

Abstract
The development of alternative architectures for genetic information-encoding systems offers the possibility of new biotechnological tools as well as basic insights into the function of the natural system. In order to examine the potential of benzo-expanded DNA (xDNA) to encode and transfer biochemical information, we carried out a study of the processing of single xDNA pairs by DNA Polymerase I Klenow fragment (Kf, an A-family sterically rigid enzyme) and by the Sulfolobus solfataricus polymerase Dpo4 (a flexible Y-family polymerase). Steady-state kinetics were measured and compared for enzymatic synthesis of the four correct xDNA pairs and twelve mismatched pairs, by incorporation of dNTPs opposite single xDNA bases. Results showed that, like Kf, Dpo4 in most cases selected the correctly paired partner for each xDNA base, but with efficiency lowered by the enlarged pair size. We also evaluated kinetics for extension by these polymerases beyond xDNA pairs and mismatches, and for exonuclease editing by the Klenow exo+ polymerase. Interestingly, the two enzymes were markedly different: Dpo4 extended pairs with relatively high efficiencies (within 18–200-fold of natural DNA), whereas Kf essentially failed at extension. The favorable extension by Dpo4 was tested further by stepwise synthesis of up to four successive xDNA pairs on an xDNA template.
Introduction
A primary goal of biomimetic chemistry is to generate designed molecules that function as much as possible like their natural congeners.1 The purpose of this mimicry is multifold: first, it tests our knowledge of the mechanisms by which complex biomolecules function, and second, if these designed molecules function well, such molecules can be useful as probes and as tools in biological and biomedical applications. In the realm of nucleic acids, much work has been directed to the design of DNA analogs in which the sugar-phosphate backbone is replaced with other scaffolds.2 Although a number of such analogs have proven quite successful in helix formation, relatively few have been shown to be competent in replication by acting as substrates for polymerase enzymes.3
As compared with the voluminous literature on DNA backbone variations, much less work has focused on biomimetic chemistry of the DNA bases, even though it is these heterocycles that encode the biological information of cellular function. However, several laboratories have recently demonstrated novel DNA base and base pair structures that can support not only base pair formation and double helix stabilization, but also the ability to function with polymerase enzymes.4 To date, nearly all of this work has focused on the design of base pairs that can function within the context of the natural genetic system; that is, the natural double helix architecture and natural replicating enzymes. Some of the goals of such work have been to develop useful probes for basic science,4g,n new base pairs that can add to the genetic alphabet,4e,i and useful biotechnological tools.4m
One may ask, however, whether there are other base pair and genetic system architectures that may also function as DNA does. To address this question, we have developed a size-expanded DNA (xDNA) design (Fig. 1), in which all base pairs are ca. 2.4 A° larger than Watson–Crick pairs as a result of benzo-fusion of the natural heterocycles.5,6 Studies have shown that xDNA forms antiparallel helices with hydrogen-bonded and stacked base pairs analogous to Watson–Crick pairs. These helices are more stable than B-DNA because of the superior stacking properties of the larger, more hydrophobic bases.6d,4n In addition to this favorable stability, the sequence selectivity and inherent fluorescence of xDNAs may be useful in the development of tools and probes in biochemical and biological systems.5,7
Fig. 1.
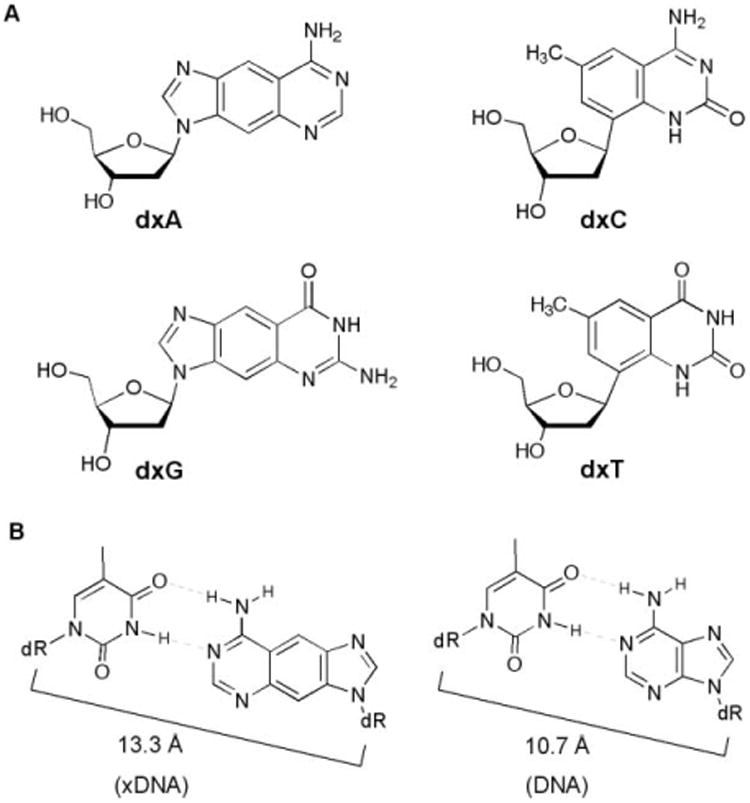
Nucleosides and base pairs used in this study. (A) The four xDNA monomers. (B) Comparison of xDNA and DNA base pairs, showing difference in C1′–C1′ distances.
Very recently, a number of other size-expanded base pairing designs have been reported from other laboratories as well, underscoring the general interest in design of genetic sets with novel architectures.8 Matsuda has described base pairs designed to form four hydrogen bonds; in some pairing arrangements these are expected to form base pairs with larger-than-natural size.8a,b Inouye recently reported an alkyne–DNA design in which DNA bases were extended outward from deoxyribose by an ethynyl bridge.8c In addition to these, Jovin,8d Battersby8e and Switzer8f have performed experiments with purine–purine pairing architectures, using pairs such as G–isoG and A–hypoxanthine. Although structures are not yet available in those other systems, all are expected to have a similar magnitude of structural expansion as xDNA, which has been measured in recent structural studies to have a glycosidic C1′–C1′-distance across the base pair of ca. 13.3 A° (Fig. 1B), nearly 25% greater than that of natural DNA.9 In addition, evidence of an even larger base pair architecture based on naphtho-expansion of bases has been reported.10
With respect to its biomimetic properties, xDNA exhibits many of the successful structural and biophysical properties of natural DNA. However, for true biochemical and biological function, a designed genetic system must encode sequence information and catalyze its transfer to a copy. This is a challenge for biomimicry because natural polymerase enzymes, which are highly specific, evolved to carry out replication using the natural purine–pyrimidine base pair architecture. Indeed, many replicative enzymes are highly sensitive to steric size and shape of base pairs.11 Despite this expected structural bias against noncanonical DNA, however, it is important first to evaluate how flexible these enzymes are to such expanded designs; it is possible that a small amount of activity may ultimately be improved through enzyme modification.12 Moreover, in a recent study, two xDNA bases were reported to be surprisingly efficient in replication bypass in living E. coli cells;13 thus it is of interest to evaluate how isolated bacterial enzymes of different classes can process xDNA.
Here we describe the first detailed studies of the in vitro enzymatic function of size-expanded DNA as an encoder of genetic information, by studying the substrate capabilities of xDNA pairs with a natural replicative polymerase, E. coli DNA polymerase I (Klenow fragment, Kf), and with a repair polymerase, Dpo4 from Solfolobus solfataricus. We find that both enzymes possess useful but distinct capabilities in replicating individual xDNA pairs. Furthermore, their properties are complementary, in that the former enzyme can synthesize and edit xDNA pairs correctly, while the latter extends the pairs efficiently. The results suggest strategies for in vitro replication of xDNA and other large-sized genetic sets, and they also shed light on the recent experiments in bacteria.
Results
Nucleotide insertion by Kf and Dpo4 polymerases
The two enzymes for these experiments were chosen on the basis that (a) they have been widely used for in vitro DNA synthesis studies; (b) they are both bacterial enzymes and are thus most relevant to the recent cellular replication studies, and (c) they are quite different from one another enzymatically and biologically. The Kf enzyme is a well-studied, relatively high fidelity enzyme of the pol A group;15 steric probing has been revealed to be highly sensitive to varied nucleobase sizes.16 A preliminary study of nucleotide insertion opposite xDNA bases showed some success with Kf, albeit with low efficiency.13 In contrast, the Dpo4 enzyme is a Y-family repair enzyme, and steric probing experiments have revealed it to be sterically much more flexible, consistent with its open and uncrowded structure around the active site.17,18 Dpo4 has not yet been tested with xDNA, and no enzyme has been tested for its ability to extend beyond xDNA pairs, a step that is often a major block for unnatural base pairs. In addition, the possibility of polymerase proofreading of this unnatural pair geometry has not been explored.
We carried out single nucleotide insertion experiments using a 28mer DNA containing single xDNA bases at one position. A 32P-labeled 23mer DNA primer was paired with it such that the xDNA base was located immediately downstream of the primer 3′ terminus. Enzymatic incorporation of an additional nucleotide opposite the xDNA base was carried out in the presence of varied concentrations of added dNTP, and the results were measured by quantitative analysis after denaturing polyacrylamide gel electrophoresis.
Values of Vmax/Km for single-nucleotide insertions are plotted in Fig. 2, showing the data for Dpo4 compared with previous data for Kf exo-.13 The full numerical data are tabulated in Table S1 (ESI†). The experiments revealed that the Dpo4 enzyme most efficiently inserted the correct partner for three of four xDNA bases (xA, xC, xG) but showed no selectivity with xT in the template, where dATP, dCTP and dTTP were all inserted approximately equally efficiently. The overall efficiencies for synthesizing the four correct xDNA pairs were lower than thatof a natural DNA pair by factors of 280 to 480 in Vmax/Km. For the three cases in which the correct pair was successfully chosen, the selectivity was quite low (and lower than that of Kf (see Fig. 2)), ranging from factors of 1.4– 21 for the right nucleotide over the next-closest incorrect one. Overall, the results for Dpo4 pol are not greatly different from those obtained previously for Kf, except that average efficiencies were higher for the latter.
Fig. 2.
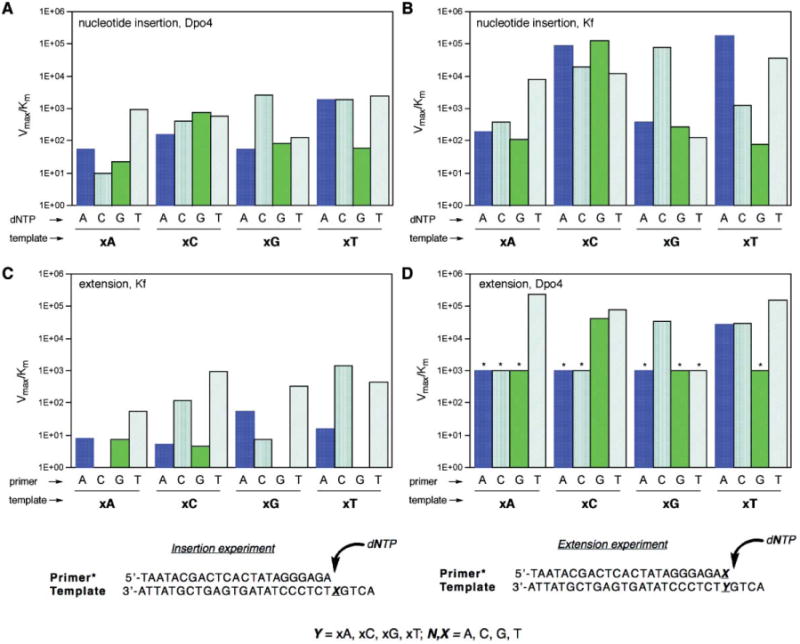
Plots (top) comparing kinetic efficiencies and selectivities for xDNA pair synthesis and extension for Dpo4 and Kf exo- enzymes and primer: templates (bottom) used in single nucleotide insertion and extension experiments. (A) Nucleotide insertion by Dpo4; (B) nucleotide insertion by Kf exo- (data from ref. 13); (C) primer/template base pair extension by Kf exo- (extending various pairs/mismatches shown); (D) base pair extension by Dpo4 (* = estimated maximum value; see Table S3†). In all graphs, units on Vmax/Km = min−1·μM−1.
Base pair extension by Kf and Dpo4 polymerases
Next we evaluated the properties of the Kf and Dpo4 enzymes in extending a single xDNA base pair or mismatch. To carry out these measurements we used the same four 28mer templates as above, each containing one xDNA base. To these we hybridized radiolabeled DNA primers that were one nucleotide longer (24nt) than for the above base pair synthesis studies. The 3′-terminal base of the primer was thus hybridized opposite an xDNA base, forming a terminal xDNA base pair or mismatch. We then measured the proficiencies of the enzymes in inserting an additional nucleotide at the primer terminus, thus extending the xDNA pair by a normal DNA base pair. We evaluated steady-state kinetics for both enzymes, using all sixteen combinations of matched and mismatched termini. The data are plotted graphically for comparison in Fig. 2C,D. The numerical data are available in Tables S2 (for Kf) and S3 (Dpo4).†
Unlike the xDNA pair synthesis studies, where the two enzyme activities were not greatly different, the extension studies revealed striking differences between these two polymerases. The kinetics data show that Kf pol almost completely failed at extending xDNA pairs, whether correctly matched or not (Fig. 2C, Table S2†). The efficiencies of extension (as judged by Vmax/Km values) were 4– 5 orders of magnitude below those for extending canonical base pairs. Moreover, only one case of four (extension of T–xA) was selective for a correctly matched pair over mismatches. The most readily extended mismatches involved x-pyrimidines paired with pyrimidines (specifically, T–xC and T–xT mismatches). They were extended approximately as readily as a natural T–G mismatch.
In contrast to this, the kinetics data for extension by Dpo4 show relatively efficient extension of xDNA pairs. This large difference is readily seen by comparing Fig. 2C and 2D. Extension efficiency with Dpo4 was lower than that of natural pairs by only relatively small factorsof 18-200. These efficiencies were essentially the same in magnitude as for extension of T–G/G–T mismatches by this enzyme, except for T–xA, which was more efficient by an order of magnitude. The Dpo4 enzyme also showed better selectivity in extension than did Kf: In two cases (xA, xG–C) the correctly matched pair was extended most readily. In the third case (xC– G) one of the mismatches was as well extended as the matched pair. Only in the case of xT was a mismatched pair (T–xT) more efficiently extended than the matched pair.
Synthesis and extension of multiple xDNA pairs by Dpo4 pol
The above results showed relatively facile extension of a large-sized xDNA pair by Dpo4. To test the limits of this steric permissiveness further, we carried out new experiments aimed at possible synthesis of multiple consecutive xDNA pairs. We used a 20mer xDNA template strand and DNA primers of varying length (14–17 nt) (Fig. 3). Each primer was tested for extension in the presence of each of the four natural dNTP's, to examine sequence selectivity; the template contained a different expanded base at each successive position downstream, thus allowing us to test xA, xC, xT, xG individually as template bases. Results showed clearly that xDNA primer–templates could be extended well by Dpo4 at each successive position. In addition, although some misincorporation was observable, the expected complementary nucleotide was incorporated considerably more efficiently than the mismatches in all four cases.
Fig. 3.
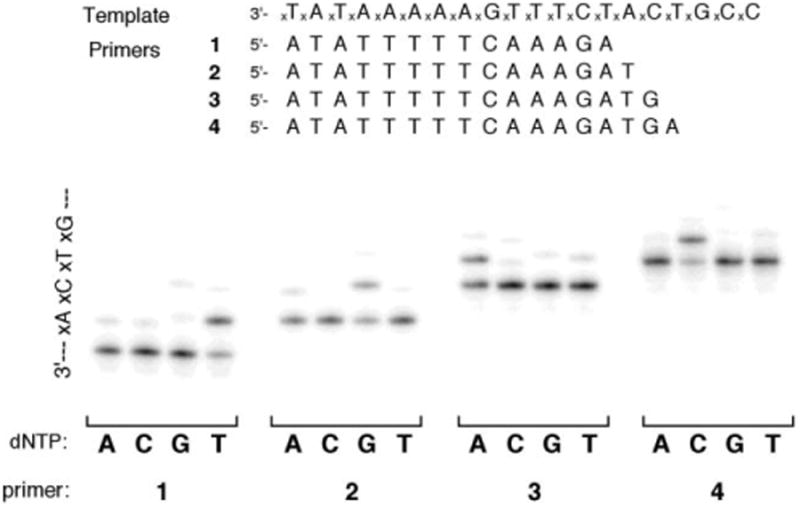
Multistep replication on an xDNA template by Dpo4. Conditions: 100 nM template–primer duplex, 40 mM Tris-HCl (pH = 8.0), 5 mM MgCl2, 10 mM DTT, 250 μg mL−1 bovine serum albumin (BSA), 100 μM single dNTP, 500 nM Dpo4, 0.3 unit mL−1 pyrophosphatase, incubated at 37 °C for 5 min in a reaction volume of 5 μL.
Terminal base pair editing by Kf exo+ polymerase
Replicative polymerases increase their fidelity by editing the terminal pairs just synthesized: mismatched nucleotides at the primer 3′-end are removed more rapidly than correctly matched ones.19 To begin to explore this third polymerase activity with expanded DNA pairs, we tested the ability of 3′-exonuclease-proficient Klenow polymerase (Kf exo+) to discriminate between matched and mismatched xDNA pairs at the terminus of a primer– template duplex. This was done as above, with radiolabeled primer, but in the absence of added dNTPs, to allow editing to proceed in the absence of base pair synthesis. We monitored removal of the 3′-terminal primer nucleotide by gel electrophoresis.
The steady-state kinetics data are given in Table 1. The data show that for each of the four xDNA bases in the template, the Kf enzyme edited mismatched primer termini more readily than the correctly matched ones. The differences were small, ranging from factors of 1.5–2 in Vmax/Km. This selectivity is smaller than for natural matches and mismatches, where the selectivity was 5– 6-fold in favor of editing mismatched pairs in these experiments. The overall efficiency of editing correctly matched pairs xDNA pairs was similar to that of matched natural base pairs.
Table 1. Steady-state kinetics data for Kf (exo+)-catalyzed editing of a single primer strand base from each xDNA pair/mismatch showna.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Template | Primer | Vmax/nM min−1 | Km/nM | 109 × Vmax/Km/min−1 | f edit (rel) |
| xA | A | 50.4 ± 16.4 | 118 ± 41 | 0.43 ± 0.21 | 0.44 |
| xA | C | 404 ± 66 | 719 ± 120 | 0.56 ± 0.13 | 0.58 |
| xA | G | 101 ± 9.6 | 254 ± 18 | 0.40 ± 0.05 | 0.41 |
| xA | T | 89.2 ± 22.6 | 295 ± 92 | 0.31 ± 0.12 | 0.31 |
| xC | A | 64.8 ± 10.4 | 91.9 ± 15.8 | 0.71 ± 0.17 | 0.73 |
| xC | C | 132 ± 23 | 165 ± 38 | 0.81 ± 0.23 | 0.83 |
| xC | G | 82.6 ± 11.0 | 211 ± 40 | 0.40 ± 0.09 | 0.41 |
| xC | T | 101 ± 18 | 162 ± 38 | 0.63 ± 0.19 | 0.65 |
| xG | A | 68.6 ± 1.9 | 84 ± 6.1 | 0.82 ± 0.06 | 0.85 |
| xG | C | 97.6 ± 2.4 | 150 ± 10 | 0.65 ± 0.05 | 0.67 |
| xG | G | 70.4 ± 13.6 | 93 ± 18 | 0.76 ± 0.21 | 0.78 |
| xG | T | 70.0 ± 5.7 | 103 ± 9 | 0.68 ± 0.08 | 0.71 |
| xT | A | 65.5 ± 13.6 | 110 ± 26 | 0.60 ± 0.19 | 0.62 |
| xT | C | 106 ± 4 | 150 ± 4 | 0.71 ± 0.03 | 0.73 |
| xT | G | 76.0 ± 2.9 | 78.5 ± 3.3 | 0.97 ± 0.06 | 1.0 |
| xT | T | 128 ± 16 | 200 ± 30 | 0.64 ± 0.13 | 0.66 |
| C | G | 49.9 ± 17.0 | 335 ± 135 | 0.15 ± 0.08 | 0.16 |
| T | A | 46.4 ± 15.0 | 307 ± 142 | 0.16 ± 0.09 | 0.16 |
| T | G | 76.4 ± 7.8 | 91.6 ± 12.0 | 0.84 ± 0.14 | 0.87 |
| A | A | 79.7 ± 1.6 | 91.3 ± 4.6 | 0.88 ± 0.05 | 0.90 |
Conditions: 0.5 μM template–primer duplex, 100 mM Tris-HCl (pH = 7.5), 10 mM MgCl2, 1 mM dithiothreitol and 100 μg mL−1 BSA, varied concentrations of dCTP, incubated at 37 °C in a reaction volume of 10 μL. Data are averaged from triplicates; standard deviations are shown. Vmax is normalized to 0.005 unit μL−1 enzyme concentration.
Discussion
Our data show that there is one large functional difference between the Kf and Dpo4 enzymes in processing xDNA pairs, and that is in the base pair extension step. Dpo4 extends single xDNA pairs relatively well, with Vmax/Km values only 18–200-fold below those for extension of natural Watson–Crick pairs. In marked contrast, Kf essentially does not extend these pairs, giving extension frequencies 4–5 orders of magnitude below that of natural DNA. This likely reflects the different roles of these enzymes in cellular replication and repair. While Kf participates in replication of chromosomal DNA as part of normal cellular DNA synthesis, the Dpo4 enzyme and its E. coli relatives are repair enzymes. Dpo4 in particular is a translesion synthesis enzyme, being responsible for extension of mismatches and alkylated base damage.20,21 Based on this biological role, it is logical that Dpo4 might be functionally better suited to extending xDNA pairs, which, like most mismatched and damaged pairs, are sterically large and geometrically distinct from natural pairs.
Our data for Dpo4-catalyzed synthesis of multiple consecutive xDNA pairs (Fig. 3) show that this polymerase is not restricted to a single xDNA pair in its ability to synthesize and extend this unnatural structure. The experiments confirm that the enzyme can bind a template-primer when it is fully composed of the expanded pairs, and can make four successive correct xDNA pairs. The ability of the enzyme to process this enlarged helix is interesting, and we know of no prior examples in which an enzyme has been shown to correctly process a fully substituted unnatural base pair architecture. NMR structural studies of xDNA helices have shown that they are right-handed and resemble B-DNA in backbone conformation;9 this resemblance to the native structure is likely to aid in the polymerase binding. However, the groove widths and depths are quite different than natural DNA, which suggests that this enzyme's steric flexibility must be important to its activity in this context.
A closer look at the data for cases in which the two enzymes exhibited incorrect selectivity in extension (i.e., a preference for an xDNA mismatch) is instructional. The experiments showed frequent extension of T–xT and T–xC mispairs (Fig. 2C,D). We hypothesize that these mispairs are favored because they most closely adopt a structure resembling that of a canonical DNA pair (Fig. 3). Examination of likely hydrogen-bonded mismatch structures shows that the T–xT mismatch is expected to be able to pair with a structure closely analogous to a T–G wobble-type mismatch (Fig. 4, left). Interestingly, both enzymes extend T–xT mismatches with efficiency close to that of T–G/G–T mismatches (Tables S2 and S3†). A second example is the relatively efficient T–xC mismatch, which might adopt a hydrogen-bonded structure analogous to an A–T pair. In addition, tautomerization of xC to further increase complementarity to T cannot be ruled out. In both these cases, the mismatches appear to be able to form a structure more analogous in size to natural DNA, which may explain their relative efficiency. Structural studies would be needed to confirm whether these specific mispaired structures are indeed formed.
Fig. 4.
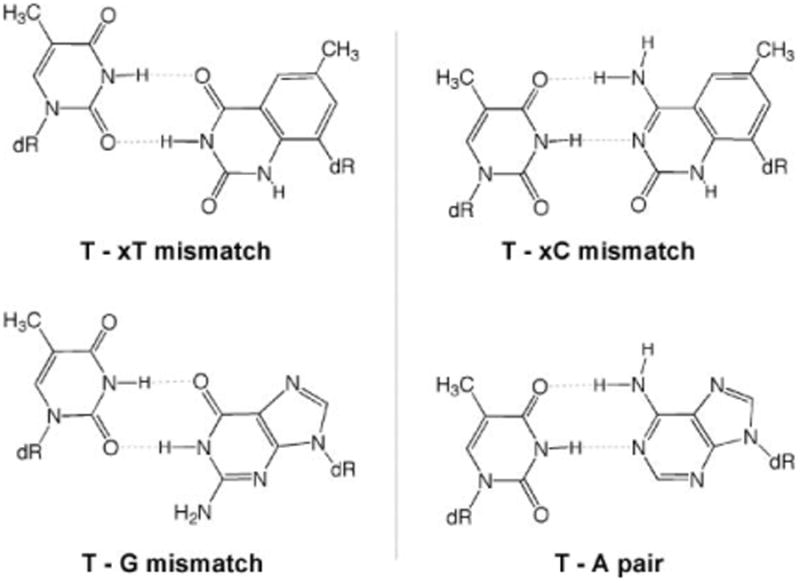
Comparisons showing hypothesized geometric similarity of possible xDNA mispair structures with structures of a T–G wobble-type mismatch and a T–A pair.
Recent studies by Matsuda and coworkers8b also provide evidence of this polymerase selectivity for structures that approximate the natural DNA pair size and geometry. Using the Klenow fragment (exo-), they explored single base pair synthesis involving pairs having four hydrogen bonds. They observed that enzymatic synthesis of naphthyridine : imidazopyridopyrimidine pairs, which were closely analogous to Watson–Crick pairs in glycosidic distance, was efficient. However, pairs that were also complementary in hydrogen bonding, but which were considerably larger by addition of a ring, were much less efficient. This is consistent with our observation of frequent mismatching of xT and xC with pyrimidines, which also may form hydrogen bonds and provide a pair size much closer to the natural one as compared with the intended xDNA pairs.
Our observation of correct editing of xDNA pairs by Kf exo+ (Table 1) is interesting, and suggests that this activity would contribute positively to the fidelity of xDNA synthesis. The frequency by which a polymerase successfully incorporates a given nucleotide overall into a growing primer depends on insertion, extension, and editing probabilities.22 The current data show that Kf can synthesize and edit xDNA correctly. However, the enzyme, which has low processivity, would very likely stall and dissociate before extending terminal xDNA pairs. On the other hand, Dpo4 extends such termini with relatively high efficiency. This suggests that a combination of these two enzymes (or related ones) might be able to replicate xDNA strands with higher efficiency and fidelity than either enzyme alone. An enzyme combination (Kf and pol b) was used recently by Romesberg and Schultz to synthesize and extend hydrophobic base pairs in an effort to address the lack of minor groove hydrogen bonding groups.23 The current results suggest that a different enzyme combination might be useful in addressing a sterically demanding base pair geometry.
These results have some interesting implications in interpreting recent observations of cellular bypass of single xDNA bases in E. coli.13 Those experiments, which involved replication of M13 single-stranded phage DNA containing one xDNA base, showed that xA and xT were highly efficiently bypassed, while xC and xG were bypassed with moderate efficiency. Induction of SOS (repair) response resulted in increased bypass of all cases. Interestingly, we find here that Dpo4 (a repair enzyme in the same family as E. coli pol IV) yields the same extension preference, with xA and xT the most efficiently extended of the four. This suggests that repair polymerases with functional behavior similar to Dpo4 are largely responsible for bypass in E. coli. However, it may well be that a combination of enzymes carries out insertion, editing and extension steps in the cellular replication. Additional experiments with bacteria having specific polymerases knocked out would be instructional on this issue.
Our data suggest that for successful replication of xDNA (and other large base pair designs), one will need to address not just the synthesis of a sterically demanding base pair, but also its editing and extension as well. It will likely be challenging to find one naturally occurring enzyme that can handle all three efficiently and correctly because of the specialized nature of cellular polymerases. This might be addressed in one of two ways: either by use of multipleenzymesincombination,assuggested above,orbyfinding or evolving mutations in existing polymerases that alter the natural preference for the size of DNA base pairs.
Experimental
Nucleoside phosphoramidite derivatives of dxA, dxG, dxT, dxC
Syntheses of these four compounds were carried out as previously reported.6b,c
Oligonucleotide synthesis
Oligodeoxynucleotides were synthesized on an Applied Biosys-tems 394 DNA/RNA synthesizer on a 1 mmol scale. Coupling employed standard β-cyanoethyl phosphoramidite chemistry, but with extended coupling time (600 s) for nonnatural nucleotides. All oligomers were deprotected in concentrated ammonium hydroxide (55 ◦C, 14–16 h), purified by preparative 20% denaturing polyacrylamide gel electrophoresis, and isolated by excision and extraction from the gel, followed by dialysis against water. The recovered material was quantified by absorbance at 260 nm with molar extinction coefficients determined by the nearest neighbor method. Molar extinction coefficients for unnatural oligomers were estimated by adding the measured value of the molar extinction coefficient of the unnatural nucleoside (at 260 nm)tothe calculated value for the natural DNA fragments. Previous studies have shown that xDNA bases have very low hypochromicity in xDNA oligomers.6a,d Molar extinction coefficients for xDNA nucleosides used were as follows: dxA, ε260 = 19 800 M-1 cm-1; dxG, ε260 = 8100 M-1 cm-1; dxT, ε260 = 1200 M-1 cm-1; dxC, ε260 = 5800 M-1 cm-1. Nonnatural oligomers were characterized by MALDI-TOF mass spectrometry: 5′-ACT GxAT CTC CCT ATA GTG AGT CGT ATT A-3′ (calcd. 8604; obsd. 8603); 5′-ACT GxGT CTC CCT ATA GTG AGT CGT ATT A-3′ (cald. 8620; obsd. 8619); 5′-ACT GxCT CTC CCT ATA GTG AGT CGT ATT A-3′ (calcd. 8595; obsd. 8594); 5′-ACT GxTT CTC CCT ATA GTG AGT CGT ATT A-3′ (calcd. 8595; obsd. 8595).
Polymerase kinetics methods
Kf polymerase
The 5′-terminus of the primer was labeled using [(γ-32 P]ATP and T4 polynucleotide kinase. The labeled primer was annealed to the template in a buffer of 100 mM Tris-HCl (pH 7.5), 20 mM MgCl2, 2 mM DTT, and 0.1 mg mL-1 acetylated BSA. A typical polymerase reaction was started by mixing equal volumes of solution A containing the DNA–enzyme complex and solution B containing dNTP substrates. Solution A was made by adding Klenow fragment (exo-) (Amersham) diluted in annealing buffer to the annealed duplex DNA and incubating for 2 min at 37 ◦C. The reaction was terminated by addition of 1.5 volumes of stop buffer (95% formamide, 120 mM EDTA, 0.05% xylene cyanol and bromophenol blue). Steady state kinetics measurements for standing start single nucleotide insertions were carried out as described.2 The final DNA template–primer complex concentration varied in each assay (described in the legends of Tables S1–S3†). Final concentration of triphosphates (0.5–500) μM, amount of polymerase used (0.005– 0.1 unit μL−1) and reaction time (1–60 min) were adjusted to give <20% conversion. Extents of reaction were determined by running quenched reaction sampleson20% denaturing polyacrylamide gel. Relative velocities were calculated as extent of reaction divided by reaction time and normalized to 0.005 unit μL−1 enzyme concentration.
Dpo4 polymerase
E. coli cells expressing Dpo4 were a gift from Dr R. Woodgate (National Institutes of Health). The protein was purified according to the published method.14 The concentration was quantitated by the Bradford method. Primer 5′ termini were labeled using [γ-32 P]ATP (Amersham Bioscience) and T4 polynucleotide kinase (Invitrogen). The labeled primer, template and unlabeled primer were mixed with 2× reaction buffer and water to give a total concentration of primer–template complex of 20 μM. 1× reaction buffer contained 40 mM Tris-HCl (pH = 8.0), 5 mM MgCl2, 100 μM nucleoside triphosphate, 10 mM dithiothreitol, 250 μg mL-1 bovine serum albumin (BSA), 2.5% glycerol. The primer–template duplexes were annealed by heating to 90 °C, and cooling slowly to 4 °C over 1 h. The aforementioned duplex solution (2.5 μL) was mixed with Dpo4 (2.5 μL), and the reaction was initiated by adding a solution of the appropriate dNTP (5 μL). Enzyme concentration (0.01– 1.0 μM), reaction time (2–60 min), and dNTP concentration (1– 500 μM) were adjusted in different dNTP reactions to give less than 20% incorporation. Reactions were quenched by 15 μL of 95% formamide/10 mM EDTA containing 0.05% xylene cyanol and 0.05% bromophenol blue. Extents of reaction were determined by running quenched reaction samplesona20% polyacrylamide/7 M urea gel. Radioactivity was quantified using a Phosphorimager (Molecular Dynamics) and the ImageQuant Program. Reaction velocity v (M min-1) was defined as v = [S]·Iext/[ (Iprim+Iext)·t], where [S] is the concentration of triphosphate, Iext and Iprim are the intensities of the extended product and the remaining primer, respectively. The kcat (Vmax) and Km values were obtained from Hanes–Woolf plots (Table 1) or Eadie–Hofstee plots (Tables S1– S3†).
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM63587) for support. H. Liu and J. Gao were supported by Stanford Graduate Fellowships. H. Lu was supported by a fellowship from Abbott Laboratories. We thank Dr R. Woodgate (National Institutes of Health) for providing bacteria expressing the Dpo4 enzyme.
Footnotes
Electronic supplementary information (ESI) available: Detailed kinetics data in three tables. See DOI: 10.1039/c002766a
References
- 1.(a) Breslow R. Pure Appl Chem. 1998;70:267–270. [Google Scholar]; (b) Kool ET, Waters ML. Nat Chem Biol. 2007;3:70–73. doi: 10.1038/nchembio0207-70. [DOI] [PubMed] [Google Scholar]; (c) Benner SA, Sismour AM. Nature. 2003;421:118. doi: 10.1038/421118a. [DOI] [PubMed] [Google Scholar]
- 2.(a) Neilsen PE, Egholm M, Berg RH, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]; (b) Gryaznov SM, Lloyd DH, Chen JK, Schultz RG, DeDionisio LA, Ratmeyer L, Wilson WD. Proc Natl Acad Sci U S A. 1995;92:5798–802. doi: 10.1073/pnas.92.13.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Summerton J, Weller D. Antisense Nucleic Acid Drug Dev. 1997;7:187–95. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]; (d) Nielsen P, Pfundheller HM, Wengel J. Chem Commun. 1997:825–826. [Google Scholar]; (e) Schöning K, Scholz P, Guntha S, Wu X, Krishnamurthy R, Eschenmoser A. Science. 2000;290:1347–1351. doi: 10.1126/science.290.5495.1347. [DOI] [PubMed] [Google Scholar]; (f) Lescrinier E, Esnouf R, Schraml J, Busson R, Heus H, Hilbers C, Herdewijn P. Chem Biol. 2000;7:719–731. doi: 10.1016/s1074-5521(00)00017-x. [DOI] [PubMed] [Google Scholar]; (g) Zhang L, Peritz A, Meggers E. J Am Chem Soc. 2005;127:4174–4175. doi: 10.1021/ja042564z. [DOI] [PubMed] [Google Scholar]
- 3.(a) Pochet S, Kaminski PA, Van Aerschot A, Herdewijn P, MarliËre P. C R Biol. 2003;326:1175–1184. doi: 10.1016/j.crvi.2003.10.004. [DOI] [PubMed] [Google Scholar]; (b) Chaput JC, Szostak JW. J Am Chem Soc. 2003;125:9274–9275. doi: 10.1021/ja035917n. [DOI] [PubMed] [Google Scholar]; (c) Shaw BR, Dobrikov M, Wang X, Wan J, He K, Lin JL, Li P, Rait V, Sergueeva Z, Sergueev D. Ann N Y Acad Sci. 2003;1002:12–29. doi: 10.1196/annals.1281.004. [DOI] [PubMed] [Google Scholar]; (d) Jung KH, Marx A. Cell Mol Life Sci. 2005;62:2080–2091. doi: 10.1007/s00018-005-5117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Veedu RN, Vester B, Wengel J. Nucleosides, Nucleotides Nucleic Acids. 2007;26:1207–1210. doi: 10.1080/15257770701527844. [DOI] [PubMed] [Google Scholar]; (f) Heuberger BD, Switzer C. J Am Chem Soc. 2008;130:412–413. doi: 10.1021/ja0770680. [DOI] [PubMed] [Google Scholar]
- 4.(a) Rappaport HP. Nucleic Acids Res. 1988;16:7253–7267. doi: 10.1093/nar/16.15.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Switzer C, Moroney SE, Benner SA. J Am Chem Soc. 1989;111:8322–8323. [Google Scholar]; (c) Tor Y, Dervan PB. J Am Chem Soc. 1993;115:4461–4467. [Google Scholar]; (d) Moran S, Ren RXF, Rumney S, Kool ET. J Am Chem Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Benner SA, Battersby TR, Eschgfaller B, Hutter D, Kodra JT, Lutz S, Arslan T, Baschlin DK, Blattler M, Egli M, Hammer C, Held HA, Horlacher J, Huang Z, Hyrup B, Jenny TF, Jurczyk SC, Konig M, von Krosigk U, Lutz MJ, MacPherson JJ, Moroney SE, Muller E, Nambiar KP, Piccirilli JA, Switzer CY, Vogel JJ, Richert C, Roughton AL, Schmidt J, Schneider KC, Stackhouse J. Pure Appl Chem. 1998;70:263–266. doi: 10.1351/pac199870020263. [DOI] [PubMed] [Google Scholar]; (f) McMinn DL, Ogawa AK, Wu YQ, Liu JQ, Schultz PG, Romesberg FE. J Am Chem Soc. 1999;121:11585–11586. [Google Scholar]; (g) Kool ET. Acc Chem Res. 2002;35:936–943. doi: 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]; (h) Mitsui T, Kitamura A, Kimoto M, To T, Sato A, Hirao I, Yokoyama S. J Am Chem Soc. 2003;125:5298–5307. doi: 10.1021/ja028806h. [DOI] [PubMed] [Google Scholar]; (i) Henry AA, Romesberg FE. Curr Opin Chem Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]; (j) Paul N, Nashine VC, Hoops G, Zhang P, Zhou J, Bergstrom DE, Davisson VJ. Chem Biol. 2003;10:815–825. doi: 10.1016/j.chembiol.2003.08.008. [DOI] [PubMed] [Google Scholar]; (k) Kincaid K, Beckman J, Zivkovic A, Halcomb RL, Engels JW, Kuchta RD. Nucleic Acids Res. 2005;33:2620–2628. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Zhang X, Lee I, Berdis AJ. Biochemistry. 2005;44:13101–13110. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]; (m) Hirao I. Curr Opin Chem Biol. 2006;10:622–627. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]; (n) Krueger AT, Kool ET. Chem Biol. 2009;16:242–248. doi: 10.1016/j.chembiol.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonard NJ. Acc Chem Res. 1982;15:128–135. [Google Scholar]
- 6.(a) Liu H, Gao J, Lynch S, Maynard L, Saito D, Kool ET. Science. 2003;302:868–871. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]; (b) Liu H, Gao J, Saito D, Maynard L, Kool ET. J Am Chem Soc. 2004;126:1102–1109. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]; (c) Liu H, Gao J, Kool ET. J Org Chem. 2005;70:639–647. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]; (d) Liu H, Gao J, Kool ET. J Am Chem Soc. 2005;127:1396–1402. doi: 10.1021/ja046305l. [DOI] [PubMed] [Google Scholar]; (e) Gao J, Liu H, Kool ET. Angew Chem Int Ed. 2005;44:3118–3122. doi: 10.1002/anie.200500069. [DOI] [PubMed] [Google Scholar]
- 7.Krueger AT, Kool ET. J Am Chem Soc. 2008;130:3989–3999. doi: 10.1021/ja0782347. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hikishima S, Minakawa N, Kuramoto K, Fujisawa Y, Ogawa M, Matsuda A. Angew Chem Int Ed. 2005;44:596–598. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]; (b) Minakawa N, Ogata S, Takahashi M, Matsuda A. J Am Chem Soc. 2009;131:1644–1645. doi: 10.1021/ja807391g. [DOI] [PubMed] [Google Scholar]; (c) Doi Y, Chiba J, Morikawa T, Inouye M. J Am Chem Soc. 2008;130:8762–8768. doi: 10.1021/ja801058h. [DOI] [PubMed] [Google Scholar]; (d) Evertsz EM, Rippe, Jovin TM. Nucleic Acids Res. 1994;22:3293–3303. doi: 10.1093/nar/22.16.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Battersby TR, Albalos M, Friesenhahn MJ. Chem Biol. 2007;14:525–531. doi: 10.1016/j.chembiol.2007.03.012. [DOI] [PubMed] [Google Scholar]; (f) Heuberger BD, Switzer C. Chem Bio Chem. 2008;9:2779–2783. doi: 10.1002/cbic.200800450. [DOI] [PubMed] [Google Scholar]
- 9.(a) Liu H, Lynch SR, Kool ET. J Am Chem Soc. 2004;126:6900–6905. doi: 10.1021/ja0497835. [DOI] [PubMed] [Google Scholar]; (b) Lynch SR, Liu H, Gao J, Kool ET. J Am Chem Soc. 2006;128:14704–14711. doi: 10.1021/ja065606n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee AHF, Kool ET. J Am Chem Soc. 2006;128:9219–9230. doi: 10.1021/ja0619004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Kunkel TA, Bebenek K. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]; (b) Kool ET. Annu Rev Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]; (c) Di Pasquale F, Fischer D, Grohmann D, Restle T, Geyer A, Marx A. J Am Chem Soc. 2008;130:10748–10757. doi: 10.1021/ja8028284. [DOI] [PubMed] [Google Scholar]
- 12.(a) Patel PH, Loeb LA. Proc Natl Acad Sci U S A. 2000;97:5095–5100. doi: 10.1073/pnas.97.10.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghadessy FJ, Ong JL, Holliger P. Proc Natl Acad Sci U S A. 2001;98:4552–4557. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xia G, Chen L, Sera T, Fa M, Schultz PG, Romesberg FE. Proc Natl Acad Sci U S A. 2002;99:6597–6602. doi: 10.1073/pnas.102577799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sauter KB, Marx A. Angew Chem Int Ed. 2006;45:7633–7635. doi: 10.1002/anie.200602772. [DOI] [PubMed] [Google Scholar]
- 13.Delaney JC, Gao J, Liu H, Shrivastav N, Essigmann JM, Kool ET. Angew Chem Int Ed. 2009;48:4524–4527. doi: 10.1002/anie.200805683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Travaglini EC, Mildvan AS, Loeb LA. J Biol Chem. 1975;250:8647–8656. [PubMed] [Google Scholar]
- 16.Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc Natl Acad Sci U S A. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizukami S, Kim TW, Helquist SA, Kool ET. Biochemistry. 2006;45:2772–2778. doi: 10.1021/bi051961z. [DOI] [PubMed] [Google Scholar]
- 18.Ling H, Boudsocq F, Woodgate R, Yang W. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 19.Fersht AR, Knill-Jones JW, Tsui WC. J Mol Biol. 1982;156:37–51. doi: 10.1016/0022-2836(82)90457-0. [DOI] [PubMed] [Google Scholar]
- 20.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. J Biol Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 21.Rechkoblit O, Malinina, Cheng, Geacintov NE, Broyde, Patel DJ. Structure. 2009;17:725–736. doi: 10.1016/j.str.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodman MF, Fygenson KD. Genetics. 1998;148:1475–1482. doi: 10.1093/genetics/148.4.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tae EL, Wu Y, Xia G, Schultz PG, Romesberg FE. J Am Chem Soc. 2001;123:7439–7440. doi: 10.1021/ja010731e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.