

Abstract
We have performed μs molecular dynamics simulation (MDS) to identify structural mechanisms for sarcolipin (SLN) uncoupling of Ca2+ transport from ATP hydrolysis for the sarcoplasmic reticulum Ca2+-ATPase (SERCA). SLN regulates muscle metabolism and energy expenditure to provide resistance against diet-induced obesity and extreme cold. MDS demonstrates that the cytosolic domain of SLN induces a salt bridge-mediated structural rearrangement in the energy-transduction domain of SERCA. We propose that this structural change uncouples SERCA by perturbing Ca2+ occlusion at residue E309 in transport site II, thus facilitating cytosolic Ca2+ backflux. Our results have important implications for designing muscle-based therapies for human obesity.
Graphical abstract
The sarcoplasmic reticulum Ca2+-ATPase (SERCA) is the membrane protein responsible for transporting Ca2+ from the cytosol into the sarcoplasmic reticulum (SR) lumen of myocytes, causing muscle to relax and establishing the Ca2+ store to be released for subsequent contraction.1 SERCA pumps two Ca2+ ions per catalytic cycle using energy derived from hydrolysis of one ATP molecule and counter-transport of two to three proton atoms.2 In skeletal and cardiac muscle, SERCA activity is reversible regulated by two membrane phosphoproteins, the 31-residue sarcolipin (SLN) and the 52-residue phospholamban (PLB). SLN is expressed predominantly in fast-twitch skeletal muscle and atria, whereas PLB is expressed predominantly in ventricles and slow-twitch skeletal muscle.3,4 Spectroscopy and crystallography show that SLN and PLB bind SERCA in 1:1 heterodimeric regulatory complexes,5–7 with distinct functional outcomes. Unphosphorylated SLN and unphosphorylated PLB suppress SERCA activity by inhibiting calcium affinity (KCa = Ca2+ concentration at half-maximal activation), which is relieved by phosphorylation of SLN or PLB, and by micromolar cytosolic Ca2+,8,9 thus regulating SERCA activity and muscle contractility during rest and work. Furthermore, SLN uncouples Ca2+ transport from ATP hydrolysis by SERCA, stimulating unproductive ATP hydrolysis and heat production,10,11 thereby contributing to non-shivering thermogenesis in skeletal muscle.12
X-ray crystal structures of SERCA show two bound Ca2+ ions in the transmembrane (TM) domain13 and one catalytic nucleotide-binding site in the cytosolic headpiece14. The coupling ratio of SERCA, defined as the number of Ca2+ ions transported per ATP molecule hydrolyzed, has a maximum value of two under ideal conditions, such as SR vesicles empty of Ca2+ and extravesicular pH ~7.0.15–17 In mouse transgenic models, SLN enhances energy expenditure and heat production to provide resistance against diet-induced obesity and extreme cold.18 While SLN decreases SERCA coupling, both SLN and PLB reduce the Ca2+ affinity of SERCA19 by populating a similar ion-free E1 intermediate protonated at residue Glu771 (E1•H+771) in TM transport site I.20,21 Thus, SLN must induce structural changes in SERCA distinct from those induced by PLB, but the structural mechanism(s) responsible for internal SERCA uncoupling by SLN remain unclear.
Here we point out that published x-ray crystal structures of SERCA‒SLN (Protein Data Bank (PDB) 3w5a6 and 4h1w7) and SERCA‒PLB (PDB 4kyt22) reveal unique effects of the two regulatory subunits on SERCA structure. The two crystal structures of SLN-bound SERCA were superposed on the crystal structure of PLB-bound SERCA for measurement of the relative root-mean-square deviation (RMSD) for each TM helix (Figure 1). Except for helices M1 and M4, the relative RMSD values are small (<0.1 nm) (Figure 1A), indicating that the structures of most TM helices are virtually identical for SERCA bound to SLN versus PLB. M1 does not interact directly with either regulatory subunit, so the relatively high RMSD value of 0.16 nm is probably due to the inherent flexibility of this helix.25 M4 shows the highest relative RMSD (0.19 nm) of all SERCA TM helices. RMSD analysis of M4, as divided into luminal and cytosolic segments R290-A306 and P312-K329, reveals that the predominant relative change in the position of M4 occurs in the cytosolic half of this helix and extends up into the cytosolic stalk helix 4 (M4S4, residues P312-K329) (Figure 1A, inset). Helix M4S4 exhibits a 17° increase in the axial tilt angle, relative to the membrane normal, in SLN-bound SERCA structures compared to PLB (Figure 1).6,7 The Ca2+ binding residue glutamate 309 (E309) in transport site II is positioned at the central break of the M4 helix in SLN- and PLB-bound SERCA structures (Figure 1).
Figure 1. Comparative analysis of SERCA‒SLN and SERCA‒PLB crystal structures.
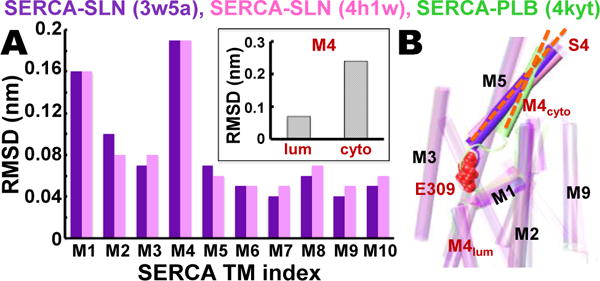
(A) RMSD for each TM helix in SERCA‒SLN crystal structures 3w5a6 (purple) and 4h1w7 (pink) was calculated relative to TM helices in the SERCA‒PLB crystal structure 4kyt22. The inset shows RMSD of M4S4 divided into luminal and cytosolic segments R290-A306 (lum) and P312-K329 (cyto). (B) Superposed crystal structures of SERCA‒SLN (purple, pink) and SERCA‒PLB (green) reveal that SLN, but not PLB, mediates a large increase in the axial tilt angle (orange dashes) of M4S4. SERCA residue E309 (red spacefill) is the Ca2+ binding residue in transport site II in the TM domain that transmits the ‘full-occupancy’ signal to initiate ATP hydrolysis and phosphoenzyme formation at D351 in the phosphorylation domain.23,24
M4S4 is in close proximity to the N-terminus of both PLB and SLN, so it is possible that a change in M4S4 tilt is due to SERCA-SERCA interactions in the crystal lattice. However, this is unlikely because the structural arrangement of SERCA is similar between SLN- and PLB-bound structures (RMSD≤0.4 nm). Therefore, we searched for intermolecular contacts that could be responsible for control of SERCA coupling. Detailed structural analysis revealed that both SERCA‒SLN crystal structures6,7 show the presence of electrostatic interactions between residue E2 of SLN and residues R324 and K328 of M4S4, forming a bifurcated salt bridge between these three residues (Figure 2A). These salt bridge interactions are not present in the SERCA‒PLB complex (Figure S1), so which suggests that these interactions are responsible for the SLN-induced changes in M4S4. However, it is uncertain whether these SERCA‒SLN intermolecular contacts (M4S4‒N-terimnus) represent native contacts in vivo, or if they are artifacts due to non-physiological crystallographic conditions. Therefore, we used the co-crystal structure of SLN bound to SERCA in the Mg2+-bound E1 state (rabbit isoforms of SLN and SERCA1a; PDB 3w5a6) to perform three independent 1-μs MDS of the native complex in a hydrated lipid bilayer. Based on our previous studies, 20,21,26 we simulated all SERCA‒SLN systems at appropriate physiological conditions by removing Mg2+ from the Ca2+ transport sites, and by modeling transport-site residues E309 (M4) and D800 (M6) as ionized, and E771 (M5) and E908 (M8) as protonated. We used the NAMD 2.11 program27 and the CHARMM36 force field to simulate SERCA‒SLN for 1 μs. Detailed computational protocols are in Supporting Information.
Figure 2. Intermolecular salt bridges between the N-terminus of SLN and M4S4 of SERCA.
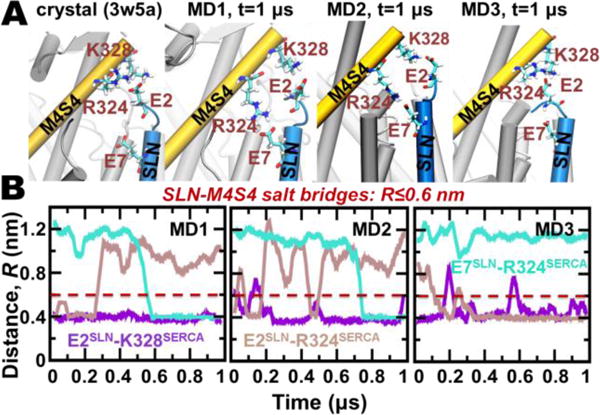
A) Salt bridges between SLN residues E2/E7 and SERCA residues R324/K328 are identified in crystal structure 3w5a6 (left) and at the end of each 1-μs MDS trajectories (right). SERCA and SLN are shown as cartoons, and residues participating in salt-bridge interactions are shown as sticks. (B). MDS distance evolution is shown between E2SLN‒K328SERCA, E2SLN‒R324SERCA, and E7SLN‒R324SERCA. Distances between E‒K and E‒R residue pairs were calculated between Cδ‒Cζ and Cδ‒Nζ atoms, respectively. The cutoff distance for salt bridge interaction using these distance definitions is 0.6 nm.30
MDS trajectory analysis showed that the salt bridge between residues E2 (SLN) and K328 (SERCA) found in the crystal structures is stable in all three MDS in a solvated lipid bilayer (Figure 2B, purple line). Conversely, the salt bridge E2‒R324 in the crystal structure is stable only in a single trajectory (MD3) and is completely disrupted in trajectories MD1 and MD2 at t=0.26 and t=0.5 μs (Figure 2B, brown). Instead, R324 forms a new salt bridge with residues E7 of SLN, as observed in two of three trajectories (MD1 and MD2 in Figure 2B, cyan). The new salt bridge E7‒R324 is formed at t=0.6–0.7 μs, and remains stable for the remainder of the 1-μs simulation (Figure 2B). Thus, these simulations indicate that initial salt bridges from E2 to R324 and K328 in crystal structures are relatively stable, yet are replaced preferentially by two separate salt bridges, E2‒R324 and E7‒ K328, in MDS. Here we find that SLN residue R3 does not interact with M1 residue E45 of SERCA, as predicted by ns MDS of mouse E7C-SLN and rabbit SERCA.28 SLN residue E7 and SERCA residues R324 and K328 are highly conserved in mammalian species; E2 of SLN is less conserved and not encoded by human SLN (Figure S2).
We predict that disrupting salt bridge(s) E2‒K328, E2–R324, and/or E7‒R324 would reverse SLN-induced tilt of M4S4. To test this hypothesis, we performed three independent 1-μs MDS of SERCA bound to SLN point mutant E7C or N-terminal mutants truncated after residue R6 (Δ6-SLN) or E7 (Δ7-SLN). As with WT-SLN, we used the crystal structure of SERCA‒SLN 3w5a6 as the starting point to model mutant SLN-SERCA constructs. Detailed computational protocols are in Supporting Information. The truncated mutants were chosen for MDS analysis because deletion of SLN N-terminus leads to loss of uncoupling function while retaining SLN binding to SERCA,28 whereas E7C-SLN was chosen because it has been studied extensively.28,29 Comparative analysis of the trajectories shows that the TM domain of SERCA undergoes a small change (<0.15 nm) in RMSD, and the RMSD values remained virtually constant during 1 μs for all SERCA-SLN constructs (Figure S3). Root-mean-square fluctuation (RMSF) and secondary structure analyses indicate that E7C-SLN, Δ6-SLN and Δ7-SLN show similar structural properties (RMSF) as WT-SLN bound to SERCA (Figures S4 and S5). Cross-linking experiments showed that SLN truncated at the N-terminus (Δ2–6) constitutively binds to SERCA, indicating that SLN binding to SERCA is mediated mostly by TM interactions.28,29 This experimental evidence is corroborated by our MDS, which determined that deletion of intermolecular SERCA-SLN salt bridges does not alter the stability of the SERCA-SLN complex.
After establishing that SERCA‒SLN complexes are stable for 1 μs MDS, we measured the effects of WT-SLN, E7C-SLN, Δ6-SLN, and Δ7-SLN on the structural arrangement of M4S4 (Figure 3). The axial tilt angle of M4S4 in SERCA‒SLN complexes was measured relative to the SERCA‒PLB crystal structure (Figure 3B, purple). In SERCA bound to WT-SLN, M4S4 maintains high tilt in all three 1-μs trajectories (Figure 3A, red); the average tilt angle for this segment is 17.5 ± 1.4° (mean ± standard error, n=3) by MDS, in excellent agreement with the 17° tilt calculated from crystal structures of SERCA‒SLN. Thus, SLN-induced M4S4 tilt is not an artifact of crystal packing. We found that M4S4 is also tilted in trajectories of SERCA bound to Δ6-SLN and E7C-SLN (Figure 3 blue and orange); the calculated tilt angle for this segment is 17.7 ± 1.5° (n=3) and 16.9 ± 1.9° (n=3) for Δ6-SLN and E7C-SLN, respectively, indicating that the presence of either E2 or E7 is sufficient to induce M4S4 tilt similar to WT-SLN. Conversely, in SERCA bound to Δ7-SLN, the tilt angle of M4S4 is similar to that calculated from the crystal structure of SERCA‒PLB (Figure 3A, green vs. magenta)22 and 1-μs trajectory of SERCA‒PLB (Figure 3B, magenta). In trajectories MD1 and MD2, this loss of tilt angle occurs at t=0.32 μs and t=0.51 μs; in both cases, the tilt angle of M4S4 settles at a plateau of ~3° for the remainder of 1 μs simulation (Figure 3B). In the MD3 trajectory, the tilt angle also decreases, but stepwise at t=0.40 μs and t≥0.66 μs to a value of ~7° (Figure 3B, green).
Figure 3. Effect of SLN mutation and truncation on M4S4 tilt angle.
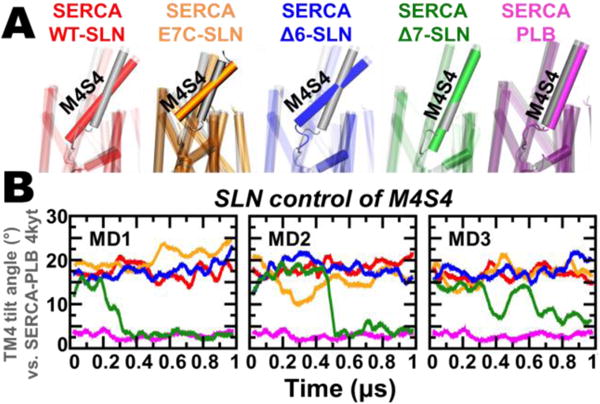
(A) Superposition of representative SERCA‒SLN constructs and SERCA‒PLB from MDS (colored) onto the crystal structure of SERCA‒ PLB (gray).22 (B) Time-dependent evolution of M4S4 tilt angle for SERCA‒SLN constructs and SERCA‒PLB. The tilt angle of M4S4 was calculated using backbone alignment, with SERCA‒PLB crystal structure 4kyt22 as reference.
The tilted orientation of M4S4 in SERCA bound to Δ6-SLN and E7C-SLN correlates with the stability of salt bridges E2‒K328/E2‒R328 and E7‒R328, respectively (Figure S6). Although E–K interaction is weaker than E–R in solution,31 we found that E2‒K328/E2–R324 salt bridges between E7C-SLN and SERCA are sufficient to induce M4S4 tilt (Figure 3B, orange). Only Δ7-SLN, which lacks both E2 and E7, reverses this structural change, with M4S4 adopting an orientation similar to that in the SERCA‒ PLB complex. We note that these structural shifts correlate with in vitro functional data showing that (a) mouse SLN constructs having one of the two glutamate residues in the N-terminus, such as E7C-SLN (i.e., E2-only SLN), produce SERCA uncoupling similar to WT-SLN28,29 and (b) SLN lacking both E2 and E7 (i.e., Δ2–6-E7C-SLN) leads to loss of uncoupling.28 These results are important, since human SLN has only one acidic residue (E7), with the N-terminal sequence 1MGINTRELFL10 (Figure S2), and because human SLN has not been tested for uncoupling function. Since human SLN protein contains E7, we predict that human SLN is sufficient to mediate M4S4 tilt and SERCA uncoupling, and thereby plays a role in human metabolism.
SLN plays a dual role in SERCA regulation: inhibition of calcium binding and uncoupling of calcium transport from ATP hydrolysis. We have recently shown that PLB and SLN inhibit the KCa of SERCA by distorting the atomic geometry of the two transport sites,20,21 so here we investigated whether changes in the tilt angle of M4S4 are independent of KCa regulation. We find that E7C-SLN and Δ6-SLN produce a transport site geometry that is very similar to that produced by WT-SLN and PLB (Figure S7), while maintaining M4S4 tilt. On the other hand, Δ7-SLN reverses the change in the tilt angle of M4S4 (Figure 3), while maintaining Ca2+ transport site geometry (Figure S7) associated with KCa inhibition.20,21 These findings indicate that SLN regulation of SERCA coupling ratio and calcium affinity operates through two separate structural mechanisms mediated by the cytosolic N-terminus (Figure 3) and the TM/luminal domain of SLN32,33, respectively.
Site-directed mutagenesis studies have identified 29 SERCA residues as critical for coupling Ca2+ transport to ATP hydrolysis: 14 residues on the entire M4S4 (between L297-K329), 12 residues in the TM domain (M5, M6), 2 residues in the lumen (loop 78), and 1 residue in the P domain (C344) (summarized in Figure S8).34–40 None of these mutants have been reported to show Ca2+-independent ATPase activity. Together, these 29 residues comprise the energy-transduction domain of SERCA (Figure S8).34,36–39 The cytosolic half of M4S4 links E309 (transport site II) in the TM domain to D351 (phosphoenzyme formation site) in the P domain, and mutation of M4S4 residues P312, I315, L319, L321, and K329 result in uncoupling.37 Here we find that the N-terminus of SLN has direct salt-bridge interactions with M4S4 to control tilt angle, and this structural change, which is not induced by PLB, is reversed by deletion of SLN residues E2 and E7 (Figure 3).We propose that salt bridge-mediated change in M4S4 tilt angle is directly responsible for SLN uncoupling of SERCA.
By what structural mechanism does SLN uncouple SERCA? Here we summarize our primary hypothesis. M4S4 is directly connected to Ca2+ binding residue E309 on M4, which is part of transport site II. E309 plays a central role in coupling ATPase activity and Ca2+ transport, as well as phosphoenzyme dephosphorylation and proton counter-transport, acting as the gate-keeper residue that transmits the ‘Ca2+ occupancy’ signal to initiate ATP hydrolysis and SERCA phosphoenzyme formation at D351. 23,24 Ca2+ binding by E309 in transport site II is required to activate ATP hydrolysis, which is the same requirement for both SLN-bound and SLN-free SERCA.23,24,36,39 Our MDS indicate that SLN-mediated change in M4S4 tilt shifts E309 position by 0.35–0.50 nm (Figure S9). We propose that SLN-induced M4S4 tilt perturbs Ca2+ occlusion in by E309, such that Ca2+ bound in site II is not transported in every catalytic cycle, thus allowing Ca2+ backflux to the cytosol. Based on compelling kinetic and mutagenesis analyses of SERCA gating,41,42 we propose that SLN uncoupling occurs before transport site interconversion in slippage intermediate(s) E1P•ADP•2Ca2+ and/or E1P•2Ca2+, prior to E2P•2Ca2+ formation, i.e., before Y122 hydrophobic cluster formation and luminal M2 gate opening.41,42
Do additional mechanism(s) play a role in SLN uncoupling of SERCA? Here we discuss five alternative possibilities. (1) M1 of SERCA: a previous ns MDS of E7C-SLN suggested that E2 of SLN interacts with M1 of SERCA;28 but our μs simulations indicate that SLN (WT or E7C) does not interact with helix M1. X-ray crystal structures6,7 and our MDS consistently identify SLN N-terminus interaction with M4S4. (2) Phosphorylation domain of SERCA: M4S4 is directly connected to the phosphorylation (P) domain of SERCA, with the auto-phosphorylation site D351 directly connected to S4, so it is possible that SLN-induced changes in P domain orientation are linked to SERCA uncoupling. However, we rule out this possibility, because MDS indicate that the nucleotide and phosphorylation domains adopt catalytically competent structural arrangement in SERCA bound to WT-SLN, E7C-SLN, and Δ6-SLN, e.g., RMSD<0.35 nm (Figure S3) relative to the crystal structure of Ca2+ and ATP-bound SERCA,43 and because SLN binding to SERCA has no significant effect on the rate of ATP hydrolysis during uncoupling of Ca2+ transport.28,29 (3) Phosphorylation of SLN: Phosphorylation of SLN at S4 or T5 relieves KCa inhibition, but the effect on coupling ratio is unknown. It is likely that phosphorylation of SLN will affect SERCA coupling ratio by introducing new charge-mediated interactions with M4S4. (4) Acylation of SLN: C9 of SLN is reportedly acylated in rabbit and pig skeletal muscle, which decreases the coupling ratio of SERCA.44 However, acylation of C9 is not observed in crystal structures of rabbit SERCA–SLN, and acylation conditions do not disrupt S4M4–SLN interaction nor anchor the SLN N-terminus to the membrane.44 Other species do not encode C9 in SLN and are instead substituted with F9 (e.g., SLN from human, mouse, rat, chicken, frog, and fish).29 (5) High luminal Ca2+: It is well-established that the experimentally-measured coupling ratio of SERCA decreases as intravesicular Ca2+ rises in SR vesicles.16,45 This decrease in apparent transport efficiency has been attributed to a variety of pathways, including Ca2+ leak across the membrane, Ca2+ slippage through SERCA, and enhancement of SLN uncoupling function by luminal Ca2+.45
In conclusion, we have used all-atom μs MDS to identify the structural mechanism for SLN uncoupling of SERCA. These findings indicate that SLN induces a unique salt bridge-mediated structural rearrangement of M4S4 in the energy-transduction domain of SERCA, and that SLN-mediated uncoupling is structurally distinct from KCa inhibition by SLN and PLB. The salt bridges responsible for uncoupling involve primarily residues E2 and E7 of SLN and R324 and K328 of SERCA. MDS shows that SLN missing either E2 or E7 causes a similar structural effect on M4S4 tilt as WT-SLN, and that only deletion of both E2 and E7 prevents this structural rearrangement. MDS also shows that SLN-induced change in the tilt angle of M4S4 leads to removal of E309 from transport site II, and this effect is maintained in SLN with E2 and/or E7, but not in SLN missing both. We propose that altered Ca2+ occlusion by E309 at transport site II, which is induced by salt bridge-mediated tilt of M4S4, constitutes the primary mechanism by which SLN uncouples SERCA. Our MDS provides novel hypotheses for M4S4 tilt that can be tested by functional mutagenesis and site-directed spectroscopy. For example, additional information may be gained by using cysteine engineering to introduce labeling sites on M4S4 for a rigidly-attached, bifunctional spin label (BSL) that measures orientation, distance, and dynamics with high-precision,46 using a heterologous system that allows co-expression of SLN and PLB.5 We propose that the energy-transduction segment M4S4 is molecular switch that responds to SLN binding, thus enabling tissue-specific uncoupling of SERCA through electrostatic control of the extended energy-transduction domain. These findings have important implications for therapeutic approaches to human obesity (e.g., through discovery of small-molecule compounds that enhance SERCA‒SLN binding).
Supplementary Material
Acknowledgments
We extensively used Minnesota Supercomputing Institute facilities. We thank Bengt Svensson for assistance with figures.
Funding Sources
This work was supported by grants to L.M.E.-F. (NIH R01GM120142, AHA 12SDG12060656) and D.D.T. (NIH R01GM27906).
Footnotes
Supporting Information
Figures S1–S9 and Methods. This material is available free of charge via the Internet at http://pubs.acs.org
Notes
The authors declare no competing financial interests.
References
- 1.Moller JV, Olesen C, Winther AM, Nissen P. Q Rev Biophys. 2010;43:501–566. doi: 10.1017/S003358351000017X. [DOI] [PubMed] [Google Scholar]
- 2.Yu X, Carroll S, Rigaud JL, Inesi G. Biophys J. 1993;64:1232–1242. doi: 10.1016/S0006-3495(93)81489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babu GJ, Bhupathy P, Carnes CA, Billman GE, Periasamy M. J Mol Cell Cardiol. 2007;43:215–222. doi: 10.1016/j.yjmcc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vangheluwe P, Schuermans M, Zador E, Waelkens E, Raeymaekers L, Wuytack F. Biochem J. 2005;389:151–159. doi: 10.1042/BJ20050068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Autry JM, Rubin JE, Pietrini SD, Winters DL, Robia SL, Thomas DD. J Biol Chem. 2011;286:31697–31706. doi: 10.1074/jbc.M111.246843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toyoshima C, Iwasawa S, Ogawa H, Hirata A, Tsueda J, Inesi G. Nature. 2013;495:260–264. doi: 10.1038/nature11899. [DOI] [PubMed] [Google Scholar]
- 7.Winther AM, Bublitz M, Karlsen JL, Moller JV, Hansen JB, Nissen P, Buch-Pedersen MJ. Nature. 2013;495:265–269. doi: 10.1038/nature11900. [DOI] [PubMed] [Google Scholar]
- 8.Gramolini AO, Trivieri MG, Oudit GY, Kislinger T, Li W, Patel MM, Emili A, Kranias EG, Backx PH, Maclennan DH. Proc Natl Acad Sci U S A. 2006;103:2446–2451. doi: 10.1073/pnas.0510883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhupathy P, Babu GJ, Ito M, Periasamy M. J Mol Cell Cardiol. 2009;47:723–729. doi: 10.1016/j.yjmcc.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mall S, Broadbridge R, Harrison SL, Gore MG, Lee AG, East JM. J Biol Chem. 2006;281:36597–36602. doi: 10.1074/jbc.M606869200. [DOI] [PubMed] [Google Scholar]
- 11.Smith WS, Broadbridge R, East JM, Lee AG. Biochem J. 2002;361:277–286. doi: 10.1042/0264-6021:3610277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bal NC, Maurya SK, Sopariwala DH, Sahoo SK, Gupta SC, Shaikh SA, Pant M, Rowland LA, Bombardier E, Goonasekera SA, Tupling AR, Molkentin JD, Periasamy M. Nature Med. 2012;18:1575–1579. doi: 10.1038/nm.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 14.Jensen AM, Sorensen TL, Olesen C, Moller JV, Nissen P. EMBO J. 2006;25:2305–2314. doi: 10.1038/sj.emboj.7601135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zafar S, Hussain A, Liu Y, Lewis D, Inesi G. Arch Biochem Biophys. 2008;476:87–94. doi: 10.1016/j.abb.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sehgal P, Olesen C, Moller JV. Methods Mol Biol. 2016;1377:157–160. doi: 10.1007/978-1-4939-3179-8_15. [DOI] [PubMed] [Google Scholar]
- 17.Karon BS, Nissen ER, Voss J, Thomas DD. Anal Biochem. 1995;227:328–333. doi: 10.1006/abio.1995.1288. [DOI] [PubMed] [Google Scholar]
- 18.Bal NC, Maurya SK, Singh S, Wehrens XH, Periasamy M. J Biol Chem. 2016;291:17247–17257. doi: 10.1074/jbc.M116.728188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odermatt A, Becker S, Khanna VK, Kurzydlowski K, Leisner E, Pette D, MacLennan DH. J Biol Chem. 1998;273:12360–12369. doi: 10.1074/jbc.273.20.12360. [DOI] [PubMed] [Google Scholar]
- 20.Espinoza-Fonseca LM, Autry JM, Ramirez-Salinas GL, Thomas DD. Biophys J. 2015;108:1697–1708. doi: 10.1016/j.bpj.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espinoza-Fonseca LM, Autry JM, Thomas DD. Biochem Biophys Res Commun. 2015;463:37–41. doi: 10.1016/j.bbrc.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akin BL, Hurley TD, Chen Z, Jones LR. J Biol Chem. 2013;288:30181–30191. doi: 10.1074/jbc.M113.501585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inesi G, Ma H, Lewis D, Xu C. J Biol Chem. 2004;279:31629–31637. doi: 10.1074/jbc.M403211200. [DOI] [PubMed] [Google Scholar]
- 24.Clausen JD, Bublitz M, Arnou B, Montigny C, Jaxel C, Moller JV, Nissen P, Andersen JP, le Maire M. EMBO J. 2013;32:3231–3243. doi: 10.1038/emboj.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi M, Kondou Y, Toyoshima C. Proc Natl Acad Sci U S A. 2007;104:5800–5805. doi: 10.1073/pnas.0700979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Espinoza-Fonseca LM, Autry JM, Thomas DD. PLoS One. 2014;9 doi: 10.1371/journal.pone.0095979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahoo SK, Shaikh SA, Sopariwala DH, Bal NC, Bruhn DS, Kopec W, Khandelia H, Periasamy M. J Biol Chem. 2015;290:14057–14067. doi: 10.1074/jbc.M115.636738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahoo SK, Shaikh SA, Sopariwala DH, Bal NC, Periasamy M. J Biol Chem. 2013;288:6881–6889. doi: 10.1074/jbc.M112.436915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donald JE, Kulp DW, DeGrado WF. Proteins. 2011;79:898–915. doi: 10.1002/prot.22927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandell DJ, Chorny I, Groban ES, Wong SE, Levine E, Rapp CS, Jacobson MP. J Am Chem Soc. 2007;129:820–827. doi: 10.1021/ja063019w. [DOI] [PubMed] [Google Scholar]
- 32.Asahi M, Sugita Y, Kurzydlowski K, De Leon S, Tada M, Toyoshima C, MacLennan DH. Proc Natl Acad Sci U S A. 2003;100:5040–5045. doi: 10.1073/pnas.0330962100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorski PA, Glaves JP, Vangheluwe P, Young HS. J Biol Chem. 2013;288:8456–8467. doi: 10.1074/jbc.M112.446161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Sumbilla C, Lewis D, Inesi G. FEBS Lett. 1993;335:261–264. doi: 10.1016/0014-5793(93)80742-d. [DOI] [PubMed] [Google Scholar]
- 35.Andersen JP. J Biol Chem. 1995;270:908–914. doi: 10.1074/jbc.270.2.908. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Sumbilla C, Lewis D, Summers S, Klein MG, Inesi G. J Biol Chem. 1995;270:16283–16290. [PubMed] [Google Scholar]
- 37.Chen L, Sumbilla C, Lewis D, Zhong L, Strock C, Kirtley ME, Inesi G. J Biol Chem. 1996;271:10745–10752. doi: 10.1074/jbc.271.18.10745. [DOI] [PubMed] [Google Scholar]
- 38.Garnett C, Sumbilla C, Belda FF, Chen L, Inesi G. Biochemistry. 1996;35:11019–11025. doi: 10.1021/bi960718k. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Lewis D, Strock C, Inesi G, Nakasako M, Nomura H, Toyoshima C. Biochemistry. 2000;39:8758–8767. doi: 10.1021/bi000185m. [DOI] [PubMed] [Google Scholar]
- 40.Miyauchi Y, Daiho T, Yamasaki K, Takahashi H, Ishida-Yamamoto A, Danko S, Suzuki H, Iizuka H. J Biol Chem. 2006;281:22882–22895. doi: 10.1074/jbc.M601966200. [DOI] [PubMed] [Google Scholar]
- 41.Daiho T, Yamasaki K, Danko S, Suzuki H. J Biol Chem. 2014;289:31241–31252. doi: 10.1074/jbc.M114.584086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamasaki K, Daiho T, Danko S, Suzuki H. J Biol Chem. 2015;290:27868–27879. doi: 10.1074/jbc.M115.693770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toyoshima C, Mizutani T. Nature. 2004;430:529–535. doi: 10.1038/nature02680. [DOI] [PubMed] [Google Scholar]
- 44.Montigny C, Decottignies P, Le Marechal P, Capy P, Bublitz M, Olesen C, Moller JV, Nissen P, le Maire M. J Biol Chem. 2014;289:33850–33861. doi: 10.1074/jbc.M114.590307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bombardier E, Smith IC, Vigna C, Fajardo VA, Tupling AR. FEBS Lett. 2013;587:1687–1692. doi: 10.1016/j.febslet.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 46.Binder BP, Cornea S, Thompson AR, Moen RJ, Thomas DD. Proc Natl Acad Sci U S A. 2015;112:7972–7977. doi: 10.1073/pnas.1500625112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.