

SUMMARY
Gene expression burdens cells by consuming resources and energy. While numerous studies have investigated regulation of expression level, little is known about gene design elements that govern expression costs. Here, we ask how cells minimize production costs while maintaining a given protein expression level and whether there are gene architectures that optimize this process. We measured fitness of ~14,000 E. coli strains, each expressing a reporter gene with a unique 5′ architecture. By comparing cost-effective and ineffective architectures, we found that cost per protein molecule could be minimized by lowering transcription levels, regulating translation speeds, and utilizing amino acids that are cheap to synthesize and that are less hydrophobic. We then examined natural E. coli genes and found that highly expressed genes have evolved more forcefully to minimize costs associated with their expression. Our study thus elucidates gene design elements that improve the economy of protein expression in natural and heterologous systems.
In Brief
While numerous studies have investigated regulation of expression level, Frumkin et al. study gene design elements that govern expression costs and allow cells to minimize such costs while maintaining a given protein expression level.
INTRODUCTION
In nature, cells must express different genes in a regulated manner. On one hand, genes must be expressed at levels that maximize their benefit, and on the other, cells need to minimize the genes’ production costs (Dekel and Alon, 2005; Wagner, 2005). Costs of expression originate from spending cellular resources, such as building blocks (amino acids and nucleotides), from allocation of cellular machineries (RNA polymerase and ribosome), and from energy and reducing power consumption (Bienick et al., 2014; Glick, 1995; Ibarra et al., 2002; Rang et al., 2003). Even after their production, proteins might still impose costs when degraded or by exerting toxicity, e.g., due to aggregation (Geiler-Samerotte et al., 2011). Understanding what molecular processes determine expression cost, its relation to cellular growth and gene regulation, and how costs evolutionarily shape the genome are key aspects of cell biology that remain largely elusive. While numerous studies investigated molecular mechanisms and gene sequence architectures that regulate expression level (Gingold and Pilpel, 2011; Kudla et al., 2009; Qian et al., 2012; Sharp et al., 1986; Subramaniam et al., 2013), very little is known about design elements that govern expression costs.
Different works have studied expression costs in unicellular organisms by imposing the expression of an unneeded protein (Bentley et al., 1990; Dekel and Alon, 2005; Dong et al., 1995; Kafri et al., 2016; Rang et al., 2003; Scott et al., 2010). The production of such unneeded proteins diverts resources from synthesis of the cell’s own proteins, thus decreasing cellular fitness (Emilsson and Kurland, 1990; Marr, 1991; Vind et al., 1993). Central to these studies is the characterization of the correlation between the imposed expression levels of the unneeded proteins to the cost. Yet, ultimately natural selection dictates the expression level of natural genes according to the required concentration of each protein. Thus, a fundamental question, which has not been addressed before, is how cells can achieve a specific expression level of a gene while minimizing its expression costs.
Addressing this question is challenging because changes in sequence could affect both expression level and expression costs. To disentangle expression level and expression costs and reveal mechanisms that affect cost per protein molecule, we utilized a synthetic reporter library of ~14,000 different sequence variants, each fused upstream to a GFP gene (Goodman et al., 2013). We then combined competition assays and deep sequencing to measure the fitness of all variants in parallel. This procedure allowed us to elucidate gene architectures that minimize expression cost at a given protein expression level. We show that various molecular mechanisms, such as protein/mRNA ratios, ribosome early elongation pauses, amino acid synthesis costs, and peptide hydrophobicity, determine the cost per protein molecule. We then generated a model that predicts the cost effectiveness of gene architectures and applied it to natural E. coli genes. We found that highly expressed genes have evolved more forcefully to be encoded by cost-minimizing mechanisms. Our observations indicate that natural selection has shaped genes’ architectures to reduce cost of gene expression.
RESULTS
5′ Gene Architecture Affects Cost of Gene Expression
Our question is whether different gene sequence elements can minimize cost of expression per protein molecule and hence increase cellular fitness. To focus on sequence features at the 5′ region of a gene, we utilized a previously published synthetic gene library (Goodman et al., 2013) composed from ~14,000 different variants expressing a GFP gene. Each variant holds a unique variable 5′ gene architecture that includes a promoter, a ribosome binding site (RBS), and an 11-amino-acid-long N terminus fusion (Figure 1A; Experimental Procedures).
Figure 1. 5′ Gene Architectures Affect Cost of Gene Expression at a Given Expression Level.

(A) We utilized a synthetic library of ~14,000 E. coli strains, each expressing a GFP construct with a unique 5′ architecture that includes a promoter, ribosome binding site (RBS), and an 11-amino-acid-fused peptide. There were two different promoter types, four RBSs, and 137 amino acid fusions that were each synonymously re-coded to 13 different versions (see Goodman et al., 2013 for full details).
(B) FitSeq methodology to measure relative fitness of strains in a pooled synthetic library. First, the library was grown six independent times for ~84 generations, and samples were taken at generations 0, ~28, ~56, and ~84. Then, unique 5′ gene architectures were simultaneously amplified and sent for deep sequencing, which allowed to follow the frequency of each variant in the population over the course of the experiment. Finally, a relative fitness score was assigned for each variant based on its frequency dynamics.
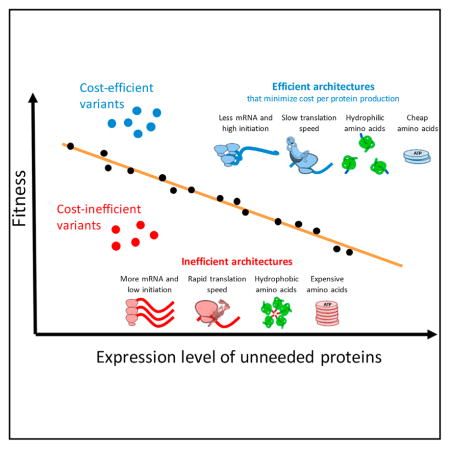
(C) GFP expression level (as measured by Goodman et al., 2013; x axis) versus fitness effect (based on results of repetition C; y axis) of each variant in the library (Pearson correlation, r = −0.79, p < 10−200). Fitness effect comes from the burden of expressing unneeded proteins on cellular growth and is calculated by analyzing the frequency dynamics of each variant (see Experimental Procedures). We defined fitness residual as the difference between a variant’s observed and expected fitness. The expected fitness is calculated from the regression line between GFP expression and fitness (black line). Some variants consistently demonstrated positive (blue dots, n = 975) or negative (red dots, n = 815) fitness residual sign. Other variants showed extremely low fitness residual, and we termed those variants as “underachievers” (purple dots, n = 80). The group size of positive, negative, and underachiever variants are significantly much higher than expected by chance (Supplemental Information). These results suggest that certain 5′ gene architectures can increase or reduce the cost of gene expression. See also Figure S1A. Inset: positive (blue violin plot) and negative (red violin plot) fitness residual variants come from the same distribution of GFP expression level (Wilcoxon rank-sum, p = 0.46). Black line represents the median value. Thus, the effect of GFP levels on fitness was successfully factored out, thus allowing us to elucidate other molecular mechanisms that tune expression cost at given expression levels.
(D) Fitness and fitness residuals demonstrate different distributions. While most variants showed negative fitness values, fitness residual is more similar to a normal distribution, though with a negative tail.
To reveal the expression cost of each variant, we measured relative fitness of all variants in parallel in a competition assay in six independent repeats. We then deep sequenced the variable region of the pool of variants and calculated relative fitness of each variant (Figure 1B; see Experimental Procedures).
We regressed fitness values against GFP expression levels and observed a negative, linear correlation (Figure 1C, Pearson correlation, r = −0.79, p < 10−200; Figure S1A). The linear decline in fitness with expression is in agreement with previous studies (Kafri et al., 2016; Scott et al., 2010). The regression line, which outlines the relations between fitness and expression, allowed us to estimate the expected fitness for each library variant according to its GFP expression level. Variants whose fitness does not deviate consistently across repeats from this regression line are deduced not to utilize mechanisms that enhance or reduce the production cost per protein molecule.
Yet, many variants did deviate from the linear regression line, demonstrating fitness that is higher or lower than expected given their GFP expression levels. We hypothesized that variants that repeatedly deviated from the expected fitness might utilize gene architectures that either reduce or increase the cost of GFP production per protein molecule. Hence, we calculated each variant’s “fitness residual,” which we defined as the difference between the actual fitness that we measured for the variant and the fitness expected for it according to its GFP expression level and the linear regression (Figure 1C). A positive fitness residual means that a given variant showed higher fitness than expected given its GFP expression level, suggesting that it can produce this GFP level with lower costs. A negative fitness residual means that the variant showed lower fitness than expected given its GFP expression level.
We then classified each variant as either positive or negative according to its fitness residual sign (Figure 1C, blue and red dots; see Experimental Procedures). Since the observed fitness residual is sensitive to biological noise (i.e., drift during competition) and experimental errors (i.e., sampling errors), we only classified variants as positive or negative if their fitness residual sign was identical in at least five out of the six repeats of the experiments in each of the two final sampling points of the competition (see Experimental Procedures and Supplemental Experimental Procedures). This approach resulted in 975 positive and 815 negative variants (significantly higher than expected by chance even at very high levels of measurement errors; Supplemental Experimental Procedures). Classification into either positive or negative fitness residual groups allowed us to eliminate the effect of GFP expression level on fitness as these two groups demonstrate the same expression distribution (Figure 1C, inset).
We also noticed a set of 80 library variants, which we termed “underachievers,” whose fitness residual scores were repeatedly at the bottom 5% of the entire library (Figure 1C, purple dots; see Experimental Procedures). We hypothesized that these underachiever variants show extremely low fitness residuals because they produce GFP even more wastefully, and we expected them to show stronger usage of low-efficiency gene architectures compared to the negative fitness residual group. There appeared to be no “overachievers” in these data.
Production of More Proteins per mRNA Molecule Is an Economic Means to Minimize Expression Costs
We first hypothesized that reaching the same GFP level with lower levels of mRNA of the GFP gene could be beneficial. While positive and negative fitness residual variants come from the same distribution of GFP expression levels (Figure 1C, inset), we compared their GFP mRNA levels and found positive variants to have lower levels compared to negative variants (Figure 2A; Wilcoxon rank-sum, p = 1.6 × 10−9, effect size = 58.26%; see Experimental Procedures). This difference was independent of GFP level: binning the data according to GFP levels, we observed the reduced levels of mRNA for positive variants in all expression bins (Figure S1B).
Figure 2. Higher Ratio of GFP Protein/mRNA Minimizes Cost of Gene Expression.

(A) Although coming from the same distribution of GFP levels, positive variants (blue violin plot) demonstrate lower mRNA levels of the GFP gene compared to negative variants (red violin plot) (effect size = 58.26%, Wilcoxon rank-sum, p = 1.6 × 10−9). Consistently, underachiever variants (purple violin plot) show higher mRNA levels compared to negative variants (effect size = 68.04%, Wilcoxon rank-sum, p = 9.6 × 10−8). Black line represents the median value.
(B) Positive variants show higher translation initiation rates compared to negative variants (effect size = 61.9%, Wilcoxon rank-sum, p = 3.7 × 10−18).
(C) Positive variants demonstrate higher translation efficiencies (protein/mRNA) compared to negative variants (effect size = 55.67%, Wilcoxon rank-sum, p = 3.4 × 10−5). Consistently, underachiever variants (purple violin plot) further show lower translation efficiencies compared to negative variants (effect size = 63.06%, Wilcoxon rank-sum, p = 1.1 × 10−4).
Statistically significant differences (p < 0.05) are marked with an asterisk. See also Figures S1B and S2A.
The observation that positive variants have equal GFP protein levels but lower GFP mRNA levels indicates that they are able to produce more GFP proteins per mRNA molecule. We postulated that high translation initiation rate could be a mechanism for maintaining the same GFP levels despite low mRNA levels in positive variants. We calculated initiation rates for all library variants using the “Ribosome Binding Site Calculator” (Salis, 2011) and observed that indeed positive variants had higher initiation rates (Figure 2B; effect size = 61.9%, Wilcoxon rank-sum, p = 3.7 × 10−18). This observation holds true when examining mRNA level versus translation initiation rate at the individual variant level (Figure S2A). Indeed, when examining translation efficiency per variant (using measured protein levels divided by mRNA levels), positive variants demonstrated higher translation efficiencies than negative fitness residual variants (Figure 2C; effect size = 55.67%, Wilcoxon rank-sum, p = 3.4 × 10−5). Moreover, we found that underachiever variants demonstrated even higher mRNA levels and lower translation efficiencies compared to the negative variants (Figures 2A and 2C; effect size = 68.04% and 63.06%, Wilcoxon rank-sum, p = 9.6 × 10−8 and 1.1 × 10−4, respectively). Thus, by increasing translation efficiency, cells reduce transcription costs and hence also cost per protein.
Slower Translation Speed at Early Elongation of Coding Region, Achieved by Diverse Means, Reduces Expression Costs
We next aimed to elucidate other cellular mechanisms that directly regulate the translation machinery and that might reduce expression costs. We first examined codon decoding speeds by the ribosome. Codon adaptation of transcripts to the cellular tRNA pool has been shown to be a regulatory mechanism for translation elongation (Goodarzi et al., 2016; Higgs and Ran, 2008; Kudla et al., 2009; Plotkin and Kudla, 2011; Shah and Gilchrist, 2011; Weinberg et al., 2016; Yona et al., 2013). Specifically, the prevalence of slowly translated codons at the 5′ of open reading frames (ORFs) has been suggested to support the efficiency of gene translation (Tuller et al., 2010a). This “ramp model” proposes that delaying ribosomes at the beginning of the elongation phase decreases downstream ribosomal pauses and collisions, which can therefore reduce ribosome jamming, and perhaps also ribosomal abortion events.
Although contradicting evidence were reported for the existence and relevance of this mechanism to expression level (Charneski and Hurst, 2014; Dana and Tuller, 2014; Heyer and Moore, 2016; Ingolia et al., 2009; Shah et al., 2013; Tuller and Zur, 2015), the main prediction of the model—that 5′ ramping reduces cost of expression at a given expression level—has not been tested so far. Here, we had the first opportunity to test this hypothesis as only the 5′ variable region of the GFP varied in the library, while all other parameters remained constant. Thus, we asked whether slow 5′ translation speed is associated with positive fitness residual. We used “mean of the typical decoding rates” (MTDR) (Dana and Tuller, 2014), a measure of codon decoding time derived empirically from ribosome profiling data in E. coli (see Experimental Procedures), to calculate translation speed for each library variant. We reasoned that if translational ramp is beneficial, then low MTDR scores, i.e., low ribosome speeds, should be more prevalent among the positive fitness residual variants. Indeed, our results showed that positive variants demonstrate significantly lower translation speeds at the N-terminal fusion (Figure 3A; effect size = 59.55%, Wilcoxon rank-sum, p = 3 × 10−12) and further for the underachievers (effect size = 64.79%, Wilcoxon rank-sum, p = 1.2 × 10−5).
Figure 3. Slow Translation Speed at Early Elongation, Achieved by Diverse Molecular Means, Reduces Expression Cost.

(A, C, and D) Positive variants show lower values of codon decoding speed (A), stronger mRNA structures (C), and lower speeds due to higher anti-Shine Dalgarno affinities (D) compared to negative variants (effect size = 59.55%, 65.03%, and 63.82%, Wilcoxon rank-sum, p = 3 × 10−12, 5.4 × 10−28, and 6.3 × 10−24, respectively). Statistically significant differences (p < 0.05) are marked with an asterisk. See also Figure S1B.
(B) Mean folding energy of mRNA secondary structure according to window’s start position for positive (blue curve) and negative (red curve) variants; error bars represent SEM. Dashed lines mark different positions along the variable region upstream to the GFP. Black vertical line marks the beginning of window with the largest observed difference, which is found at nucleotide positions +4 of the ORF, just after the first AUG codon. The distributions at this window position are seen in (C). See also Figure S2B.
Though in the original ramp model ribosome attenuation was proposed to be obtained by codons that correspond to rare tRNAs, additional mechanisms that can slow down the ribosome at early elongation regions could serve in ramping. These mechanisms include, in particular, tight mRNA secondary structure (Goodman et al., 2013; Tholstrup et al., 2012; Tuller et al., 2010b; Wen et al., 2008) and high affinity to the anti-Shine Dalgarno (aSD) motif of the ribosome (Li et al., 2012). We thus examined each of these factors separately and asked whether they are associated with positive or negative fitness residual.
When we computed folding energies for segments of mRNA nucleotides on a sliding window along the variable region of each variant, we found that positive fitness residual variants demonstrated tighter secondary structures compared to negative variants along many different window positions (Figure 3B; Figure S2B for different window sizes). Strikingly, the maximum difference in folding energy is observed when the window’s start position is at the beginning of the translated region of the ORF, excluding the upstream 5′ UTR (Figure 3C; effect size = 65.03%, Wilcoxon rank-sum, p = 5.4 × 10−28). Hence, these results, together with previous ones, reveal the dual role of mRNA folding: on one hand, loose mRNA structure at the RBS is associated with high expression level (Goodman et al., 2013), and on the other hand, utilization of a strong secondary structure at the 5′ end of the ORF can reduce per-protein costs.
It was previously suggested that elongating ribosomes in E. coli dwell longer on sequences that have high affinity to the aSD motif in the ribosome (Li et al., 2012). However, this observation has been recently questioned (Mohammad et al., 2016). We next examined the effects of Shine Dalgarno-mediated ribosomal pauses on fitness residuals. We calculated affinities to the aSD along the sequence of each variant, derived a ribosome speed estimation based on these affinities (see Experimental Procedures) and found that positive fitness residual variants are characterized by low ribosome speed early in the ORF (Figure 3D; effect size = 63.82%, Wilcoxon rank-sum test, p = 6.3 × 10−24).
We thus provide the first experimental evidence for a set of three gene architecture factors—codon decoding time, mRNA structure, and affinity to the anti-Shine Dalgarno motif—that could each implement 5′ ramping by slowing down ribosomes and, by that, allow cells to reduce the cost of gene expression at a given expression level.
Another means of reducing translation speed that was recently demonstrated (so far in yeast) is the incorporation of positively charged amino acids (Charneski and Hurst, 2013) or proline residues (Artieri and Fraser, 2014) in newly synthesized peptides. Yet, we did not detect any difference in frequency of such amino acids between the positive and negative fitness residual groups.
Amino Acid Synthesis Cost and Hydrophobicity Affect Cost of Gene Expression
So far we have examined features that are based on the nucleotide sequence and how it associates with fitness residual. Next, we aimed to explore the possibility that the amino acid composition of the N terminus fusion to the GFP associates with cellular fitness.
Amino acids differ by the metabolic costs associated with their biosynthesis—predominantly energy and reducing power determinants invested in their metabolic production (Akashi and Gojobori, 2002). We thus hypothesized that usage of energetically expensive amino acids may cause a heavier burden at a given expression level. Indeed, lower cost of the N terminus fusions were found to associate with positive fitness residual variants (Figure 4A; effect size = 72.74%, Wilcoxon rank-sum, p = 7.4 × 10−62). Here, as well, underachiever variants show more expensive amino acid usage compared to the negative group (Figure 4A; effect size = 72.75%, Wilcoxon rank-sum, p = 1.7 × 10−11).
Figure 4. Usage of Expensive-to-Synthetize, Lowly Available, and Hydrophobic Amino Acids Decreases Fitness Residual.

(A) N terminus amino acid fusions of negative variants are more expensive to synthesize compared to positive variants (effect size = 72.74%, Wilcoxon rank-sum, p = 7.4 × 10−62). Underachievers utilize even more expensive amino acids (effect size= 72.75%, Wilcoxon rank-sum, p = 1.7 × 10−11). See also Figures S1B andS2C.
(B) The frequency ratio of amino acids between positive and negative variants is negatively correlated with the energetic cost of amino acids (Pearson correlation, r = −0.54, p = 0.01). Each amino acid is marked according to its one-letter code.
(C) The frequency ratio of amino acids between positive and negative variants is negatively correlated with the demand/supply ratio of amino acids (Pearson correlation, r = −0.82, p = 10−4). Demand comes from occupancy of ribosomes on each transcript (see Experimental Procedures), and supply is the cellular concentration of each amino acid (Bennett et al., 2009).
(D) Amino acid availability and energetic cost are correlated (Pearson correlation, r = −0.72, p = 1.8 × 10−3).
(E) N terminus amino acid fusions of negative variants are more hydrophobic than positive variants (effect size = 69.11%, Wilcoxon rank-sum, p = 3.2 × 10−44). N terminus fusion of underachievers are even more hydrophobic (effect size = 81.67%, Wilcoxon rank-sum, p = 7.7 × 10−21). See also Figures S1B and S2C.
We further examined the relation between fitness residual and amino acid energetic cost by calculating the frequency ratio of each individual amino acid between the positive and negative fitness residual groups (see Experimental Procedures). Remarkably, this frequency ratio was found to negatively correlate with the metabolic cost of each amino acid (Figure 4B; Pearson correlation, r = −0.54, p = 0.01). These observations suggest that expensive-to-synthesize amino acids burden cells during their costly production due to a potential feedback that increases their synthesis in response to consumption.
In addition to direct metabolic cost, the incorporation of amino acids that appear in low cellular concentrations could reduce fitness indirectly as it might disturb the synthesis of other native proteins. We used ribosome profiling data (Li et al., 2012) to calculate amino acid demands and utilized previously measured cellular concentrations as amino acid supplies (Bennett et al., 2009) (see Experimental Procedures). Indeed, we found that amino acids with low demand-to-supply ratios are more prevalent in positive variants (Figure 4C; Pearson correlation, r = −0.82, p = 10−4). This observation implies that utilization of amino acids that are less available to the cell (either due to high demand or low supply) increase expression cost and are associated with negative fitness residual variants. Since metabolic cost of amino acids and their cellular supplies are correlated (Figure 4D; Pearson correlation, r = −0.72, p = 1.8 × 10−3), we could not evaluate which mechanism—cost or availability—contributes more to fitness residual.
We next reasoned that an additional factor by which a protein could affect fitness is its toxicity, e.g., due to aggregation. As aggregation is driven by hydrophobic interactions, we turned to a conventional measure of amino acid hydrophobicity (Kyte and Doolittle, 1982) to examine whether it is predictive of fitness residuals. We found that positive fitness residual variants tended to have significantly less hydrophobic amino acids fused to the GFP (Figure 4E; effect size = 69.11%, Wilcoxon rank-sum, p = 3.2 × 10−44). Underachievers showed an even more pronounced effect (Figure 4E; effect size = 81.67%, Wilcoxon rank-sum, p = 7.7 × 10−21). This negative effect of hydrophobic residues in cytosolic proteins could indeed be derived from post-synthesis costs, but it could also reflect an equally interesting possibility: that aggregation-prone peptides reduce the functional level of the GFP (and similarly the fraction of the active form of native proteins). According to this possibility, aggregation is wasteful and must be compensated by further costly production to reach the required expression level of the protein.
We further found that the higher the GFP expression, the more beneficial it should be to utilize cheap or hydrophilic amino acids (Figure S2C).
All Sequence Parameters Contribute Independently to Fitness
We have revealed, so far, a set of mechanisms that affect expression costs and therefore cellular fitness. Although these mechanisms are different in their nature, it is possible that variants that score highly on one of these parameters tend to score highly on others. For example, anti-Shine Dalgarno affinity could correlate with the energy of the secondary structure of the mRNA, as both parameters are influenced by Guanine content. To check this possibility, we computed the correlation among the variants in the library between each pair of sequence parameters: codon decoding speed, mRNA secondary structure, anti-Shine Dalgarno affinity, hydrophobicity, and amino acid energy cost. Reassuringly, no strong correlation was found between any two parameters (Figure 5). Nonetheless, for feature pairs that did demonstrate non-negligible correlations (Pearson correlation, r > 0.1), we asked whether the signal of one feature is still observed while controlling for variation in the other. We found that each factor contributed directly to the signal, even upon controlling for other factors as potential confounders (see Figure S3).
Figure 5. Each Feature Affects Fitness Residual Independently.

Correlation plots of each feature pair show lack of correlation in most cases and only weak correlations in other cases. For feature pairs with Pearson correlation of r > 0.1, we compared the difference in one feature while controlling for the second and vice versa. See also Figure S3. Black lines are the regression curves between each feature pair. Number at upper-left corner is the Pearson correlation.
Expression Costs Can be Minimized Even at Specified Amino Acid Sequences
Since maintaining a protein’s function usually requires keeping its specific amino acid sequence, we next asked whether the mechanisms that we found here can reduce expression costs for a specified peptide sequence by using alternative nucleotide sequences. We defined “Δfitness-residual” as the difference between a variant’s fitness residual and the average fitness residual of all library variants who share with that variant the same amino acid sequence. Then, we compared the various architectural features between variants with above-average Δfitness-residual to variants with below-average Δfitness-residual (see Experimental Procedures).
Figures 6A–6E depict, for each of the analyzed features, the difference in feature value between variants with above- or below-average Δfitness-residual. Interestingly, for each feature, the above- and below-average sub-groups had significantly different feature scores, reflecting the same trends as observed in all earlier analyses. For example, mRNA levels tend to be higher in the below-average sub-group in most of the 137 N terminus fusions (t test, p values for GFP mRNA levels = 6.2 × 10−3, initiation rates = 7 × 10−9, codon decoding speeds = 4.3 × 10−2, mRNA folding = 3.5 × 10−16, and aSD velocity = 7.6 × 10−7). The conclusion from this analysis is that although amino acid features affect fitness residuals, the other features provide sufficient degrees of freedom to minimize costs even at a specified amino acid sequence.
Figure 6. Variant with Same N Terminus Amino Acid Fusion Demonstrate a Range of Fitness Residuals.

(A–E) Each dot represents one of the 137 N terminus fusions in the library. The x axis and the y axis represent the mean value of a feature for the variants with either below-average or above-average Δfitness-residual, respectively. The vertical and horizontal error bars represent standard errors for each of the axes. A statistical difference for deviance from the X = Y line was observed for all features, suggesting that even at a given amino acid sequence, these mechanisms affect fitness residual and can minimize expression costs (t test, p values: A, mRNA levels, 6.2 × 10−3; B, initiation rates, 7 × 10−9; C, codon decoding speeds, 4.3 × 10−2; D, mRNA folding, 3.5 × 10−16; and E, aSD velocity, 7.6 × 10−7). d is Cohen’s d that calculates the effect size.
A Regression Model Calculates Relative Contribution of Each Feature and Predicts Fitness Residual Scores
So far, we have examined fitness residual as a binary classification, namely categorizing variants with either positive or negative fitness residual. Complementing this binary analysis, in Figure S4A, we show that each feature correlates significantly with actual fitness residual values. We next aimed to predict actual fitness residual values of the library variants from their gene architecture features using a multiple linear regression model. We trained the model on a randomly chosen subset of 70% of the library variants, cross validated it on all other variants by comparing their predicted and observed fitness residual, and found a good correlation (see Experimental Procedures; Figure 7A; r = 0.53, p < 10−200).
Figure 7. A Model that Predicts Fitness Residual Accurately Reveals that Fitness Residual of Natural Bacterial Genes Is Correlated with Their Expression Level.

(A) A linear regression model based on all eight features predicts fitness residual accurately in a cross-validation test (Pearson correlation, r = 0.53, p < 10−200). See also Figure S4.
(B) The weighted coefficients of each feature in the regression model demonstrating the relative contribution of each feature to fitness residual (p value for regression coefficient of mRNA level = 3.5 × 10−11, initiation rate = 2.5 × 10−12, TEGFP protein/mRNA = 2.7 × 10−9, codon decoding speed = 8.7 × 10−3, mRNA folding energy = 1.5 × 10−50, aSD velocity = 8.7 × 10−3, hydrophobicity < 10−200, and amino acid synthesis cost = 5.4 × 10−80). The sign of the contribution of each coefficient shows whether a feature is associated positively or negatively with fitness residuals. Error bars represent standard error of the coefficient estimation.
(C) Predicted fitness residuals of E. coli genes according to the regression model are correlated with their expression levels (Pearson correlation, r = 0.25, p = 2 × 10−53), suggesting that natural selection shapes 5′ gene architectures in order to minimize costs of gene expression.
(D) Distribution of fitness residual scores for E. coli genes as predicted by regression model that was trained on either experimental or mock data. The experimentally based model predicts a significant, higher range of fitness residuals (p < 10−5), suggesting that the mechanisms that we elucidate with the synthetic library also apply on natural genes.
(E) Predicted fitness residuals of B. subtilis genes according to the regression model are correlated with their expression levels (Pearson correlation, r = 0.33, p = 10−93), suggesting that our model also applies for other bacteria species.
(F) Same as (D), only for B. subtilis genes.
When the regression was performed on a scrambled library, which randomly links feature values and variants, the correlation between observed and predicted fitness residual was practically eliminated (Figure S4B; r = 0.02). We performed 105 such randomizations, and all of them demonstrated such extremely weak correlations. This negative control demonstrates that we obtained a genuine means to predict fitness residual values based on computable gene architecture parameters. We concluded that a gene architecture that utilizes more of the features that we discovered and that, to a greater extent, typically gives rise to higher fitness residuals as expression costs are further minimized.
Additionally, this regression model allowed us to calculate the relative contribution of each feature by comparing the coefficients assigned by the regression model (Figure 7B). This analysis revealed that the features contributing to fitness residual the most are hydrophobicity and metabolic cost of the N terminus fusion, while codon decoding speed contributes the least. To avoid over-fitting of our model on the library data, we performed feature selection using the Lasso algorithm (see Experimental Procedures). This validation resulted in the exclusion of only codon decoding speed from the model, suggesting that its contribution to fitness residual is indeed lower compared to other features.
Highly Expressed Natural Bacterial Genes Have Evolved Gene Architectures that Minimize Their Production Costs
With these findings from the synthetic library, we next asked whether the mechanisms that we revealed as cost reducing were also utilized by natural selection to optimize E. coli’s native genes. We thus calculated each E. coli gene’s score with respect to the relevant features and used the regression model to predict its fitness residual score (see Experimental Procedures and Table S4, related to Figure 7). Since a higher expression level results in higher expression cost, we next hypothesized that E. coli genes with higher expression levels are more likely to be endowed with cost-reducing architectures. Indeed, we found a significant correlation between predicted fitness residual of E. coli genes and their protein expression levels (Figure 7C; r = 0.25, p = 2 × 10−53), demonstrating a stronger selection for optimizing the 5′ gene architecture for highly expressed genes. We obtained similar results when predicting fitness residuals for all genes in the Gram positive B. subtilis, pointing to the generality of the model (Figure 7E; r = 0.33, p = 10−93; see Experimental Procedures and Table S4, related to Figure 7).
Interestingly, the range of fitness residuals predicted by our model for the E. coli and B. subtilis genes was significantly larger than the range predicted by a mock regression model that was trained on randomly scrambled data of the synthetic library (see Experimental Procedures; Figures 7D and 7F; p < 10−5). This observation suggests that the model that we trained on the library data is able to expose the expression-cost optimality of natural 5′ gene architectures.
DISCUSSION
In this study, we found architectures and motifs that govern expression costs and reveal their function even beyond a direct effect on the process of expression. We show that regulating initiation and mRNA levels affects expression cost, as increasing the number of proteins that are produced per mRNA is associated with a positive fitness residual. This architecture could be beneficial because it reduces energy and resource consumption that are devoted to mRNA production. If cost reducing, why do genomes not further utilize the strategy of low transcription and mRNA abundance, combined with high translation initiation? One potential reason is that too low of mRNA levels might lead to increased expression noise (Taniguchi et al., 2010) or increased response time to an environmental signal (Gasch and Werner-Washburne, 2002). It is thus expected that natural genes would show a tradeoff between cost-reducing architectures and designs that satisfy other requirements, such as controlled noise and short response times.
The “translational ramp” theory predicted an effect of ribosome speed at early elongation on expression cost at a given expression level (Tuller et al., 2010a). The theory was never tested as such, since fitness reduction upon expression of an unneeded protein was not systematically measured for different gene sequences at various expression levels. We demonstrate here that slow translation speed at the 5′ end is beneficial in terms of reduced expression cost and increased cellular growth rate. We show that in addition to codon decoding times, there are at least two additional ramping means that are likely beneficial: occurrence of Shine-Dalgarno-like sequences and strong secondary structures.
Recent works showed that 5′ mRNA secondary structure governs expression level of transcripts in bacteria (Goodman et al., 2013; Kudla et al., 2009; Shah et al., 2013). Here, we observed that tight mRNA structures are enriched in positive variants. Consequently, it seems that mRNA structure plays a more complex role than previously thought. On one hand, 5′ mRNA structure, specifically upstream of the AUG start codon, regulates expression levels as it governs initiation rates (Goodman et al., 2013; Salis, 2011). On the other hand, tight structures at the beginning of the ORF, which were previously observed in E. coli genes (Tuller et al., 2011), are shown here to be beneficial in minimizing expression cost.
We revealed that the amino acid composition of a gene can also affect expression cost at a given expression level by showing that hydrophobic amino acids reduce fitness residual, perhaps due to their increased tendency to form toxic aggregates in the cytoplasm. In agreement with this, it was shown that mis-folded proteins impose growth reduction to yeast cells in a dosage-dependent manner (Geiler-Samerotte et al., 2011). It is interesting to postulate that hydrophobic residues that promote aggregation can reduce the portion of properly folded, functional protein. Such futile protein synthesis might need to be compensated for by further costly production in order to reach the needed functional level of a certain protein.
We further demonstrate that there are sufficient degrees of freedom for a gene to evolve a cost-reducing architecture, even when its amino acid sequence is constant. Hence, our study suggests design elements that could be utilized both for better heterologous gene expression and by natural selection for the optimization of natural genes.
As such, our observations are also relevant to biotechnology and synthetic biology. Many times in such non-natural systems, there is a need to express a foreign gene, whose expression could deprive resources from the hosting cell. Our results allow the design of an optimized nucleotide sequence version for heterologous expression that minimizes the cost of production and, by that, reduces the burden on the cell while not compromising expression level.
EXPERIMENTAL PROCEDURES
See Supplemental Experimental Procedures for full description.
Library Architecture
The synthetic library was provided to us by Goodman et al. (2013) and is fully described there. In short, each variant in the library harbors a unique 5′ gene architecture that is composed of a promoter, a ribosome binding site, and an N′ terminus amino acid fusion of 11 amino acids followed by a super-folder GFP (sfGFP) gene. The library as a whole includes two promoters with either high or low transcription rates; three synthetic RBSs with strong, medium, or low translation initiation rates, as well as 137 different genomic RBSs that were defined as the 20 bp upstream to the ORF of 137 E. coli genes; and, finally, 137 coding sequences (CDSs) consisting of the first 11 amino acids from the same genes. Each CDS appears in the library in 13 different nucleotide sequences representing alternative synonymous forms. All combinations amounted in 14,234 distinct library variants.
Competition Assay
Competition experiment was carried out by serial dilution. The library was grown on 1.2 mL of Lysogeny broth (LB) and 50 μg/mL kanamycin at 30°C, the exact same conditions that were used in Goodman et al. (2013) to measure GFP expression level. We grew six parallel, independent lineages, and each was diluted daily by a factor of 1:120 into fresh media (resulting in ~6.9 generations per dilution). This procedure was repeated for 12 days, and samples were taken from each lineage every 4 days (~27 generations), mixed with glycerol, and kept at −80°C.
Fitness and Fitness Residual Estimations
Fitness effect is derived from the following equation:
where f is the variant frequency, t is the generation number, and s is the fitness effect.
To extract fitness effect, we took two independent approaches. First, we took the logarithm of the ratio between the frequency of a variant at a certain time point and its frequency at time zero. We then divided this value by the number of generations. This calculation was performed for both generation ~84 and generation ~56. See Supplemental Experimental Procedures for description of fitness calculation based on maximum likelihood. The two fitness-estimation methods were highly correlated (Figures S5A and S5B; r = 0.99, p < 10−200) and resulted in the same conclusions throughout our analyses.
We then defined “fitness residual” of a variant as the difference between the observed fitness by FitSeq and the fitness predicted by a linear model given the variant’s GFP expression level (see Supplemental Experimental Procedures for further details).
Model for Estimating Translation Velocity Based on Anti-Shine Dalgarno Affinity
The Shine-Dalgarno affinity was calculated identically to Li et al. (2012). In short, for each position, we calculated the affinity of 8–11 bp upstream of that position (the distance between the ribosome A site and the aSD site) to the anti-Shine Dalgarno motif. The free energy of interaction between the aSD motif and the mRNA sequence (ΔG) was calculated for all possible 10-mer sequences for that position using the RNA annealing function from the ViennaRNA package algorithm (Lorenz et al., 2011), and the highest affinity (lowest energy) score was used. We calculated the affinity for all positions for which the annealing with the aSD motif resides in the 11 amino acid fusion (positions 19–33) and then transformed all affinities of a given variable sequence to estimated ribosomal velocity, as follows.
We converted the ΔG estimates into the equilibrium constant of the interaction, K, which represents the equilibrium between association (kf) and dissociation (kb). The elongation velocity (v) as the ribosome moves from current site n to the n + 1 site is given by the harmonic mean of the dissociation reaction of site n and the association reaction of site n + 1:
| Equation 1 |
| Equation 2 |
We further assume that the association reaction rate is not dependent on the sequence, therefore, for every n, kfn = kf, and that differences in affinity thus only reflect differences in dissociation constant displayed by various sequences. We then get a term for the ribosomal velocity at a specific position by the anti-Shine Dalgarno affinity:
| Equation 3 |
To calculate the average ribosomal velocity across the entire N terminus fusion sequence of each library variant, we calculated the harmonic mean of the velocity values for all positions. See Supplemental Experimental Procedures for full description.
Supplementary Material
Highlights.
Microorganisms can minimize expression cost with diverse molecular means
Some design elements can produce more unneeded proteins but maintain high fitness
Such elements optimize use of production machineries and utilize cheap materials
Natural highly expressed genes evolved more forcefully to lower expression costs
Acknowledgments
We are grateful to Daniel B. Goodman and George M. Church for providing us with the E. coli library and to Daniel B. Goodman for helpful discussions along the way. We are thankful to Tamir Tuller and Gilad Shaham for fruitful discussions about the MTDR concept. We thank Naama Barkai, Maya Schuldiner, Moshe Oren, Tal Galili, Tslil Ast, Avihu Yona, and Hila Gingold for helpful discussions. Our gratitude goes to Shlomit Gilad and Sima Benjamin from the Nancy & Stephen Grand Israel National Center for Personalized Medicine (G-INCPM) for assistance with high-throughput data. I.F. thanks the Azrieli Foundation for the Azrieli Ph.D. Fellowship award. S.F.L. is supported by The Louis and Beatrice Laufer Center and NIH grants R01 HG008354 and U01 HL127522. This study was supported by the Minerva Foundation, which funded the “Minerva Center for Live Emulation of Evolution in the Lab” and a Minerva grant to Y.P.
Footnotes
ACCESSION NUMBERS
The accession number for all sequencing data reported in this paper is SRA: SRP092267.
Supplemental Information includes Supplemental Experimental Procedures, five figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.11.007.
AUTHOR CONTRIBUTIONS
I.F., D.S., A.R., and Y.P. conceived and designed the study. I.F., D.S., A.R., F.L., L.Z., E.M., O.A., S.W., and S.F.L. acquired the data. I.F., D.S., A.R., and Y.P. analyzed and interpreted the data. I.F., D.S., and Y.P. wrote the manuscript.
References
- Akashi H, Gojobori T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc Natl Acad Sci USA. 2002;99:3695–3700. doi: 10.1073/pnas.062526999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Fraser HB. Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res. 2014;24:2011–2021. doi: 10.1101/gr.175893.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS. Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng. 1990;35:668–681. doi: 10.1002/bit.260350704. [DOI] [PubMed] [Google Scholar]
- Bienick MS, Young KW, Klesmith JR, Detwiler EE, Tomek KJ, Whitehead TA. The interrelationship between promoter strength, gene expression, and growth rate. PLoS ONE. 2014;9:e109105. doi: 10.1371/journal.pone.0109105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneski CA, Hurst LD. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 2013;11:e1001508. doi: 10.1371/journal.pbio.1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneski CA, Hurst LD. Positive charge loading at protein termini is due to membrane protein topology, not a translational ramp. Mol Biol Evol. 2014;31:70–84. doi: 10.1093/molbev/mst169. [DOI] [PubMed] [Google Scholar]
- Dana A, Tuller T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014;42:9171–9181. doi: 10.1093/nar/gku646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekel E, Alon U. Optimality and evolutionary tuning of the expression level of a protein. Nature. 2005;436:588–592. doi: 10.1038/nature03842. [DOI] [PubMed] [Google Scholar]
- Dong H, Nilsson L, Kurland CG. Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J Bacteriol. 1995;177:1497–1504. doi: 10.1128/jb.177.6.1497-1504.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Kurland CG. Growth rate dependence of transfer RNA abundance in Escherichia coli. EMBO J. 1990;9:4359–4366. doi: 10.1002/j.1460-2075.1990.tb07885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Werner-Washburne M. The genomics of yeast responses to environmental stress and starvation. Funct Integr Genomics. 2002;2:181–192. doi: 10.1007/s10142-002-0058-2. [DOI] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y. Determinants of translation efficiency and accuracy. Mol Syst Biol. 2011;7:481. doi: 10.1038/msb.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BR. Metabolic load and heterologous gene expression. Biotechnol Adv. 1995;13:247–261. doi: 10.1016/0734-9750(95)00004-a. [DOI] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016;165:1416–1427. doi: 10.1016/j.cell.2016.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman DB, Church GM, Kosuri S. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013;342:475–479. doi: 10.1126/science.1241934. [DOI] [PubMed] [Google Scholar]
- Heyer EE, Moore MJ. Redefining the translational status of 80S monosomes. Cell. 2016;164:757–769. doi: 10.1016/j.cell.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Higgs PG, Ran W. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol Biol Evol. 2008;25:2279–2291. doi: 10.1093/molbev/msn173. [DOI] [PubMed] [Google Scholar]
- Ibarra RU, Edwards JS, Palsson BO. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature. 2002;420:186–189. doi: 10.1038/nature01149. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafri M, Metzl-Raz E, Jona G, Barkai N. The cost of protein production. Cell Rep. 2016;14:22–31. doi: 10.1016/j.celrep.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr AG. Growth rate of Escherichia coli. Microbiol Rev. 1991;55:316–333. doi: 10.1128/mr.55.2.316-333.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Woolstenhulme CJ, Green R, Buskirk AR. Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 2016;14:686–694. doi: 10.1016/j.celrep.2015.12.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Yang JR, Pearson NM, Maclean C, Zhang J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012;8:e1002603. doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang C, Galen JE, Kaper JB, Chao L. Fitness cost of the green fluorescent protein in gastrointestinal bacteria. Can J Microbiol. 2003;49:531–537. doi: 10.1139/w03-072. [DOI] [PubMed] [Google Scholar]
- Salis HM. The ribosome binding site calculator. Methods Enzymol. 2011;498:19–42. doi: 10.1016/B978-0-12-385120-8.00002-4. [DOI] [PubMed] [Google Scholar]
- Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T. Interdependence of cell growth and gene expression: origins and consequences. Science. 2010;330:1099–1102. doi: 10.1126/science.1192588. [DOI] [PubMed] [Google Scholar]
- Shah P, Gilchrist MA. Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc Natl Acad Sci USA. 2011;108:10231–10236. doi: 10.1073/pnas.1016719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. Rate-limiting steps in yeast protein translation. Cell. 2013;153:1589–1601. doi: 10.1016/j.cell.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Tuohy TM, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Pan T, Cluzel P. Environmental perturbations lift the degeneracy of the genetic code to regulate protein levels in bacteria. Proc Natl Acad Sci USA. 2013;110:2419–2424. doi: 10.1073/pnas.1211077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tholstrup J, Oddershede LB, Sørensen MA. mRNA pseudo-knot structures can act as ribosomal roadblocks. Nucleic Acids Res. 2012;40:303–313. doi: 10.1093/nar/gkr686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Zur H. Multiple roles of the coding sequence 5′ end in gene expression regulation. Nucleic Acids Res. 2015;43:13–28. doi: 10.1093/nar/gku1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010a;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci USA. 2010b;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Veksler-Lublinsky I, Gazit N, Kupiec M, Ruppin E, Ziv-Ukelson M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011;12:R110. doi: 10.1186/gb-2011-12-11-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind J, Sørensen MA, Rasmussen MD, Pedersen S. Synthesis of proteins in Escherichia coli is limited by the concentration of free ribosomes. Expression from reporter genes does not always reflect functional mRNA levels. J Mol Biol. 1993;231:678–688. doi: 10.1006/jmbi.1993.1319. [DOI] [PubMed] [Google Scholar]
- Wagner A. Energy constraints on the evolution of gene expression. Mol Biol Evol. 2005;22:1365–1374. doi: 10.1093/molbev/msi126. [DOI] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, Bartel DP. Improved ribosome footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016;14:1787–1799. doi: 10.1016/j.celrep.2016.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, Bustamante C, Tinoco I. Following translation by single ribosomes one codon at a time. Nature. 2008;452:598–603. doi: 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona AH, Bloom-Ackermann Z, Frumkin I, Hanson-Smith V, Charpak-Amikam Y, Feng Q, Boeke JD, Dahan O, Pilpel Y. tRNA genes rapidly change in evolution to meet novel translational demands. eLife. 2013;2:e01339. doi: 10.7554/eLife.01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.