms annotated to two proteins. Next, it uses a kNN classifier to predict novel annotations of a protein. Yu et al. [18] proposed downward random walks (dRW) to predict missing (or new) functions of partially annotated proteins. Particularly, dRW applies downward random walks with restart [20] on the GO DAG, started on terms annotated to a protein, to predict additional annotations of the protein.
A protein is often engaged with several biological activities and thus is annotated with several GO terms. Each term can be regarded as a functional label, and protein function prediction can be modeled as a multilabel learning problem [21, 22]. From this viewpoint, protein function prediction using incomplete annotations can be modeled as a multilabel weak learning problem [22]. More recently, Yu et al. [23] proposed a method called PILL to replenish missing functions for partially annotated proteins using incomplete hierarchical labels information. Fu et al. [24] proposed a method called dHG to predict novel functions of proteins using a directed hybrid graph, which is consisted with GO DAG, protein-protein interaction network, and available functional associations between GO terms and proteins. These aforementioned methods (except DRBM) can be regarded as shallow machine learning approaches [25]. They do not capture deep associations between proteins and GO terms.
In this paper, we investigate the recently widely applied technique, deep learning [25], to capture deep associations between proteins and GO terms, and to replenish the missing annotations of incompletely annotated proteins. For this investigation, we apply deep restricted Boltzmann machines (DRBM) to predict functional annotations of proteins. DRBM utilizes the archived annotations of four model species (Homo sapiens, Saccharomyces cerevisiae, Mus musculus, and Drosophila) to explore the hidden associations between proteins and GO terms and the structural relationship between GO terms. At the same time, it optimizes the parameters of DRBM. After that, we validate the performance of DRBM by comparing its predictions with recently archived GO annotations of these four species. The empirical and comparative study shows DRBM achieves better results than other related methods. DRBM also runs faster than some of these comparing methods.
The structure of this paper is organized as follows. Section 2 briefly reviews some related deep learning techniques that are recently applied for protein function prediction. Section 3 introduces the restricted Boltzmann machine and deep restricted Boltzmann machine for protein function prediction. The experimental datasets, setup, and results are discussed in Section 4. Conclusions are provided in Section 5.
2. Related Work
Some pioneers have already applied deep learning for some bioinformatics problems [26], but few works have been reported for protein function prediction. Autoencoder neural networks (AE) can process complex structural data better than shallow machine learning methods [25, 27, 28]. AE has been applied in computer vision [28], speech recognition [25, 27], and protein residue-residue contacts prediction [26]. Chicco et al. [29] recently used deep AE to predict protein functions. Experiments show that deep AE can explore the deep associations between proteins and GO terms and achieve better performance than other shallow machine learning based function prediction methods, including tSVD [29].
Deep AE takes much more time in fine-tuning network; if the network is very deep, it will lead to vanishing gradient problem. In this work, we suggest to use deep restricted Boltzmann machines (DRBM), instead of AE, to predict functional annotations of proteins. DRBM has rapid convergence speed and good stability. DRBM has been used to construct the deep belief networks [30], for speech recognition [31, 32], collaborative filtering [33], computational biology [34], and other fields. Recently, Wang and Zeng [34] proposed to predict drug-target interactions using restricted Boltzmann machines and achieved good prediction performance. More recently, Li et al. [35] used conditional restricted Boltzmann machines to capture high-order label dependence relationships and facilitate multilabel learning with incomplete labels. Experiments have demonstrated the efficacy of restricted Boltzmann machines on addressing multilabel learning with incomplete labels.
To the best of our knowledge, few teams investigate DRBM for large-scale missing functions prediction. For this purpose, we study it for predicting functions of proteins of Homo sapiens, Saccharomyces cerevisiae, Mus musculus, and Drosophila and compare it with a number of related methods. The experimental results show that DRBM achieves better results than these comparing methods on various evaluation metrics.
3. Methods
In this section, we will describe the deep restricted Boltzmann machines to predict missing GO annotations of proteins.
3.1. Restricted Boltzmann Machine
A restricted Boltzmann machine (RBM) is a network of undirected graphical model with stochastic binary units [32]. As shown in Figure 1, an RBM is a two-layer bipartite graph with two types of units, a set of visible units v ∈ {0,1}, and a set of hidden units h ∈ {0,1}. Input units and hidden units are fully connected; there is no connection between nodes in the same layer. In this paper, the number of visible units is equal to the number of GO terms, and these units take the protein function association matrix as inputs.
Figure 1.
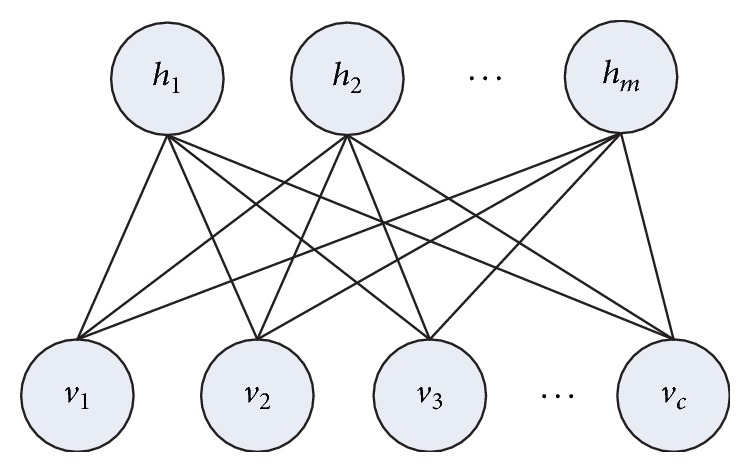
An RBM with binary hidden units (hj) representing latent features and visible units (vi) encoding observed data.
RBM is an unsupervised method; it learns one layer of hidden features. When the number of hidden units is smaller than that of visual units, the hidden layer can deal with nonlinear complex dependency and structure of data, capture deep relationship from input data [30], and represent the input data more compactly. Latent feature values are represented by the hidden units and visible units encode available GO annotations of proteins. Suppose there are c (the number of GO terms) visible units and m hidden units in an RBM. vi (i = 1,…, c) indicates the state of the ith visible unit, where vi = 1 means the ith term is annotated to the protein and vi = 0 means the ith term is not associated with the protein. Binary variable hj (j = 1,…, m) indicates the state of hidden unit, and hj = 1 denotes the jth hidden unit which is active. Let Wij be the weight associated with the connection between vi and hj. (v, h) is a joint configuration of an RBM.
The energy function capturing the interaction patterns between visual layer and hidden layer can be modeled as follows:
| (1) |
where θ = {Wij, ai, bj} are parameters of RBM, while ai and bj are biases for the visible and hidden variables, respectively. W ∈ ℝc×m encodes the weights of connection between c visual variables and m hidden variables. Then, a joint probability configuration of v and h can be defined as
| (2) |
where Z is a normalization constant or partition function, Z = ∑v,he−E(v, h). The marginal distribution over visible data is
| (3) |
There is no connection between visible units (or hidden units) in an RBM; the conditional distributions over the visible and hidden units are given by logistic functions as follows:
| (4) |
| (5) |
where σ(x) = 1/(1 + exp(−x)) is a logistics sigmoid function.
It is difficult to train an RBM with a large number of parameters. To efficiently train an RBM and to optimize the parameters, we maximize the likelihood of visible data with respect to the parameters. To achieve this goal, the derivative of log probability of the training data derived from (4) can be adopted to incrementally adjust the weights as follows:
| (6) |
where 〈·〉 indicates expectations under the distribution. It is very easy to learn the log-likelihood probability of training data:
| (7) |
where ϵ controls the learning rate. Since there are no direct connections in the hidden layer of an RBM, so we can get an unbiased sample of 〈vihj〉data easily. Unfortunately, it is difficult to compute an unbiased sample of 〈vihj〉model, since it requires exponential time. To avoid this problem, a fast learning algorithm, called Contrastive Divergence (CD) [36], is proposed by Hinton [37]. CD sets visible variables as training data. Then the binary states of hidden units are all computed in parallel using (5). Once the states have been chosen for the hidden units, a “reconstruction” is produced by setting each vi to 1 with a probability given by (4). In addition, weights are also adjusted in each training pass as follows:
| (8) |
〈vihj〉data is the average value over all input data for each update and 〈vihj〉recon is the average value over reconstruction; it is considered as a good approximation to 〈vihj〉model.
3.2. Deep RBM
In this paper, we will use a fully connected restricted Boltzmann machine and consider learning a multilayer RBMs (as shown in Figure 2). In the network structure, each layer captures complicated correlations between hidden layer and its beneath layer.
Figure 2.
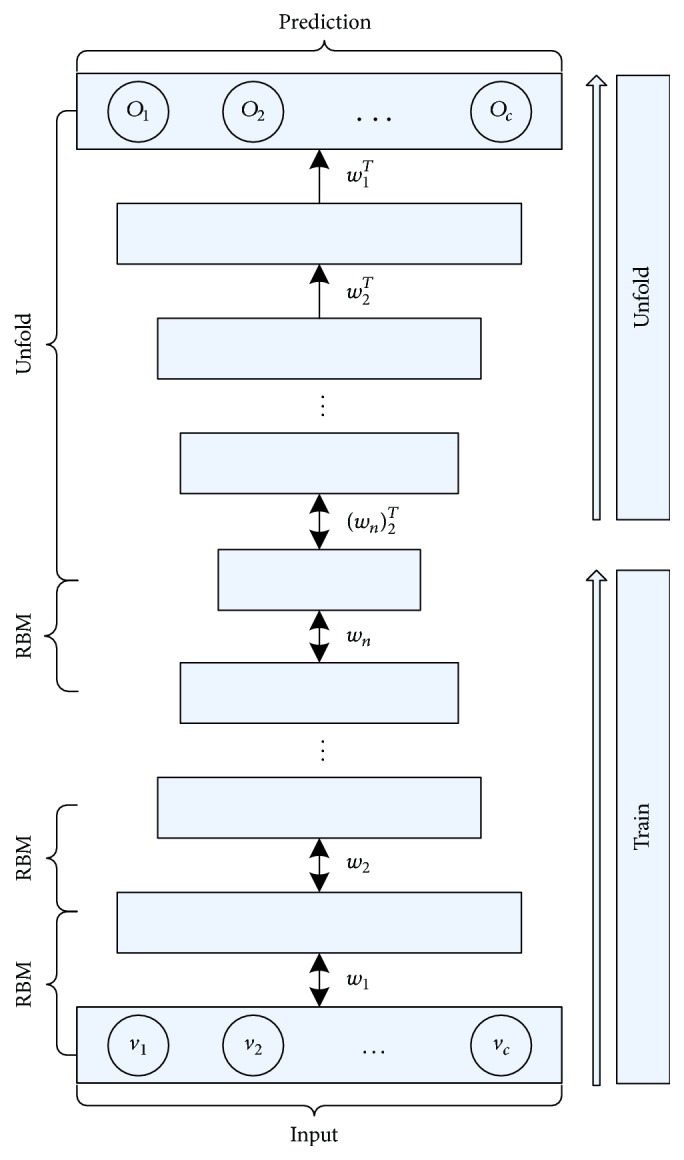
Network architecture of DRBM.
DRBM is adopted for several reasons [38]. Firstly, DRBM, like deep belief networks, has the potential of learning internal representations that become increasingly complex; it is regarded as a promising way to solve complex problems [30]. Second, high-level representations can be built from large volume incomplete sensory inputs and scarce labeled data and then be used to unfold the model. Finally, DRBM can well propagate the uncertainty information and hence robustly deal with ambiguous inputs. Hinton et al. [30] introduced a greedy, layer-by-layer unsupervised learning algorithm that consists of learning a stack of RBMs. After the stacked RBMs have been learned, the whole stack can be viewed as a single probabilistic model. In this paper, we use that greedy algorithm to optimize the parameters of DRBM. DRBM greedily trains a stack of more than two RBMs, and the modification only needs to be used for the first and last RBMs in the stack. Retraining consists of learning a stack of RBMs; each RBM has only one layer of feature detectors. The learned feature activation of one RBM is used as the input data to train the next RBM in the stack. After that, these RBMs are popped up (or unfolded) to create a DRBM. Through the above training, we can optimize the parameters of DRBM and then take the outputs of the network as the results of protein function prediction.
4. Result and Discussion
4.1. Datasets and Experimental Setup
To study the performance of DRBM on predicting missing GO annotations of incompletely annotated proteins. We downloaded the GO file (http://geneontology.org/page/download-ontology) (archived date: 2015-10-22), which describes hierarchical relationships between GO terms using a DAG. These GO terms are divided into three branches, describing molecular functions (MF), cellular component (CC), and biological process (BP) functions of proteins. We also downloaded the Gene Ontology Annotation (GOA) (archived date: 2014-10-27) files (http://geneontology.org/page/download-annotations) of Saccharomyces cerevisiae, Homo sapiens, Mus musculus, and Drosophila. We preprocessed the GO file to exclude the GO terms tagged “obsolete.” To avoid circular prediction, we processed the GOA file to exclude the annotations with evidence code “IEA” (inferred from Electronic Annotation). The missing annotations of a protein often correspond to the descendants of the terms currently annotated to the protein. So the terms corresponding to these missing annotations are located at deeper level than their ancestor terms, and these terms characterize more specific biological functions of proteins than their ancestors. These specific terms are usually annotated to no more than 30 proteins; they are regarded as sparse functions. On the other hand, root terms, GO:0008150 for BP, GO:0003674 for MF, and GO:0005575 for CC, are annotated to majority of proteins; the prediction on these terms is not interesting, so we removed these three root terms. We kept the terms annotated at least one protein in the GOA file for experiments. The statistics of preprocessed GO annotations of proteins in these four model species are listed in Table 1.
Table 1.
Statistics of experimental datasets. The data in the third column (N) is the number of proteins annotated with at least 1 term for a particular subontology. C is the number of involved GO terms; Avg ± Std is the average number of annotations of a protein and its standard deviation.
| Dataset | N | C | Avg ± Std | |
|---|---|---|---|---|
| Homo sapiens | BP | 11628 | 12514 | 60.24 ± 60.83 |
| CC | 12523 | 1574 | 20.17 ± 12.28 | |
| MF | 11628 | 3724 | 10.97 ± 8.81 | |
|
| ||||
| Mus musculus | BP | 10990 | 13500 | 56.26 ± 61.08 |
| CC | 10549 | 1592 | 15.73 ± 10.25 | |
| MF | 9906 | 3775 | 9.59 ± 7.30 | |
|
| ||||
| Saccharomyces cerevisiae | BP | 4671 | 4909 | 44.13 ± 31.41 |
| CC | 4128 | 970 | 20.67 ± 10.30 | |
| MF | 4291 | 2203 | 9.60 ± 6.60 | |
|
| ||||
| Drosophila | BP | 6188 | 6645 | 48.53 ± 48.97 |
| CC | 4851 | 1097 | 15.10 ± 10.27 | |
| MF | 4489 | 2255 | 9.05 ± 5.75 | |
We also downloaded recently archived GOA files (date: 2015-10-12) of these four species to validate the performance of DRBM and processed these GOA files in a similar way. We use the data archived in 2014 to train DRBM and then use the data archived in 2015 for validation.
In order to comparatively evaluate the performance of DRBM, we compare it with SVD [15], NtN [16], dRW [18], and AE [29]. SVD, NtN, and dRW are shallow machine learning algorithms. AE and DRBM are deep machine learning methods. DRBM is set with a learning rate of 0.01 for 25 iterations [29]. L2 regularization is used on all weights, which are initialized randomly from the uniform distribution between 0 and 1. We set the hidden unit function as sigmoid and the number of hidden units as half of visible units and the number of the second hidden layer as half of the first hidden layer and so on. The number of hidden layers is 5. In the following experiments, to prevent overfitting, we used weight-decay and dropout. Weight-decay adds an extra term to the normal gradient. This extra term is the derivative of a function that penalizes large weights. We used the simplest L2 penalty function. As well as that, dropout is a regularization technique for reducing overfitting in neural networks by preventing complex coadaptations on training data [39].
The accuracy of protein function prediction can be evaluated by different evaluation metrics, and the performance of different prediction models is affected by the adopted evaluation metrics. To do a fair and comprehensive comparison, we used four evaluation metrics, MacroAvgF1, AvgROC, RankingLoss, and Fmax. These evaluation metrics measure the performance of protein function prediction from different aspects. The first three metrics have been applied to evaluate the results of multilabel learning [40]. AvgROC and Fmax are recommended metrics for evaluating protein function prediction [6, 41]. MacroAvgF1 gets the F1-Score of each term and then takes the average of F1-score across all the terms. AvgAUC firstly calculates the area under receiver operating curve of each term and then takes the average value of these areas as whole to measure the performance. Fmax [6] is the overall maximum harmonic mean of recall and precision across all possible thresholds on the predicted protein function association matrix. RankingLoss computes the average fraction of wrongly predicted annotations ranking ahead of ground-truth annotations of proteins. To be consistent with other evaluation metrics, we use 1 − RankLoss instead of RankingLoss. Namely, the higher the value of these metrics is, the better the performance is. The formal definition of these metrics can be found in [6, 22, 40]. Since these metrics capture different aspects of a function prediction method, it is difficult for an approach to consistently outperform the others across all the evaluation metrics.
4.2. Experimental Results
Based on the experimental protocols introduced above, we conduct experiments to investigate the performance of DRBM on protein function prediction. In Table 2, we report the experimental results on proteins of Homo sapiens annotated with BP, CC, and MF terms, respectively. The results on Mus musculus, Saccharomyces cerevisiae, and Drosophila are provided in Tables 3–5. In these tables, the best results are in boldface.
Table 2.
Experimental results on Homo sapiens.
| MacroAvgF1 | AvgROC | 1 − RankLoss | Fmax | ||
|---|---|---|---|---|---|
| BP | NtN | 0.0107 | 0.7498 | 0.6920 | 0.1712 |
| dRW | 0.6902 | 0.9044 | 0.8737 | 0.9301 | |
| SVD | 0.7313 | 0.9053 | 0.9349 | 0.9206 | |
| AE | 0.5341 | 0.9049 | 0.8495 | 0.5617 | |
| DRBM | 0.8378 | 0.9109 | 0.9883 | 0.9217 | |
|
| |||||
| CC | NtN | 0.0036 | 0.6569 | 0.6641 | 0.1063 |
| dRW | 0.6806 | 0.8999 | 0.9186 | 0.9516 | |
| SVD | 0.7139 | 0.8942 | 0.9592 | 0.9157 | |
| AE | 0.8081 | 0.8932 | 0.9629 | 0.8819 | |
| DRBM | 0.7982 | 0.9192 | 0.9955 | 0.9437 | |
|
| |||||
| MF | NtN | 0.3891 | 0.7767 | 0.8450 | 0.0121 |
| dRW | 0.7909 | 0.9130 | 0.9208 | 0.9529 | |
| SVD | 0.8022 | 0.8022 | 0.9526 | 0.9480 | |
| AE | 0.7683 | 0.9047 | 0.8186 | 0.5604 | |
| DRBM | 0.8517 | 0.9085 | 0.9898 | 0.9470 | |
Table 3.
Experimental results on Mus musculus.
| MacroAvgF1 | AvgROC | 1 − RankLoss | Fmax | ||
|---|---|---|---|---|---|
| BP | NtN | 0.0154 | 0.6950 | 0.7055 | 0.1542 |
| dRW | 0.5666 | 0.8155 | 0.8296 | 0.9049 | |
| SVD | 0.6169 | 0.8220 | 0.9130 | 0.8914 | |
| AE | 0.4573 | 0.8139 | 0.8219 | 0.5340 | |
| DRBM | 0.7221 | 0.8476 | 0.9841 | 0.8962 | |
|
| |||||
| CC | NtN | 0.0055 | 0.6244 | 0.6436 | 0.1062 |
| dRW | 0.4913 | 0.8001 | 0.7857 | 0.8694 | |
| SVD | 0.5415 | 0.7847 | 0.8856 | 0.8539 | |
| AE | 0.6548 | 0.7933 | 0.9139 | 0.8694 | |
| DRBM | 0.6676 | 0.8412 | 0.9813 | 0.8644 | |
|
| |||||
| MF | NtN | 0.7338 | 0.9135 | 0.9401 | 0.0111 |
| dRW | 0.8742 | 0.9493 | 0.9474 | 0.9693 | |
| SVD | 0.7408 | 0.9466 | 0.9703 | 0.9188 | |
| AE | 0.9035 | 0.9461 | 0.9724 | 0.7044 | |
| DRBM | 0.9133 | 0.9492 | 0.9906 | 0.9652 | |
Table 4.
Experimental results on Saccharomyces cerevisiae.
| MacroAvgF1 | AvgROC | 1 − RankLoss | Fmax | ||
|---|---|---|---|---|---|
| BP | NtN | 0.0072 | 0.7026 | 0.7027 | 0.1172 |
| dRW | 0.8042 | 0.9268 | 0.9337 | 0.9649 | |
| SVD | 0.7794 | 0.9199 | 0.9659 | 0.9440 | |
| AE | 0.6990 | 0.9179 | 0.9252 | 0.5032 | |
| DRBM | 0.8524 | 0.9256 | 0.9905 | 0.9555 | |
|
| |||||
| CC | NtN | 0.0072 | 0.7026 | 0.7027 | 0.1172 |
| dRW | 0.8112 | 0.9264 | 0.9612 | 0.9771 | |
| SVD | 0.7408 | 0.9274 | 0.9767 | 0.9198 | |
| AE | 0.8595 | 0.9262 | 0.9851 | 0.9771 | |
| DRBM | 0.8722 | 0.9278 | 0.9948 | 0.9744 | |
|
| |||||
| MF | NtN | 0.7338 | 0.9135 | 0.9401 | 0.0111 |
| dRW | 0.8742 | 0.9493 | 0.9474 | 0.9693 | |
| SVD | 0.7408 | 0.9466 | 0.9703 | 0.9188 | |
| AE | 0.9035 | 0.9461 | 0.9724 | 0.7044 | |
| DRBM | 0.9133 | 0.9492 | 0.9906 | 0.9652 | |
Table 5.
Experimental results on Drosophila.
| MacroAvgF1 | AvgROC | 1 − RankLoss | Fmax | ||
|---|---|---|---|---|---|
| BP | NtN | 0.7724 | 0.8450 | 0.8958 | 0.9416 |
| dRW | 0.6875 | 0.8525 | 0.9011 | 0.9455 | |
| SVD | 0.6852 | 0.8516 | 0.9479 | 0.9371 | |
| AE | 0.5882 | 0.8486 | 0.9049 | 0.5772 | |
| DRBM | 0.7699 | 0.8601 | 0.9877 | 0.9382 | |
|
| |||||
| CC | NtN | 0.0101 | 0.6475 | 0.7808 | 0.1957 |
| dRW | 0.6599 | 0.8425 | 0.9210 | 0.9553 | |
| SVD | 0.6446 | 0.8222 | 0.9585 | 0.9156 | |
| AE | 0.7331 | 0.8251 | 0.9678 | 0.9553 | |
| DRBM | 0.7438 | 0.8558 | 0.9922 | 0.9448 | |
|
| |||||
| MF | NtN | 0.5071 | 0.7640 | 0.9065 | 0.0700 |
| dRW | 0.7346 | 0.8206 | 0.9309 | 0.9610 | |
| SVD | 0.7131 | 0.8125 | 0.9631 | 0.9549 | |
| AE | 0.7558 | 0.8133 | 0.9639 | 0.6429 | |
| DRBM | 0.7719 | 0.8187 | 0.9895 | 0.9499 | |
From these tables, we can see that DRBM achieves better results than NtN, dRW, SVD, and AE in most cases. We further analyze the differences between DRBM and these comparing methods by Wilcoxon signed rank test [42, 43], we find that DRBM performs significantly better than NtN, dRW, and SVD on the first three metrics (where p values are all smaller than 0.004), and it also gets better performance than deep AE across these four metrics (p value smaller than 0.001). dRW often obtains larger Fmax than DRBM; the possible reason is that dRW utilizes threshold to filter out some predictions and thus increases the true positive rate.
dRW applies downward random walks with restart on the GO directed acyclic graph to predict protein function; dRW takes into count the hierarchical structure relationship between GO terms and achieves better results than NtN and SVD. This observation confirms that the hierarchical relationship between terms plays important roles in protein function prediction. Although dRW utilizes the hierarchical structure relationship between terms, it is still a shallow machine learning method and it does not capture the deep associations between proteins and GO terms as DRBM does, so it is often outperformed by DRBM.
The results of NtN and SVD are always lower than those of AE and DRBM. The possible reason is that singular value decomposition on sparse matrix is not suitable for this kind of protein function prediction problem, in which there are complex hierarchical relationships between GO terms. NtN uses the ontology hierarchy to adjust the weights of protein function associations, but it does not get better results than SVD. The reason is that NtN gives large weights to specific annotations but small weights to shallow annotations. From the true path rule, ancestor terms are generally annotated to more proteins than their descendant terms. For this reason, NtN is often outperformed by SVD and say nothing of AE and DRBM. Both AE and DRBM are deep machine learning techniques, but DRBM frequently performs better than AE. That is because the generalization ability of AE is not as well as that of DRBM, and AE is easy to fall into local optimal. In summary, these results and comparisons demonstrate that DRBM can capture deep associations between proteins and GO terms, and thus it achieves better performance than other related methods across different evaluation measures. DRBM is an effective alternative approach for protein function prediction.
4.3. Runtime Analysis
Here, we study runtime (include training phase and test phase) cost of these comparing methods on Homo sapiens and Mus musculus in GO BP subontology, since this subontology includes much more annotations and GO terms. The experimental platform is Windows Server 2008, Intel Xeon E7-4820, 64 GB RAM. The recorded runtime for these comparing methods is reported in Table 6.
Table 6.
Runtime cost (seconds) on Homo sapiens and Mus musculus in BP subontology.
| NtN | dRW | SVD | AE | DRBM | |
|---|---|---|---|---|---|
| Homo sapiens | 30180 | 27660 | 1200 | 15840 | 6180 |
| Mus musculus | 24180 | 28020 | 1260 | 33780 | 7500 |
From this table, we can see that DRBM is faster than these comparing methods, except SVD. NtN and dRW spend a lot of time to compute semantic similarity between GO terms, so they take more time than others. In contrast, SVD directly applies matrix decomposition on the protein function association matrix and the matrix is sparse, so SVD takes fewer time than DRBM. AE employs back propagation neural networks to tune parameters; it costs a large amount of time. DRBM utilizes Contrastive Divergence, which is a fast learning algorithm, to optimize the parameters, so its runtime is fewer than AE. This comparison further confirms DRBM is an efficient and effective alternative solution for protein function prediction.
5. Conclusions
In this paper, we study how to predict additional functional annotations of annotated proteins. We investigate deep restricted Boltzmann machines (DRBM) for this purpose. Our empirical study on the proteins of Saccharomyces cerevisiae, Homo sapiens, Mus musculus, and Drosophila shows that DRBM outperforms several competitive related methods, especially shallow machine learning models. This paper will drive more research on using deep machine learning techniques for protein function prediction. As part of our future work, we will integrate other types of proteomic data with DRBM to further boost the prediction performance.
Acknowledgments
This work is partially supported by Natural Science Foundation of China (no. 61402378), Natural Science Foundation of CQ CSTC (nos. cstc2014jcyjA40031 and cstc2016jcyjA0351), Science and Technology Development of Jilin Province of China (20150101051JC and 20160520099JH), Science and Technology Foundation of Guizhou (Grant no. QKHJC20161076), the Science and Technology Top-Notch Talents Support Project of Colleges and Universities in Guizhou (Grant no. QJHKY2016065), and Fundamental Research Funds for the Central Universities of China (nos. XDJK2016B009 and 2362015XK07).
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- 1.Roberts R. J. Identifying protein functiona call for community action. PLoS Biology. 2004;2(3):p. e42. doi: 10.1371/journal.pbio.0020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandey G., Kumar V., Steinbach M. Computational approaches for protein function prediction: a survey. A survey: Department of Computer Science and Engineering, University of Minnesota; 2006. pp. 6–28. [Google Scholar]
- 3.Schnoes A. M., Ream D. C., Thorman A. W., Babbitt P. C., Friedberg I. Biases in the experimental annotations of protein function and their effect on our understanding of protein function space. PLoS Computational Biology. 2013;9(5) doi: 10.1371/journal.pcbi.1003063.e1003063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas P. D., Wood V., Mungall C. J., Lewis S. E., Blake J. A. On the use of gene ontology annotations to assess functional similarity among orthologs and paralogs: a short report. PLoS Computational Biology. 2012;8(2) doi: 10.1371/journal.pcbi.1002386.e1002386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Legrain P., Aebersold R., Archakov A., et al. The human proteome project: current state and future direction. Molecular & Cellular Proteomics. 2011;10(7, article 009993) doi: 10.1074/mcp.O111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radivojac P., Clark W., Oron T., et al. A large-scale evaluation of computational protein function prediction. Nature Methods. 2013;10(3):221–227. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee D., Redfern O., Orengo C. Predicting protein function from sequence and structure. Nature Reviews Molecular Cell Biology. 2007;8(12):995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 8.Sharan R., Ulitsky I., Shamir R. Network-based prediction of protein function. Molecular Systems Biology. 2007;3:p. 88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao M., Pietras C. M., Feng X., et al. New directions for diffusion-based network prediction of protein function: Incorporating pathways with confidence. Bioinformatics. 2014;30(12):I219–I227. doi: 10.1093/bioinformatics/btu263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesa-Bianchi N., Re M., Valentini G. Synergy of multi-label hierarchical ensembles, data fusion, and cost-sensitive methods for gene functional inference. Machine Learning. 2012;88(1-2):209–241. doi: 10.1007/s10994-011-5271-6. [DOI] [Google Scholar]
- 11.Yu G., Domeniconi C., Rangwala H., Zhang G., Yu Z. Transductive multi-label ensemble classification for protein function prediction. Proceedings of the 18th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD 2012; August 2012; chn. pp. 1077–1085. [DOI] [Google Scholar]
- 12.Yu G., Fu G., Wang J., Zhu H. Predicting Protein Function via Semantic Integration of Multiple Networks. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2016;13(2):220–232. doi: 10.1109/TCBB.2015.2459713. [DOI] [PubMed] [Google Scholar]
- 13.Ashburner M., Ball C. A., Blake J. A., et al. Gene ontology: tool for the unification of biology. Nature Genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King O. D., Foulger R. E., Dwight S. S., White J. V., Roth F. P. Predicting gene function from patterns of annotation. Genome Research. 2003;13(5):896–904. doi: 10.1101/gr.440803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khatri P., Done B., Rao A., Done A., Draghici S. A semantic analysis of the annotations of the human genome. Bioinformatics. 2005;21(16):3416–3421. doi: 10.1093/bioinformatics/bti538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Done B., Khatri P., Done A., Drǎghici S. Predicting novel human gene ontology annotations using semantic analysis. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2010;7(1):91–99. doi: 10.1109/TCBB.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao Y., Sam L., Li J., Friedman C., Lussier Y. A. Information theory applied to the sparse gene ontology annotation network to predict novel gene function. Bioinformatics. 2007;23(13):i529–i538. doi: 10.1093/bioinformatics/btm195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu G., Zhu H., Domeniconi C., Liu J. Predicting protein function via downward random walks on a gene ontology. BMC Bioinformatics. 2015;16(1, article no. 271) doi: 10.1186/s12859-015-0713-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salton G., Wong A., Yang C. S. A vector space model for automatic indexing. Communications of the ACM. 1975;18(11):613–620. doi: 10.1145/361219.361220. [DOI] [Google Scholar]
- 20.Tong H., Faloutsos C., Pan J.-Y. Random walk with restart: Fast solutions and applications. Knowledge and Information Systems. 2008;14(3):327–346. doi: 10.1007/s10115-007-0094-2. [DOI] [Google Scholar]
- 21.Yu G., Rangwala H., Domeniconi C., Zhang G., Yu Z. Protein function prediction with incomplete annotations. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2013;11(3):579–591. doi: 10.1109/TCBB.2013.142. [DOI] [PubMed] [Google Scholar]
- 22.Yu G., Domeniconi C., Rangwala H., Zhang G. Protein function prediction using dependence maximization. Proceedings of the Joint European Conference on Machine Learning and Knowledge Discovery in Databases; Springer Berlin Heidelberg; pp. 574–589. [DOI] [Google Scholar]
- 23.Yu G., Zhu H., Domeniconi C. Predicting protein functions using incomplete hierarchical labels. BMC Bioinformatics. 2015;16(1, article no. 1) doi: 10.1186/s12859-014-0430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu G., Yu G., Wang J., Zhang Z. Novel protein function prediction using a direct hybrid graph. Science China-Information Science. 2016;46(4):461–475. [Google Scholar]
- 25.Deng L., Yu D. Deep learning: methods and applications. Foundations and Trends in Signal Processing. 2013;7(3-4):197–387. doi: 10.1561/2000000039. [DOI] [Google Scholar]
- 26.Eickholt J., Cheng J. Predicting protein residue-residue contacts using deep networks and boosting. Bioinformatics. 2012;28(23):3066–3072. doi: 10.1093/bioinformatics/bts598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 28.Hinton G. E., Salakhutdinov R. R. Reducing the dimensionality of data with neural networks. American Association for the Advancement of Science. Science. 2006;313(5786):504–507. doi: 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- 29.Chicco D., Sadowski P., Baldi P. Deep autoencoder neural networks for gene ontology annotation predictions. Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, ACM BCB 2014; September 2014; usa. pp. 533–540. [DOI] [Google Scholar]
- 30.Hinton G. E., Osindero S., Teh Y.-W. A fast learning algorithm for deep belief nets. Neural Computation. 2006;18(7):1527–1554. doi: 10.1162/neco.2006.18.7.1527. [DOI] [PubMed] [Google Scholar]
- 31.Fasel I., Berry J. Deep belief networks for real-time extraction of tongue contours from ultrasound during speech. Proceedings of the 20th International Conference on Pattern Recognition, ICPR 2010; August 2010; tur. pp. 1493–1496. [DOI] [Google Scholar]
- 32.Fischer A., Igel C. Progress in Pattern Recognition, Image Analysis, Computer Vision, and Applications. Vol. 7441. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. An Introduction to Restricted Boltzmann Machines; pp. 14–36. (Lecture Notes in Computer Science). [DOI] [Google Scholar]
- 33.Salakhutdinov R., Mnih A., Hinton G. Restricted Boltzmann machines for collaborative filtering. Proceedings of the 24th International Conference on Machine learning (ICML '07); June 2007; Corvallis, Oregon. pp. 791–798. [DOI] [Google Scholar]
- 34.Wang Y., Zeng J. Predicting drug-target interactions using restricted Boltzmann machines. Bioinformatics. 2013;29(13):i126–i134. doi: 10.1093/bioinformatics/btt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X., Zhao F., Guo Y. Conditional restricted boltzmann machines for multi-label learning with incomplete labels. Proceedings of the in Proceedings of 18th International Conference on Artificial Intelligence and Statistics; 2015; pp. 635–643. [Google Scholar]
- 36.Hinton G. E. Training products of experts by minimizing contrastive divergence. Neural Computation. 2002;14(8):1771–1800. doi: 10.1162/089976602760128018. [DOI] [PubMed] [Google Scholar]
- 37.Hinton G. A practical guide to training restricted Boltzmann machines. In: Montavon G., Orr G. B., Müller K.-R., editors. Neural Networks: Tricks of the Trade. 2nd. Vol. 7700. Berlin, Germany: Springer; 2012. pp. 599–619. (Lecture Notes in Computer Science). [DOI] [Google Scholar]
- 38.Salakhutdinov R., Hinton G. E. Deep Boltzmann Machines. Proceedings of the In Proceedings of 12th International Conference on Artificial Intelligence and Statistics; 2009; pp. 448–455. [Google Scholar]
- 39.Srivastava N., Hinton G., Krizhevsky A., Sutskever I., Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. The Journal of Machine Learning Research. 2014;15(1):1929–1958. [Google Scholar]
- 40.Zhang M.-L., Zhou Z.-H. A review on multi-label learning algorithms. IEEE Transactions on Knowledge and Data Engineering. 2014;26(8):1819–1837. doi: 10.1109/TKDE.2013.39. [DOI] [Google Scholar]
- 41.Jiang Y. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biology. 2016;17(1–19):1819–1837. doi: 10.1186/s13059-016-1037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilcoxon L. Individual comparison by ranking methods. Biometrics. 1945;1(6):80–83. doi: 10.2307/3001968. [DOI] [Google Scholar]
- 43.Demsar J. Statistical comparisons of classifiers over multiple data sets. The Journal of Machine Learning Research. 2006;7(1):1–30. [Google Scholar]