

Abstract
KRAS mutant non-small cell lung cancers (NSCLCs) vary in clinical outcome depending on which specific KRAS mutation is present. Shorter progression free survival has been associated with KRAS variants G12C and G12V. Cell lines with these variants depend to a greater extent on the RAS/RAF/MEK/ERK signaling pathway and become more susceptible to MEK inhibition. Because different KRAS mutations may lead to altered drug sensitivity, we aimed to determine specific KRAS mutation status in a NSCLC patient cohort at our institution. A total of 502 NSCLC samples were screened for somatic mutations using the 50 gene AmpliSeq™ Cancer Hotspot Panel v2 (CHPv2). However only samples positive for variants in the KRAS gene were included in this study. Variants identified in the KRAS genes were curated using publicly available databases. The overall mutation rate in the KRAS gene was 32.7% (164/502). The most common KRAS mutations were G12C (41%), G12V (19%), and G12D (14%) along with less frequent variants. After re-mining our sequencing data, we found that more than a half of our KRAS mutant NSCLC patients could potentially benefit from the addition of a MEK inhibitor such as selumetinib to standard chemotherapeutic agents. Due to mutated KRAS, these patients will likely fail traditional anti-EGFR therapies but be eligible for newer combination therapies.
Keywords: Somatic mutation, KRAS, lung cancer, next generation sequencing, data mining
Introduction
Personalized medicine efforts with respect to oncology are heavily dependent on somatic mutation analysis of tumor cells that identifies gene mutations associated with response, positive or negative, to novel targeted therapies. Numerous molecular technologies are available for use in such testing and in many instances a combination of techniques and/or assays is used to identify a tumor mutation profile that can then be used in the design of a management strategy for a particular patient.
As new therapeutics were introduced after clinical trials and FDA approval, the need for companion diagnostics far outpaced the single gene, single mutation types of assays that many labs were offering. Throughput of traditional multiplexed assays and Sanger sequencing were also overtaken by increasing demand for more information about additional genes representing a variety of targeted pathways. Many laboratories validated SNaPshot assays which increased numbers of detectable mutations in a single test as well as next generation sequencing (NGS) (1). Our lab initially introduced NGS for clinical testing by validating the Ion Torrent AmpliSeq Hotspot Cancer Panel, a pancancer 50 gene hotspot test that is routinely run on primary and metastatic colorectal cancers, and metastatic gliomas, melanomas, lung adenocarcinomas, and triple negative breast cancers (2).
The molecular reclassification of non-small-cell lung cancer (NSCLC) has changed the diagnostic and therapeutic approach and improved outcome for patients with EGFR mutations and ALK rearrangements as oncogenic drivers (3,4). Despite these advances, the largest known genetically defined subset of NSCLC, harboring mutation in KRAS oncogene, remains an elusive therapeutic target (5). KRAS mutations, associated with adenocarcinoma histology and tobacco use, have been linked to worse survival and lack of response to cytotoxic chemotherapy as well as anti-EGFR therapy (3–5). As compounds designed to specifically target RAS proteins, such as farnesyl transferase inhibitors, showed little efficacy, efforts have shifted to inhibit downstream effector proteins in RAS-RAF-MEK-ERK (MAPK) or PI3K/AKT/mTOR signaling pathways with promising yet variable response rates in preclinical and clinical testing (6–8). The particular challenge to effectively target the KRAS-driven cancers has been recently attributed to a greater molecular heterogeneity in tumors with mutated KRAS compared with tumors with other known oncogenic drivers (3,4,7). It has been shown that different KRAS amino acid substitutions differ in their patterns of downstream signaling pathways, suggesting that specific KRAS alleles may account for at least part of this diversity (7). Recently, Janne et al. identified a subset of KRAS mutation positive lung adenocarcinomas that showed sensitivity to the combined therapy of MEK1/MEK2 inhibitor selumetinib and docetaxel, supporting the notion that the biology of KRAS mutations differ, and the efficacy of targeted therapies may be related to the specific KRAS mutation (9,10) This study prompted us to re-review our NGS data on more than 500 lung adenocarcinomas to identify the frequency of specific KRAS mutations and the number of patients who might benefit from such combination therapy.
Materials and methods
Samples and DNA extraction
Five hundred and two formalin-fixed, paraffin-embedded (FFPE) tissues from non-small cell lung cancer (NSCLC) patients were received at Dartmouth-Hitchcock Medical Center (DHMC) between May 2013 and September 2015 for somatic mutation screening. However only samples positive for variants in the KRAS gene were included in this retrospective study.
The FFPE sample types received at DHMC included mainly surgical (50) and cytology (114). All hematoxylin and eosin (H&E) stained slides from each patient were reviewed by an attending pathologist who determined the tumor area and the percentage of tumor cells present in the tissue section. The tumor cellularity ranged from 10 to 95% in both sample types (surgical and cytology).
DNA extraction was performed using the Gentra Pure Gene Kit (Qiagen) or the QiaCube (Qiagen; after August 2015), and quantified using the Quant-iT™ PicoGreen® dsDNAAssay Kit (Invitrogen) according to the manufacturer’s recommendations. DNA quality was assessed using the KAPA hgDNA Quantification and QC Kit (KAPA Biosystems, Wilmington, MA).
Next generation sequencing (NGS)
As determined during validation, samples with (1) tumor cellularity below 10% and (2) DNA concentration below 1.7 ng/µL and DNA quality ratio below 0.4 were not screened for somatic variants. Approximately 10 ng of genomic DNA from each sample was used to create barcoded libraries using the Ion AmpliSeq™ Cancer Hotspot Panel v2 (CHPv2) (ThermoFisher). The CHPv2 panel consists of hotspot regions for 50 oncogenes and tumor suppressor genes covering approximately 2800 Catalogue of Somatic Mutations in Cancer (COSMIC) mutations. Barcoded libraries were combined to a final concentration of 100 pM using the Ion Library Quantitation Kit (ThermoFisher), and a maximum of 10 samples were sequenced on an Ion 318 Chip v2 using the Ion PGM System (ThermoFisher).
For data analysis, read mapping, alignment of sequences to hg19 (Human Genome Version 19), variant calling, and coverage analysis were performed using the Torrent Suite software (v4.0.2). Variant annotation and prediction of functional significance were performed using Golden Helix SNP & Variation Suite (SVS) software (v8.2.1). Reported variants passed the minimum reporting thresholds established during validation which includes 500× coverage, 5% allelic frequency and strand bias of 0.40–0.59.
For NSCLC, variants identified in the BRAF, EGFR, KRAS, or PIK3CA genes were characterized as clinically actionable according to NCCN Clinical Practice Guidelines in Oncology and My Cancer Genome: Genetically Informed Cancer Medicine. Curation of the variants identified in the KRAS genes was performed using publicly available databases. All reports were reviewed by a genomic informaticist before being reviewed and curated by a genomic analyst.
Results
The overall mutation rate in the KRAS gene was 32.7% among the NSCLC cases studied (164/502). The most common KRAS mutations were G12C (41%) and G12V (19%) that are both considered RAS/RAF/MEK/ERK pathway dependent. Another common subtype, G12D (14%), is known to act more through AKT phosphorylation. The remaining mutations included G12A (6%), Q61H (5%), G13C (4%), and rarer subtypes: G12R, G61L, G12S (2% each), and G13D, G13S, G12F, L19F, G13P (1% each). The different types of KRAS mutations found within our population are presented in Figure 1.
Figure 1.
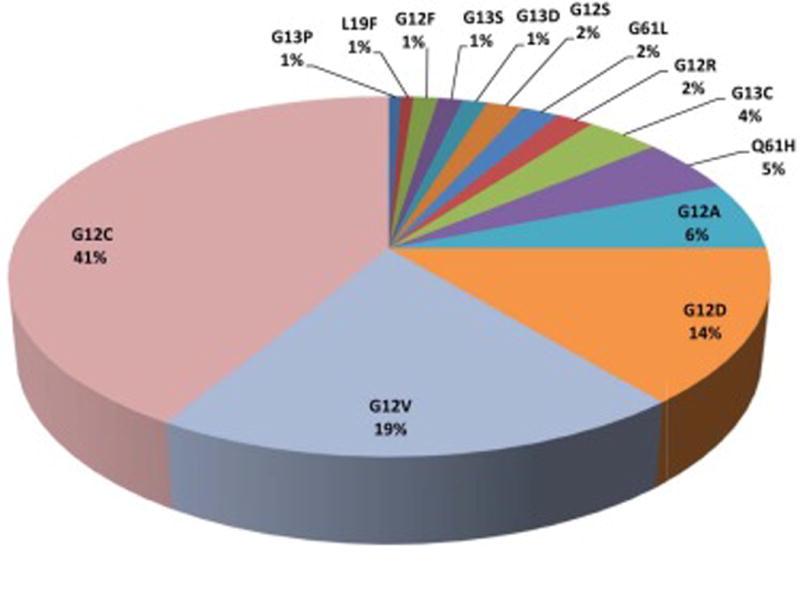
Distribution of KRAS mutation subtypes in non-small cell lung cancer in the study population. Among different exon locations of KRAS mutations, mutations in exon 2 codon G12 were detected most frequently. The most common variants were G12C and G12V (considered RAS/RAF/MEK/ERK pathway dependent) that occurred with a combined frequency of 60%.
Discussion
An unprecedented amount of molecular data is being generated by analyzing human tumors in the name of personalized medicine. While the technologies used are evolving at a fast pace with costs decreasing faster than Moore’s Law could have predicted, proven clinical utility of the generated data is lagging, in part due to the complexity of human cancers. Nonetheless, the impetus to develop novel therapeutic strategies to improve the management of the individual cancer patient drives this testing.
Single gene mutations identified in a specific tumor type, such as BRAF V600E in melanoma, have relatively straightforward interpretations with respect to response to select BRAF inhibitors. However, interpretation of somatic mutation testing quickly becomes complex when multiple mutations are identified in the same gene or different genes within the same tumor sample. What is the relevance of multiple mutations in the same tumor cell with respect to response to therapy, including the potential for resistance? Could multiple mutations detected in a tumor specimen be a sign of heterogeneity at the cellular and molecular levels? These are only a few of the biological questions that we must address for personalized medicine to be as effective as possible.
In addition to biological variables, we are seeing a variety of therapeutic variables whereby therapies are being used “off-label” in tumor types with specific molecular profiles as part of the so called “basket trials” like the NCI-MATCH trials. Just as common are the new combination therapies that include traditional cytotoxic chemotherapy with novel targeted therapy as in the case with the lung adenocarcinoma study discussed here. Clinical laboratory scientists along with oncologists must be aware of the need to re-mine NGS or other molecular data sets as new information about response to these modalities becomes available.
Activating mutations in a KRAS oncogene are found in a substantial proportion of NSCLC; however, data have been conflicting regarding their impact on treatment response and patient outcomes (11,12). The frequency of KRAS mutations (32.7%) detected in our cohort is relatively higher when compared to the estimated frequency as reported in the COSMIC database where KRAS mutations in lung cancers are reported at a frequency of approximately 15% (13). A likely contributing factor to this difference is the higher sensitivity of detection by NGS compared to other sequencing and genotyping approaches. In addition, other biological and clinical factors, outside the scope of this study that led to these patients being tested, could contribute to the higher mutation frequency. Nonetheless, our ability to retrospectively identify these patients is an important step in their management strategy.
Increasing evidence suggests that KRAS mutant lung cancer is heterogeneous and that not all KRAS mutation subtypes are alike in terms of their biology, and individual genotypes should be considered separately (14). Recent studies demonstrated a difference in clinical outcomes depending on specific KRAS mutation subtype with shorter progression free survival associated with variants G12C and G12V, compared with all other KRAS mutations or wild-type KRAS (15). In vitro analysis of cell lines carrying various KRAS mutants showed that replacement of a single amino acid results in distinct phosphorylation patterns and leads to different signal transduction cascades (7). Of the 14 KRAS mutation types that we detected in our study population, mutations in exon 2 codon G12 were found most frequently, in agreement with other cohorts (14,16,17). The most common variants were G12C and G12V that occurred with a combined frequency of 60%. The G12C transversion and G12V transition mutations are common hydrophobic alterations known to depend to a greater extent on the RAS/RAF/MEK/ERK signaling pathway. In contrast with the third most frequent variant detected in our study (14%), the G12D transition mutation results in a hydrophilic alteration and is considered to act through AKT phosphorylation. Therefore different KRAS mutations in the same codon are not biologically equivalent and may lead to different drug sensitivity (7).
While standard platinum based doublet chemotherapy remains the recommended treatment option in a large group of patients with mutated KRAS, studies are emerging highlighting different sensitivity patterns in distinct KRAS variants (14,16,17). In particular, Garassino et al. found significant differences in treatment response to cisplatin among KRAS overexpressing clones of human lung adenocarcinoma cells with G12V mutant cells responding better to cisplatin chemotherapy and G12C showing the least response (18). In a study by Mellema et al. patients with G12V had better response to taxane treatment (16). Therefore the simple definition of KRAS-mutated tumor is no longer enough to identify patients with a different response profile to chemotherapeutics.
In this study, we show the frequency of different KRAS mutation subtypes in lung adenocarcinoma patients which were once thought to be uniformly unresponsive to therapeutic options. Initially information concerning specific KRAS mutations and response to therapy was shown in metastatic chemotherapy-refractory colorectal cancer (19). Previous clinical trials consistently reported patients with KRAS-mutated tumors resistant to EGFR targeted treatment, based on KRAS wild-type vs mutant testing while grouping codons 12 and 13 mutations together, without subgroup analysis. Recent studies demonstrated survival benefit of cetuximab combined with chemotherapy specifically in patients with KRAS G13D-mutated tumors, highlighting the need for a higher resolution testing for better assignment of patients to available therapeutic options in both colon and lung carcinomas (19–21).
With a number of agents in clinical development for oncology, it is especially important to discern the prognostic and predictive role of specific KRAS mutations. Targeting KRAS downstream effector MEK1/2 has emerged as the most promising approach for advanced-stage KRAS mutant lung cancer (11,22). A placebo-controlled phase II trial (NCT00890825) of second-line selumetinib, an orally available MEK1/2 inhibitor, combined with docetaxel in patients with KRAS mutant NSCLC, proved that an addition of selumetinib was beneficial for survival but was associated with increased adverse events relative to docetaxel with placebo (9). When the study population was revisited to investigate an impact of different KRAS codon specific mutations, the greatest response to selumetinib was found in patients with G12C or G12V KRAS mutations (10). Therefore, identifying these patients is critical to assess the benefit to risk ratio and stratify patients to the appropriate treatment. It is especially important, considering that without MEK inhibition, G12C and G12V cancers seem to progress faster than all other KRAS subtypes or wildtype KRAS. Moreover, due to mutated KRAS, these patients will likely fail anti-EGFR therapies and have very limited therapeutic options.
As not all KRAS mutations are equal in their biological characteristics, re-mining somatic mutation testing of KRAS status in lung NSCLC might yield critical new prognostic and predictive information. After re-mining our sequencing data as part of this study, we found that more than a half of our KRAS mutant NSCLC patients could potentially benefit from the addition of a MEK inhibitor such as selumetinib to standard chemotherapeutic agents. Although, if KRAS G12C or G12V mutations have greater sensitivity to selumetinib compared to other variants requires further investigation in future clinical trials, this possibility clearly indicates that simply assessing for the presence or absence of KRAS mutations does not suffice as reliable biomarker for efficacy of MEK inhibitors. A randomized, double-blinded, placebo-controlled phase III trial (SELECT-1) of selumetinib plus docetaxel in second-line KRAS mutant advanced NSCLC is recruiting patients and will give more insight into the impact of specific KRAS mutations.
Acknowledgments
The authors wish to thank the staff of the Laboratory for Clinical Genomics and Advanced Technology (CGAT). The data presented in this manuscript were in part generated through CGAT in the Department of Pathology and Laboratory Medicine of the Geisel School of Medicine at Dartmouth, the Dartmouth Hitchcock Medical Center and the Norris Cotton Cancer Center.
References
- 1.Rovigatti U. Cancer modelling in the NGS era—Part I: emerging technology and initial modelling. Crit Rev Oncol Hematol. 2015;96:274–307. doi: 10.1016/j.critrevonc.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 2.Tsongalis GJ, Peterson JD, de Abreu FB, et al. Routine use of the Ion Torrent AmpliSeq Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med. 2014;52:707–714. doi: 10.1515/cclm-2013-0883. [DOI] [PubMed] [Google Scholar]
- 3.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31:1105–1111. doi: 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahearn IM, Haigis K, Bar-Sagi D, et al. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2011;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whyte DB, Kirschmeier P, Hockenberry TN, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 7.Ihle NT, Byers LA, Kim ES, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–239. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riely GJ, Johnson ML, Medina C, et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6:1435–1437. doi: 10.1097/JTO.0b013e318223c099. [DOI] [PubMed] [Google Scholar]
- 9.Janne PA, Shaw AT, Pereira JR, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 10.Janne PA, Smith I, McWalter G, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. Br J Cancer. 2015;113:199–203. doi: 10.1038/bjc.2015.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin P, Leighl NB, Tsao MS, et al. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013;8:530–542. doi: 10.1097/JTO.0b013e318283d958. [DOI] [PubMed] [Google Scholar]
- 12.Mascaux C, Iannino N, Martin B, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92:131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cserepes M, Ostoros G, Lohinai Z, et al. Subtype-specific KRAS mutations in advanced lung adenocarcinoma: a retrospective study of patients treated with platinum-based chemotherapy. Eur J Cancer. 2014;50:1819–1828. doi: 10.1016/j.ejca.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellema WW, Masen-Poos L, Smit EF, et al. Comparison of clinical outcome after first-line platinum-based chemotherapy in different types of KRAS mutated advanced non-small-cell lung cancer. Lung Cancer. 2015;90:249–254. doi: 10.1016/j.lungcan.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Hames ML, Chen H, Iams W, et al. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer. 2016;92:29–34. doi: 10.1016/j.lungcan.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garassino MC, Marabese M, Rusconi P, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235–237. doi: 10.1093/annonc/mdq680. [DOI] [PubMed] [Google Scholar]
- 19.De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–1820. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 20.Bando H, Yoshino T, Yuki S, et al. Clinical outcome of Japanese metastatic colorectal cancer patients harbouring the KRAS p.G13D mutation treated with cetuximab + irinotecan. Jpn J Clin Oncol. 2012;42:1146–1151. doi: 10.1093/jjco/hys160. [DOI] [PubMed] [Google Scholar]
- 21.Tejpar S, Celik I, Schlichting M, et al. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30:3570–3577. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 22.Caunt CJ, Sale MJ, Smith PD, et al. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]