

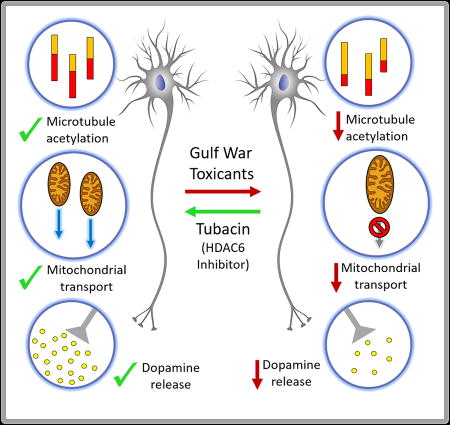
Abstract
Many veterans of the 1990–1991 Gulf War contracted Gulf War Illness, a multi-symptom disease that primarily affects the nervous system. Here we treated cultures of human or rat neurons with diisopropylfluorophosphate (DFP), an analog of sarin, one of the organophosphate toxicants to which the military veterans were exposed. All observed cellular defects produced by DFP were exacerbated by pretreatment with corticosterone or cortisol, which, in the rat and human neurons respectively, serves in our experiments to mimic the physical stress endured by soldiers during the war. To best mimic the disease, DFP was used below the level needed to inhibit acetylcholinesterase. We observed a diminution in the ratio of acetylated to total tubulin that was correctable by treatment with tubacin, a drug that inhibits HDAC6, the tubulin deacetylase. The reduction in microtubule acetylation was coupled with deficits in microtubule dynamics, which were correctable by HDAC6 inhibition. Deficits in mitochondrial transport and dopamine release were also improved by tubacin. Thus, various negative effects of the toxicant/stress exposures were at least partially correctable by restoring microtubule acetylation to a more normal status. Such an approach may have therapeutic benefit for individuals suffering from GWI or other neurological disorders linked to organophosphate exposure.
Keywords: organophosphate, stress, microtubule, neuron, axon, Gulf War Illness, acetylation, tubacin
Introduction
Gulf War Illness (GWI) is a chronic, multi-symptom disorder that afflicts 25–32% of the nearly 700,000 United States veterans who served in the 1990–1991 Gulf War1. Veterans with GWI struggle with unabated central nervous system (CNS) deficits including chronic headaches, memory and concentration problems, sleep difficulty, fatigue and mood alterations2–4. Epidemiological and intelligence data indicate that at least 100,000 GW veterans were likely exposed to the organophosphate (OP) nerve agents sarin and cyclosarin, which were released as fallout from demolitions of the ammunition depot at Khamisiyah, Iraq in March 19915–7. While acute symptoms of OP toxicity have been characterized, only recently have the neurological consequences of exposure to low levels of OPs been studied8,9.
Understanding the impact on the CNS of low level OP exposure is important because OP pesticides represent the largest group of insecticides, and their use is widespread around the world, with >5 billion pounds of pesticides applied to crops, homes, schools, parks and forests10. In addition, the threat of OP nerve agent use in terrorism and war remains a concern. Growing evidence indicates a link between OP pesticide exposure and the development of Parkinson’s and Alzheimer’s diseases and amyotrophic lateral sclerosis11–15, necessitating a deeper investigation into the underlying mechanisms.
OP nerve agents are known to cause acute life-threatening acetylcholinesterase inhibition. Current thinking is that GWI is not due to acetylcholinesterase inhibition but rather due to sub-threshold effects of OPs that produce a non-self-correcting state of neurodegeneration. Neuroinflammation, axonal transport deficits, microtubule impairments and dopaminergic changes have all been observed in animal (rodent) and cell culture models of GWI at OP levels below those needed to inhibit acetylcholinesterase16–18. Additional animal studies have shown that the effects of OP exposures are exacerbated when coupled with physical restraint or chemically-induced stressors designed to mimic the physical stress of the battlefield19.
We are interested in microtubule deficits that occur in the nervous system of veterans suffering from GWI, with the goal of developing microtubule-based therapies. OPs can affect a variety of proteins and pathways in cells, for example by covalently binding to tyrosine and lysine residues. The impacts on microtubules and microtubule-related proteins are likely to be many, including effects on molecular motor proteins and microtubule stability and dynamics, as well as the binding to microtubules of microtubule-associated proteins such as tau20–24. In light of the complexity, the question becomes whether particular microtubule-related deficits can be identified that when corrected result in marked improvement of GWI symptoms.
Here, we pre-treated cultured neurons with the stress hormone corticosterone (CORT) in the case of rat neurons, and cortisol in the case of human-derived neurons, and then exposed them to diisopropyl fluorophosphate (DFP), an OP compound used by researchers as a sarin-surrogate25. We then assessed various microtubule-related parameters such as microtubule dynamics and mitochondrial transport, using live-cell imaging, to ascertain potential deficits. We included in our studies analyses of dopamine release, which is a complex process that involves microtubules but many other factors as well26. Upon ascertaining that the toxin regimens resulted in diminished microtubule acetylation, we sought to determine whether a drug that increases microtubule acetylation could restore to normal observed deficits in microtubule dynamics and mitochondrial transport, as well as dopamine release. Improvement in dopamine release was taken as an indicator that this approach has the potential to effectively treat complex symptoms of GWI.
Results
Pretreatment with CORT or cortisol intensifies DFP-induced aberrations in microtubule acetylation in human and rat neurons
Human neural stem cells were differentiated into neurons and allowed to mature and form networks in vitro for one month. These human neurons, or rat fetal cortical neurons grown for 8–14 days in culture, were exposed to DFP at concentrations of 20 or 200 nM with or without pretreatment with 2 µM cortisol (for the human neurons) or CORT (for the rat neurons), and changes in tubulin acetylation were assessed (Fig. 1A). Human neuronal phenotype was confirmed by positive staining for βIII-tubulin, a neuron specific tubulin isotype, and Tbr1, a transcription factor specific to glutamatergic forebrain neurons (Fig. 1B). Immunostaining and Western blot analyses both revealed a reduction in the ratio of acetylated to total tubulin after DFP exposure (Fig. 1C and D and S1A and B, respectively). With the rat neurons, as described in Fig. 1A, the ratio was reduced after 20 and 200 nM DFP treatment, fortifying the finding from human neurons. Pretreatment with CORT or cortisone exacerbated the effect (Fig. 1E [top], 1F [left], 1G).
Figure 1. Pretreatment with CORT or cortisol intensifies DFP-induced aberrations in microtubule acetylation status in human and rat neurons.

A. Schematic summary of experimental design. B. Validation of neuronal differentiation by immunostaining. βIII-tubulin (Biolegend Cat. #801202), which is specific to neurons and is significantly upregulated in mature neurons, is colored red. Tbr1, a transcription factor which serves as a marker for forebrain cortical glutamatergic neurons, is colored green, and DAPI, which stains nuclei, is blue. Scale bar represents 50 µm.
C. Co-immunostaining of acetylated-tubulin (green; Sigma Cat. #T6793) and βIII-tubulin. The white checkered box represents the ROI to be analyzed with arrows pointing to the respective signal intensity values. Scale bar represents 15 µm. D. Bar graph depicting results from acetylated:total tubulin protein analysis of human neurons subjected to various conditions. E. Western blot acetylated-tubulin and βIII-tubulin bands from rat cortical neurons that after 24h with or without 10 µM tubacin treatment. F. Key identifying the conditions for each well. (for easier identification, empty lanes seen were loaded with sample buffer alone) G. Bar graph depicting results from Western blot analysis. Protein levels were normalized to cofilin (Abcam cat. # 42824) load control. Blots were cropped * - p<0.05; ** - p<0.01; N.S. – no significance found
We sought to restore the acetylation status of the microtubules by inhibiting the tubulin de-acetylating enzyme, HDAC6. Treatment with 1 µM tubacin significantly improved the ratio of acetylated to total tubulin after treatment with 20 nM DFP alone, but did not significantly restore the effects of 200 nM DFP alone or any CORT pretreatment groups. Treatment with 10 µM tubacin yielded significant improvements in the 20 and 200 nM DFP groups with and without CORT pretreatment (Fig. 1E [bottom], F [right], and G).
Exposure to DFP alters microtubule dynamics in a manner exacerbated by CORT and correctable by tubacin
To explore microtubule dynamics, neurons were treated as before, but on DIV 9, GFP-EB3, a microtubule end-binding protein that affiliates with the growing tips of microtubules, was ectopically expressed and microtubule growth assessed 24 h after DFP exposure. The excursion of GFP-EB3 at the plus end of the microtubule appears as a ‘comet’ because of the gradual dissociation of EB3 molecules from the microtubules27. When neurons were treated with DFP alone, microtubule comet number was decreased at 20 nM (Fig. 2B, C), but the rate of comet movement was not affected (Fig. 2D; Movie S1). CORT pretreatment enhanced the effect of DFP, causing a reduction in comet number at 2 and 20 nM DFP, and yielding a reduction in comet rate at 20 nM DFP. Tubacin restored comet number to control levels but not rate (Fig. 2D).
Figure 2. Exposure to DFP alters microtubule dynamics in a manner exacerbated by CORT and correctable by tubacin.

A. Still frames from live-cell imaging movie of EB3 comets in rat cortical neurons that received vehicle with no DFP. B. Scatter plot of the observed polymerization events per axon (20 µm region of interest). C. Horizontal bar graph depicting the average number of comets per 100 µm across treatments with or without tubacin treatment. D. Horizontal bar graph depicting the average rate of observed comets across treatment groups with or without tubacin Page 10 treatment. * - p<0.05; ** - p<0.01; *** - p<0.001
Toxicant/stress exposure disrupts mitochondrial transport in tubacin-correctable manner
Mitochondria are the primary site of energy production and have been suggested to be the target of non-cholinergic toxicity of OPs28. Past studies have indicated a link between exposure to higher concentrations of OP and oxidative stress and transport deficits after OP exposure29–31. Consistent with previous work, when neurons were treated with the DFP, transport of mitochondria was impaired and mitochondrial length increased, suggesting an increase in mitochondrial fusion. Mitochondria were stained with a live-cell mitochondrial dye called Mitotracker and live neurons were imaged (Fig. 3A). An increase in stalled mitochondria was observed when neurons were treated with DFP, and the contribution to this effect was greater when cells were pretreated with CORT compared to DFP alone (Fig. 3B, kymographs; Fig. 3C, stacked bar graph; movie S2). When treated with 10 µM tubacin, mitochondrial transport events were recovered (Fig. 3D).
Figure 3. Toxicant/stress exposure disrupts mitochondrial transport in tubacin-correctable manner.

A. Image of rat cortical neurons stained with Mitotracker Orange (Inset: magnified image of axon; ThermoFisher Scientific Cat. #M751). B. Representative kymographs depicting distance traveled over time for vehicle control (left) and ethanol + 20 nM DFP (right). Cells were imaged for a total of 2.5 minutes at 1 frame per second. C. 100% stacked bar graph showing the distribution of transport event types across conditions. D. Dot plot depicting the average number of transport events across conditions with or without tubacin treatment. * - p<0.05; ** - p<0.01.
Dopamine release is altered after DFP exposure in a manner exacerbated by CORT pretreatment
Dopamine and neurotransmission alterations have been reported after exposure to GW toxins1,4, which we hypothesize in part are due to changes in microtubules, which serve as tracks for synaptic vesicle transport. We cultured fetal ventral mesencephalic cells (dopamine precursor cells) and allowed them to grow for 7 days before submitting them to the treatment regimen (Fig. 4A). At DIV 9, cultures were heterogeneous, with 27±2% of control cells staining positive for tyrosine hydroxylase, a dopaminergic marker, and no significant difference across treatments (Fig. 4B; S2A). Supernatant was collected from cultures that were exposed to DFP with or without CORT pretreatment, and subsequently treated with tubacin at 1 µM and 10 µM, or with vehicle. Supernatant was analyzed for dopamine content using HPLC (Fig. 4C). Exposure to 2 nM and 20 nM DFP alone resulted in a significant reduction in extracellular dopamine (38±5% and 40±4%, respectively), and this reduction was even greater following CORT pretreatment (46±5% and 55±4%, Fig. 4D). Treatment with 10 µM but not 1 µM tubacin was able to fully restore dopamine release after DFP alone, and partially restore release when cells were exposed to DFP after CORT pretreatment (Fig. 4D). Tubacin did not significantly alter extracellular dopamine across control cells (Fig. S2B).
Figure 4. Dopamine release is altered after DFP exposure in a manner exacerbated by CORT pretreatment.

A. Chart outlining design of dopamine release experiments. B. Left: Bar graph showing TH positive cells across DFP treatments. Right: Co-immunostain of fetal ventral mesencephalon neurons. Inverted dark cells are neurons stained for βIII-Tubulin, and blue cells represent neurons that are positive for tyrosine hydroxylase (EMD Millipore cat. #AB152). C. Bar graph showing dopamine release as a percent of vehicle control. Values were normalized to total TH positive neuron count. D. Dot plot of each sample (4 duplicates per condition, represented by different shape and color dots [see plot legend]) in the presence or absence of tubacin at 1 and 10 µM concentrations. Top brackets show no significant difference between EtOH controls. Bottom bracket shows no significant improvement in EtOH DFP or CORT DFP groups after treatment with 1 µM tubacin, and treatment with 10 µM tubacin yielded significant improvement in all groups when compared to control. * - p<0.05; ** - p<0.01; *** - p<0.001; N.S. – no significance found. Scale bar represents 15 µm.
Discussion
The present studies are the first to use cultured rodent and human-derived neurons to demonstrate that hormonal changes resulting from chemically-induced stressors can exacerbate the negative consequences of OP exposure on microtubule and microtubule-related events in the axon. We used both human and rat neurons as a cross-validation because rat neurons are more amenable to established microtubule-related analyses, while human neurons are a better path toward therapy, given that some features of human neurodegeneration are not reflected in rodents32,33. In the future, an even more refined option would be to use induced pluripotent cell lines from the GW veterans themselves, given that genetic and possibly epigenetic factors potentially relevant to disease susceptibility would be preserved34. For now, the results on the human cells confirm that, as with the rat neurons, DFP causes a decrease in tubulin acetylation. The work on rat neurons indicates that pharmacologic restoration of tubulin acetylation, lowered by DFP±CORT, rescues various cellular events impaired by DFP±CORT.
While microtubule acetylation generally correlates with microtubule stability, the correlation depends upon the levels and activity of the enzyme(s) that acetylates tubulin after it becomes incorporated into the microtubule, as well as the enzyme(s) that deacetylates the tubulin after it is released from the microtubule during bouts of disassembly35–37. The live imaging of microtubule dynamics shows DFP/CORT reduces the number of assembly excursions, which is not easily explained by diminished microtubule stability. Tubacin corrected for the deficit in microtubule dynamics, presumably because various proteins, such as microtubule-severing proteins, interact differently with the microtubule, depending on its acetylation status35. The finding that a higher tubacin concentration is needed to correct the acetylation deficit produced by higher DFP concentration suggests that the DFP/CORT may affect the levels or activity of the relevant enzymes. For example, if DFP/CORT increases HDAC6 levels or activity, a higher concentration of tubacin would be needed to inhibit the available enzyme.
OPs used at sub-threshold levels undoubtedly affect a variety of proteins and pathways, some of which are obviously microtubule-related and others of which are not as direct or obvious in their relationship to microtubules. In terms of therapy for sufferers of GWI, the question is whether restoring microtubule acetylation to a more normal status will translate into improvements in cognition and sleep, and help alleviate headaches and other symptoms of the disease. Deficits in mitochondria transport could explain some GWI symptoms, such as fatigue, and hence HDAC6 inhibitors might be helpful in treating those symptoms. We are especially encouraged by our dopamine release results, which suggest that a complex process directly related to neurotransmission, and one that could potentially go awry at many different levels, can be corrected by this treatment.
Reports exist in the literature of other neurodegenerative diseases that involve microtubule deacetylation, with symptoms improved in animal models by HDAC6 inhibitors38–41. In that regard, efforts are already underway to develop therapeutics that can be translated to human patients. Our next step is to test the effectiveness of such an approach on an animal model for GWI. From there, translation to human patients using new generations of HDAC6 inhibitors will be our goal.
In conclusion, the results presented here provide new mechanistic clues to the cellular basis of GWI, and point to a therapeutic strategy that may reverse neuronal decline in sufferers.
Materials and Methods
Cell Culture
Rat Cortical Neurons
Primary cortical neurons were prepared as previously described42. Briefly, the pregnant Sprague-Dawley rat was euthanized on E18 in accordance with Public Health Services Policy on Humane Care and Use of Laboratory Animals, and fetuses were removed. The cortices from both hemispheres of each fetal rat brain were excised, dissociated, and plated at a concentration of 500,000 cells per well in 6-well plates (for Western blots) or 35,000 cells per well in glass-bottomed dishes (for live-cell imaging).
Rat Ventral Mesencephalon Neurons
The ventral mesencephalon was isolated from fetuses removed from a Sprague-Dawley rat on E14. Cells were isolated and dissociated according to a previously published protocol43.
Human neural stem cells
Human IPSC-derived neural stem cells (ax0018), differentiation media, expansion media, and maintenance media were purchased from Axol and cultures were established and differentiated into neurons according to the Axol Human IPSC-Derived Neural Stem Cell Protocol v5.0.
Treatment Preparation and Application
CORT or cortisol (Sigma), dissolved in ethanol to make a 2 mM stock solution, was diluted in culture media to make working concentrations. DFP (Sigma), dissolved in isopropanol to make a 100 µM stock solution, was diluted in culture media to make working concentrations. Experimental design is shown in Fig. 1A. Cells were assayed at DIV 9 (no tubacin) or DIV 10 (24h tubacin). The two lower concentrations of DFP used in this study are sub-threshold (below the level need to inhibit acetylcholinesterase), while the 200 nM concentration may partially inhibit acetylcholinesterase.
Live-cell Imaging
Assaying Microtubule-based Transport of Mitochondria
Mitotracker CMH2TMRos Orange was added directly into the media of primary cortical neuron cultures and incubated at 37C for 25 minutes. After incubation, dishes were washed twice with serum-free media, and Fluorobrite DMEM imaging medium was added immediately before imaging. Dishes were imaged using a Zeiss AxioObserver Z1 inverted microscope equipped with a 1.4 NA 63X oil objective and an AxioCam mRm CCD camera. Images were captured every 1 second for 2.5 minutes at a fixed exposure time across conditions. Transport was analyzed using the MultiKymograph and DifferenceTracker plugins for (FIJI) ImageJ.
Assessing Changes in Microtubule Dynamics
Rat cortical neurons were plated at a density of 35,000 cells per well, allowed to grow for 8 days, and transfected to express GFP-tagged EB3 using Lipofectamine 2000 (ThermoFisher) according to the published product protocol. Cells were imaged using a Zeiss AxioObserver Z1 inverted scope equipped with a 1.46 NA 100X oil objective and a Zeiss Axiocam 506 mono CCD camera. Images were captured every 1 second for a total of 2.5 minutes at a fixed exposure time across conditions. Movies were analyzed using the MultiKymograph plugin for FIJI (ImageJ).
Determination of Released Dopamine
after OP Exposure Extracellular dopamine was quantified by electrochemical detection high performance liquid chromatography (HPLC-ECD). Reagents, chemicals, standard preparation, and mobile phase were prepared as previously published44.
Protein Level Analysis
Whole cell lysate collection
Lysis buffer was prepared according to the Santa Cruz RIPA buffer lysis system (Santa Cruz; Cat. # sc-24948A) instructions with added phosphatase inhibitor. The media from culture wells were aspirated on DIV 10, and 200 µL of lysis buffer was added. Cells were detached using a cell scraper for 1 minute per well, and the lysis solution containing the cells was transferred to Eppendorf tubes. Lysate was incubated on ice for 45 minutes, sonicated, and centrifuged at 10,000 RPM for 10 minutes at 4°C. Supernatant was aliquoted and stored at −80 °C until use.
Protein Quantification and Western Blotting
Protein content of each sample was quantified using the Thermo Scientific Pierce BCA Protein Assay Kit (ThermoFisher Scientific; Cat. # 23227). Western blotting was performed using standard methods, developed using the Bio-Rad Chemidoc MP Imaging System, and quantified using the Bio-Rad Image Lab software. Antibody information is provided in figure legends.
Tubacin Treatment
Tubacin (Cayman Chemical; Cat. # 537049-40-4) was used at a concentration of 1 or 10 µM, as established previously45. Tubacin was added into culture media immediately after the 24 h DFP treatment and incubated with cells for 24 h. Cells were then either lysed for Western blotting or imaged to assess mitochondrial transport or microtubule dynamics. Supernatant was collected after 24 h-post tubacin.
Experimental Design and Data Analysis
Western blots and dopamine experiments were repeated four times to ensure reproducibility and power. Live-cell imaging experiments were performed using tissue from three different animals with at least 15 cells across 4 dishes per animal. Data were analyzed by blinded raters and statistical significance was assessed by student t-test, ANOVA followed by post-hoc Bonferroni test, or the Kruskal-Wallis non-parametric ANOVA. All statistical analyses were performed using the IBM SPSS program.
Supplementary Material
Acknowledgments
We thank members of the Gulf War Illness Consortium for helpful discussions. This work was funded by grants from the US DoD Congressionally Directed Medical Research Program (CDMRP) to PWB (W81XWH-15-1-0433 and W81XWH-07-1-0466) and to the GWI Consortium (W81XWH-13-2-0072) directed by KAS, with subcontracts to PWB and MMB. ANR is supported by an F31 Award from the NIH (1F31NS093748-01A1). Neurotransmitter work was partially funded by a grant from the NIH to RAE (R01DA031900). Additional funding was provided to PWB by the NIH (R01NS28785) and the Pennsylvania Department of Health CURE program to Drexel University College of Medicine.
References
- 1.(RAC) RAC. Gulf War Illness and the Health of Gulf War Veterans: Scientific Findings and Recommendations. 2008 [Google Scholar]
- 2.Sullivan K, Krengel M, Proctor SP, Devine S, Heeren T, White RF. Cognitive functioning in treatment-seeking Gulf War veterans: pyridostigmine bromide use and PTSD. Journal of Psychopathology and Behavioral Assessment. 2003;25(2):95–103. [Google Scholar]
- 3.Proctor SP, Heaton KJ, Heeren T, White RF. Effects of sarin and cyclosarin exposure during the 1991 Gulf War on neurobehavioral functioning in US army veterans. Neurotoxicology. 2006;27(6):931–939. doi: 10.1016/j.neuro.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 4.White RF, Steele L, O'Callaghan JP, et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex. 2016;74:449–475. doi: 10.1016/j.cortex.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haley RW, Tuite JJ. Epidemiologic evidence of health effects from long-distance transit of chemical weapons fallout from bombing early in the 1991 Persian Gulf War. Neuroepidemiology. 2012;40(3):178–189. doi: 10.1159/000345124. [DOI] [PubMed] [Google Scholar]
- 6.Steele L, Sastre A, Gerkovich MM, Cook MR. Complex factors in the etiology of Gulf War illness: wartime exposures and risk factors in veteran subgroups. Environmental health perspectives. 2012;120(1):112. doi: 10.1289/ehp.1003399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heaton KJ, Palumbo CL, Proctor SP, Killiany RJ, Yurgelun-Todd DA, White RF. Quantitative magnetic resonance brain imaging in US army veterans of the 1991 Gulf War potentially exposed to sarin and cyclosarin. Neurotoxicology. 2007;28(4):761–769. doi: 10.1016/j.neuro.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Phillips KF, Deshpande LS. Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War Illness. Neurotoxicology. 2016;52:127–133. doi: 10.1016/j.neuro.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Ismail AA, Bodner T, Rohlman D. Neurobehavioral performance among agricultural workers and pesticide applicators: a meta-analytic study. Occupational and environmental medicine. 2012;69(7):457–464. doi: 10.1136/oemed-2011-100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.EPA U. Pesticides Industry Sales and Usage 2006 and 2007 Market Estimates. Pesticides Industry Sales and Usage Reports. 2011 [Google Scholar]
- 11.Yadav SS, Singh MK, Yadav RS. Organophosphates Induced Alzheimer's Disease: An Epigenetic Aspect. Journal of Clinical Epigenetics. 2016 [Google Scholar]
- 12.Wang A, Cockburn M, Ly TT, Bronstein JM, Ritz B. The association between ambient exposure to organophosphates and Parkinson's disease risk. Occupational and environmental medicine. 2014 doi: 10.1136/oemed-2013-101394. oemed-2013-101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baltazar MT, Dinis-Oliveira RJ, de Lourdes Bastos M, Tsatsakis AM, Duarte JA, Carvalho F. Pesticides exposure as etiological factors of Parkinson's disease and other neurodegenerative diseases—a mechanistic approach. Toxicology letters. 2014;230(2):85–103. doi: 10.1016/j.toxlet.2014.01.039. [DOI] [PubMed] [Google Scholar]
- 14.Hayden KM, Norton MC, Darcey D, et al. Occupational exposure to pesticides increases the risk of incident AD The Cache County Study. Neurology. 2010;74(19):1524–1530. doi: 10.1212/WNL.0b013e3181dd4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merwin SJ, Obis T, Nunez Y, Re DB. Organophosphate neurotoxicity to the voluntary motor system on the trail of environment-caused amyotrophic lateral sclerosis: the known, the misknown, and the unknown. Archives of Toxicology. 2017:1–14. doi: 10.1007/s00204-016-1926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Callaghan JP, Kelly KA, Locker AR, Miller DB, Lasley SM. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J Neurochem. 2015;133(5):708–721. doi: 10.1111/jnc.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terry AV, Jr, Gearhart DA, Beck WD, Jr, et al. Chronic, intermittent exposure to chlorpyrifos in rats: protracted effects on axonal transport, neurotrophin receptors, cholinergic markers, and information processing. J Pharmacol Exp Ther. 2007;322(3):1117–1128. doi: 10.1124/jpet.107.125625. [DOI] [PubMed] [Google Scholar]
- 18.Terry AV, Jr, Callahan PM, Beck WD, et al. Repeated exposures to diisopropylfluorophosphate result in impairments of sustained attention and persistent alterations of inhibitory response control in rats. Neurotoxicol Teratol. 2014;44:18–29. doi: 10.1016/j.ntt.2014.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parihar VK, Hattiangady B, Shuai B, Shetty AK. Mood and memory deficits in a model of Gulf War illness are linked with reduced neurogenesis, partial neuron loss, and mild inflammation in the hippocampus. Neuropsychopharmacology. 2013;38(12):2348–2362. doi: 10.1038/npp.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang W, Duysen EG, Hansen H, Shlyakhtenko L, Schopfer LM, Lockridge O. Mice Treated with Chlorpyrifos or Chlorpyrifos Oxon Have Organophosphorylated Tubulin in the Brain and Disrupted Microtubule Structures, Suggesting a Role for Tubulin in Neurotoxicity Associated with Exposure to Organophosphorus Agents. Toxicological Sciences. 2010;115(1):183–193. doi: 10.1093/toxsci/kfq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seifert J, Casida JE. Possible role of microtubules and associated proteases in organophosphorus ester-induced delayed neurotoxicity. Biochemical Pharmacology. 1982;31(11):2065–2070. doi: 10.1016/0006-2952(82)90422-1. [DOI] [PubMed] [Google Scholar]
- 22.Lockridge O, Schopfer LM. Review of tyrosine and lysine as new motifs for organophosphate binding to proteins that have no active site serine. Chemico-biological interactions. 2010;187(1):344–348. doi: 10.1016/j.cbi.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gearhart DA, Sickles DW, Buccafusco JJ, Prendergast MA, Terry AV. Chlorpyrifos, chlorpyrifos-oxon, and diisopropylfluorophosphate inhibit kinesin-dependent microtubule motility. Toxicology and applied pharmacology. 2007;218(1):20–29. doi: 10.1016/j.taap.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Grigoryan H, Schopfer LM, Peeples ES, et al. Mass spectrometry identifies multiple organophosphorylated sites on tubulin. Toxicology and applied pharmacology. 2009;240(2):149–158. doi: 10.1016/j.taap.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Lein PJ, Liu C, et al. Spatiotemporal pattern of neuronal injury induced by DFP in rats: a model for delayed neuronal cell death following acute OP intoxication. Toxicol Appl Pharmacol. 2011;253(3):261–269. doi: 10.1016/j.taap.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gardiner J, Overall R, Marc J. The microtubule cytoskeleton acts as a key downstream effector of neurotransmitter signaling. Synapse (New York, NY) 2011;65(3):249–256. doi: 10.1002/syn.20841. [DOI] [PubMed] [Google Scholar]
- 27.Stepanova T, Slemmer J, Hoogenraad CC, et al. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23(7):2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masoud A, Kiran R, Sandhir R. Impaired mitochondrial functions in organophosphate induced delayed neuropathy in rats. Cellular and molecular neurobiology. 2009;29(8):1245–1255. doi: 10.1007/s10571-009-9420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soltaninejad K, Abdollahi M. Current opinion on the science of organophosphate pesticides and toxic stress: a systematic review. Med Sci Monit. 2009;15(3):RA75–90. [PubMed] [Google Scholar]
- 30.Morfini GA, Burns M, Binder LI, et al. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29(41):12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao J, Naughton SX, Wulff H, et al. Diisopropylfluorophosphate impairs the transport of membrane-bound organelles in rat cortical axons. Journal of Pharmacology and Experimental Therapeutics. 2016;356(3):645–655. doi: 10.1124/jpet.115.230839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mestas J, Hughes CCW. Of Mice and Not Men: Differences between Mouse and Human Immunology. The Journal of Immunology. 2004;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 33.Götz J, Deters N, Doldissen A, et al. A Decade of Tau Transgenic Animal Models and Beyond. Brain Pathology. 2007;17(1):91–103. doi: 10.1111/j.1750-3639.2007.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiang L, Rao AN, Mostoslavsky G, et al. Reprogramming cells from Gulf War veterans into neurons to study Gulf War illness. Neurology. 2017 doi: 10.1212/WNL.0000000000003938. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howes SC, Alushin GM, Shida T, Nachury MV, Nogales E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Molecular biology of the cell. 2014;25(2):257–266. doi: 10.1091/mbc.E13-07-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kull FJ, Sloboda Roger D. A Slow Dance for Microtubule Acetylation. Cell. 2014;157(6):1255–1256. doi: 10.1016/j.cell.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 37.Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. Journal of Cell Science. 2009;122(19):3531–3541. doi: 10.1242/jcs.046813. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Leng Y, Wang J, et al. Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of α-tubulin acetylation and FGF-21 up-regulation. Scientific Reports. 2016;6:19626. doi: 10.1038/srep19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.d'Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- 40.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin. Journal of Biological Chemistry. 2005;280(48):40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 41.Rivieccio MA, Brochier C, Willis DE, et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proceedings of the National Academy of Sciences. 2009;106(46):19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pacifici M, Peruzzi F. Isolation and culture of rat embryonic neural cells: a quick protocol. Journal of visualized experiments: JoVE. 2012;(63) doi: 10.3791/3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pruszak J, Just L, Isacson O, Nikkhah G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr Protoc Stem Cell Biol. 2009 doi: 10.1002/9780470151808.sc02d05s11. Chapter 2:Unit 2D 5. [DOI] [PubMed] [Google Scholar]
- 44.Brodnik ZD, Jaskiw GE. Effect of Mobile Phase pH on the Function of Other Optimization Parameters in an HPLC–ECD Assay of Biogenic Amines and Their Metabolites. Journal of Liquid Chromatography & Related Technologies. 2015;38(4):467–471. [Google Scholar]
- 45.Sudo H, Baas PW. Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J Neurosci. 2010;30(21):7215–7226. doi: 10.1523/JNEUROSCI.0048-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.