

Summary
Genetic improvement of fibre quality is one of the main breeding goals for the upland cotton, Gossypium hirsutum, but there are difficulties with precise selection of traits. Therefore, it is important to improve the understanding of the genetic basis of phenotypic variation. In this study, we conducted phenotyping and genetic variation analyses of 719 diverse accessions of upland cotton based on multiple environment tests and a recently developed Cotton 63K Illumina Infinium SNP array and performed a genome‐wide association study (GWAS) of fibre quality traits. A total of 10 511 polymorphic SNPs distributed in 26 chromosomes were screened across the cotton germplasms, and forty‐six significant SNPs associated with five fibre quality traits were detected. These significant SNPs were scattered over 15 chromosomes and were involved in 612 unique candidate genes, many related to polysaccharide biosynthesis, signal transduction and protein translocation. Two major haplotypes for fibre length and strength were identified on chromosomes Dt11 and At07. Furthermore, by combining GWAS and transcriptome analysis, we identified 163 and 120 fibre developmental genes related to length and strength, respectively, of which a number of novel genes and 19 promising genes were screened. These results provide new insight into the genetic basis of fibre quality in G. hirsutum and provide candidate SNPs and genes to accelerate the improvement of upland cotton.
Keywords: Gossypium hirsutum, Genome‐wide association study, fibre quality, single nucleotide polymorphism, RNA‐sequencing
Introduction
Cotton is one of the most important natural textile fibre crops and is extensively planted throughout the world (Chen et al., 2007; Wendel, 1989). The cotton genus, Gossypium, consists of approximately 46 diploid species and six allotetraploid species (Grover et al., 2014; Wendel and Cronn, 2003). Of these, Gossypium hirsutum, also known as ‘upland cotton’, is the most widely cultivated species, constituting more than 95% of the world's cotton production. Upland cotton is characterized by wide adaptability and high production. However, its low‐quality fibre requires improvement to meet human demands and progress in spinning technology.
The fibre quality of cotton is a complex quantitative trait that is controlled by multiple genes and is susceptible to environmental conditions (Paterson et al., 2003). The major fibre quality characteristics include length, strength, fineness (also called micronaire), uniformity and elongation, among which length, strength and fineness are most important for spinning yarn quality. Previous studies have dissected the genetic architecture of fibre quality through traditional quantitative trait locus (QTL) linkage mapping using biparental populations (Jamshed et al., 2016; Paterson et al., 2003; Shen et al., 2005; Yang et al., 2014b; Zhang et al., 2011). Many QTLs of fibre quality traits have been identified (Lacape et al., 2010; Said et al., 2013), providing significant insight into fibre genetics. However, most of these QTLs obtained from interspecific populations are not directly applicable to upland cotton improvement because they are localized in very large genetic regions and are often not stable across populations (Islam et al., 2016), and the molecular mechanisms underlying most of these QTLs are unknown.
Genome‐wide association study (GWAS) based on linkage disequilibrium (LD) can effectively associate genotypes with phenotypes in natural populations and simultaneously detect many natural allelic variations and candidate genes in a single study, in contrast to QTL linkage mapping (Gupta et al., 2005; Huang and Han, 2014; Nuzhdin et al., 2012). Because of its advantages, including high resolution, cost efficiency and nonessential pedigrees, GWAS has been applied to many important and complicated phenotypes in crops such as rice (Huang et al., 2010; Yano et al., 2016; Zhao et al., 2011), maize (Li et al., 2013; Thornsberry et al., 2001; Yang et al., 2014a), soybean (Han et al., 2015; Zhou et al., 2015b), sorghum (Mace et al., 2013; Morris et al., 2013) and millet (Jia et al., 2013). The identification and characterization of the genes associated with important agronomic traits is essential for understanding the genetic basis of phenotypic variation and for promoting crop improvement.
The first commercial high‐density CottonSNP63K array was recently developed; it provides a new resource for the genetic dissection of agronomically and economically important traits in cotton improvement (Hulse‐Kemp et al., 2015). Additionally, the release of the upland cotton TM‐1 genome sequence (Li et al., 2015; Zhang et al., 2015b) and the development of high‐throughput single nucleotide polymorphism (SNP) assays (Zhao et al., 2011) have enabled GWAS to explore the genetic basis of complex cotton traits. However, there are few reports of GWA mapping using large natural populations based on SNP markers in cotton.
Thus, in this study, to identify the natural allelic variation in the cotton genome and candidate genes significantly associated with fibre quality, we performed a GWAS of fibre quality using 719 upland cotton accessions and the high‐density CottonSNP63K array based on phenotypic tests in eight environments. The results further our understanding of the mechanisms underpinning fibre quality, provide molecular markers for high‐quality cotton breeding and may provide a reference for genetic dissection of other complex quantitative traits in cotton.
Results
Phenotypic variation in fibre quality traits
To evaluate the phenotypic variation in fibre quality traits in the association population, we analysed five traits in eight environments during two years. Fibre length (FL), fibre strength (FS), fibre micronaire (FM), fibre uniformity (FU) and fibre elongation (FE) varied from 22.07 to 35.56 mm, 22.69 to 36.80 cN/tex, 3.14 to 6.34, 77.48 to 89.00 % and 3.55 to 8.65 %, respectively (Table 1). The combined variance analysis based on phenotype traits in the eight environments showed significant differences among genotypes, environments and a genotype and environment interaction (Table S2). Correlation analysis of the traits showed that FL was significantly positively correlated with FS and FU, and a positive correlation was also found between FS and FU. FM was significantly negatively correlated with FL and FS (Figure S1).
Table 1.
Phenotypic variation for five fibre quality traits in the association population
| Trait | Environment | Mean | SD | Max | Min | CV(%) |
|---|---|---|---|---|---|---|
| FL(mm) | 14BD | 29.27 | 1.73 | 33.68 | 22.07 | 5.92 |
| 14HJ | 28.76 | 1.43 | 34.78 | 23.91 | 4.97 | |
| 14XJ | 30.13 | 1.41 | 35.20 | 22.73 | 4.69 | |
| 14QX | 29.52 | 1.45 | 35.56 | 24.50 | 4.91 | |
| 14HN | 29.25 | 1.25 | 33.60 | 24.50 | 4.27 | |
| 15XJ | 30.02 | 1.29 | 35.14 | 22.74 | 4.28 | |
| 15QX | 26.69 | 1.24 | 31.08 | 23.75 | 4.66 | |
| 15HN | 29.06 | 1.43 | 34.80 | 23.17 | 4.91 | |
| FS(cN/tex) | 14BD | 29.40 | 2.45 | 35.80 | 23.30 | 8.32 |
| 14HJ | 29.10 | 2.21 | 35.00 | 23.30 | 7.59 | |
| 14XJ | 30.82 | 2.13 | 36.30 | 25.20 | 6.92 | |
| 14QX | 30.09 | 2.14 | 36.80 | 24.70 | 7.10 | |
| 14HN | 28.36 | 2.11 | 36.30 | 23.10 | 7.44 | |
| 15XJ | 31.03 | 1.84 | 36.61 | 25.85 | 5.94 | |
| 15QX | 28.43 | 1.99 | 34.16 | 23.24 | 7.01 | |
| 15HN | 25.63 | 1.78 | 32.05 | 22.69 | 6.95 | |
| FM | 14BD | 5.10 | 0.46 | 6.34 | 3.50 | 8.97 |
| 14HJ | 5.21 | 0.40 | 6.21 | 3.67 | 7.72 | |
| 14XJ | 5.07 | 0.42 | 6.20 | 3.14 | 8.24 | |
| 14QX | 5.00 | 0.39 | 6.03 | 3.52 | 7.88 | |
| 14HN | 4.50 | 0.47 | 5.78 | 3.23 | 10.47 | |
| 15XJ | 4.80 | 0.34 | 5.65 | 3.93 | 7.04 | |
| 15QX | 5.36 | 0.28 | 6.15 | 4.10 | 5.31 | |
| 15HN | 4.58 | 0.43 | 6.16 | 3.16 | 9.31 | |
| FU (%) | 14BD | 84.99 | 1.25 | 88.00 | 80.60 | 1.47 |
| 14HJ | 85.09 | 1.25 | 89.00 | 80.40 | 1.46 | |
| 14XJ | 85.12 | 1.10 | 88.10 | 80.80 | 1.30 | |
| 14QX | 84.72 | 1.18 | 88.30 | 78.90 | 1.40 | |
| 14HN | 85.28 | 0.98 | 88.30 | 82.25 | 1.15 | |
| 15XJ | 85.34 | 0.99 | 88.29 | 81.31 | 1.16 | |
| 15QX | 82.76 | 1.21 | 86.01 | 77.48 | 1.46 | |
| 15HN | 85.72 | 0.96 | 88.40 | 82.75 | 1.12 | |
| FE (%) | 14BD | 6.75 | 0.07 | 7.00 | 6.50 | 1.05 |
| 14HJ | 6.75 | 0.06 | 6.90 | 6.50 | 0.94 | |
| 14XJ | 6.79 | 0.06 | 7.00 | 6.60 | 0.83 | |
| 14QX | 6.78 | 0.06 | 7.00 | 6.50 | 0.93 | |
| 14HN | 5.91 | 0.88 | 8.65 | 3.55 | 14.89 | |
| 15XJ | 7.19 | 0.24 | 8.06 | 6.47 | 3.29 | |
| 15QX | 6.35 | 0.26 | 7.23 | 5.72 | 4.07 | |
| 15HN | 6.66 | 0.24 | 8.25 | 5.97 | 3.65 |
FL, fibre length; FS, fibre strength; FM, fibre micronaire; FU, fibre uniformity; FE, fibre elongation; SD, standard deviation, which was calculated based on the measured values of the fibre traits from two replicates in 2014 and three replicates in 2015; CV, coefficient of variation.
Genetic variation based on SNPs
The genotypes of 719 accessions were examined using the Cotton 63K Illumina Infinium SNP array. All the SNP data were analysed using Illumina GenomeStudio software. First, SNPs without allele polymorphism were eliminated. Low‐quality SNP loci (call rate < 85% and minor allele frequency < 0.05) were also deleted. A final set of 10 511 high‐quality SNPs was screened and used for genetic variation and GWA analysis (Figure 1a; Table 2). These SNP markers were not evenly distributed across the entire genome, with 3923 and 6588 SNPs in the At and Dt subgenomes, respectively. Chromosome Dt08 had the maximum number of SNPs (844), and At04 had the minimum (97) (Figure 1a; Table 2). The polymorphism information content (PIC) values ranged from 0.208 to 0.312 among chromosomes, and the mean PIC of the At and Dt subgenomes was 0.287 and 0.283, respectively (Table 2).
Figure 1.
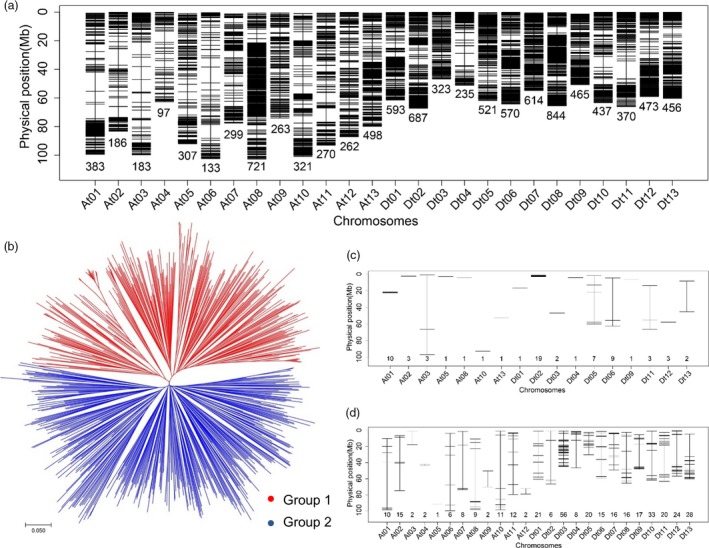
Genetic divergence of upland cotton germplasms. (a) Genome‐wide SNP density in the entire association mapping panel. Dark and white horizontal bars show genomic regions that are rich and poor in SNPs, respectively. (b) Neighbour‐joining tree of all cotton accessions in this study. Accessions in the NJ tree are represented by different colours: Group 1 (red) and Group 2 (blue). (c) Distribution of SNPs that only showed polymorphism in Group 1. (d) Distribution of SNPs that only showed polymorphism in Group 2. The number of SNP markers anchored to different chromosomes of the G. hirsutum genome is given on the horizontal axis.
Table 2.
The summary of the number of polymorphic SNPs mapped in 26 chromosomes of Gossypium hirsutum
| Chr. | No. of SNPs | Chr. Size (Mb) | Density of SNP (kb/SNP) | PIC |
|---|---|---|---|---|
| At01 | 383 | 99.9 | 260.8 | 0.310 |
| At02 | 186 | 83.5 | 448.7 | 0.288 |
| At03 | 183 | 100.3 | 547.9 | 0.294 |
| At04 | 97 | 62.9 | 648.6 | 0.312 |
| At05 | 307 | 92.1 | 299.8 | 0.307 |
| At06 | 133 | 103.2 | 776.2 | 0.277 |
| At07 | 299 | 78.3 | 261.7 | 0.300 |
| At08 | 721 | 103.7 | 143.8 | 0.208 |
| At09 | 263 | 75.0 | 285.2 | 0.308 |
| At10 | 321 | 100.9 | 314.2 | 0.272 |
| At11 | 270 | 93.3 | 345.6 | 0.246 |
| At12 | 262 | 87.5 | 333.9 | 0.296 |
| At13 | 498 | 80.0 | 160.6 | 0.308 |
| Dt01 | 593 | 61.5 | 103.6 | 0.278 |
| Dt02 | 687 | 67.3 | 97.9 | 0.311 |
| Dt03 | 323 | 46.7 | 144.6 | 0.241 |
| Dt04 | 235 | 51.5 | 218.9 | 0.283 |
| Dt05 | 521 | 61.9 | 118.9 | 0.299 |
| Dt06 | 570 | 64.3 | 112.8 | 0.247 |
| Dt07 | 614 | 55.3 | 90.1 | 0.294 |
| Dt08 | 844 | 61.9 | 73.4 | 0.277 |
| Dt09 | 465 | 51.0 | 109.7 | 0.293 |
| Dt10 | 437 | 63.4 | 145.0 | 0.295 |
| Dt11 | 370 | 66.1 | 178.6 | 0.282 |
| Dt12 | 473 | 59.1 | 125.0 | 0.293 |
| Dt13 | 456 | 60.5 | 132.7 | 0.285 |
Chr., chromosome; PIC, polymorphism information content
To understand the genetic diversity of our association panel, molecular phylogenetic analysis among these accessions was conducted based on the genetic distances of these SNPs. The neighbour‐joining (NJ) tree results showed two divergent groups (Figure 1b), designated G1 and G2, with 323 and 396 accessions, respectively (Table S1). We then compared the differences in genome‐wide SNPs between the two groups. There were 360 unique SNPs across the accessions of G2 and only 68 unique SNPs in G1 (Figure 1c, d). These results suggested the two groups had a degree of genetic differentiation at the molecular level (Coart et al., 2002).
Population structure and linkage disequilibrium
We investigated the population structure of our panel using STRUCTURE software. The LnP(K) values continuously increased with K from 1 to 10 (Figure 2a); however, the delta K value reached a sharp peak at K = 2 (Figure 2b). Therefore, this association population was clustered into two subpopulations (Figure 2c). Similarly, PCA showed two clusters for the population, despite some accessions overlapping in the two clusters (Figure 2d). Combined with the results of the normal distribution of all investigated traits (Figure S1), the varietal population in this study was considered not highly structured and could be used for GWAS as in other previously reported crops (Yano et al., 2016). The extent of LD provides a moderate resolution for genome‐wide identification for gene discovery (Flint‐Garcia et al., 2003). The LD decay of our population was approximately 0.82 Mb, where the r 2 drops to half the maximum value (Figure 2e). The overall LD decay distance in the At subgenome was significantly higher than that in the Dt subgenome, 1.30 and 0.33 Mb, respectively (Figure 2e). Considering the LD decay distances and in comparison with other crops, such as rice and maize, with LD of 100 kb‐1 Mb and 1‐100 kb, respectively (Gore et al., 2009; Nordborg et al., 2002; Remington et al., 2001; Zhao et al., 2011), we assumed 200 kb as the region of SNP‐associated candidate genes for fibre traits.
Figure 2.
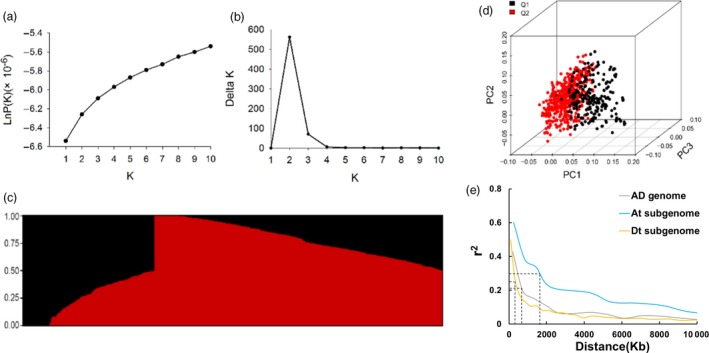
Analysis of the population structure of 719 upland cotton accessions. (a) Estimated LnP(K) of possible clusters (K) from 1 to 10. (b) Delta K based on the rate of change of LnP(K) between successive K. (c) Population structure based on STRUCTURE when K = 2. (d) Principal component analysis showing the population structure in the diversity panel. Two subpopulations are designated Q1 (black) and Q2 (red). (e) Genome‐wide average LD decay estimated in the AD genome (grey), At subgenome (blue) and Dt subgenome (yellow).
Genome‐wide association mapping
To explore the genetic factors associated with the fibre quality, we conducted a GWAS, which took into account the population structure and familial relatedness (Yu et al., 2006) using all traits analysed in 2014 and 2015. A total of 46 significantly associated SNPs were detected in at least one environment (Table 3). These SNPs were located on chromosomes At01, At07, At08, At10, At12, At13, Dt01, Dt03, Dt04, Dt05, Dt06, Dt07, Dt10, Dt11 and Dt13. In addition, some were detected repeatedly across multiple environments, which was similar to the results for other plants, showing more stable SNPs linked with traits (Xu et al., 2016; Zhao et al., 2011).
Table 3.
The summary of SNPs significantly associated with fibre quality traits
| Traits | SNPs | Chr. | Site | Allele | MAF | ‐Log10(P) | R2(%) | Environment |
|---|---|---|---|---|---|---|---|---|
| FL | i39753Gh | At07 | 72067994 | G/A | 0.09(G) | 4.78 | 3.32 | E4 |
| i02033Gh | At07 | 72193182 | G/T | 0.12(G) | 4.05–4.15 | 2.70–2.73 | E4,E5 | |
| i02034Gh | At07 | 72198802 | A/G | 0.07(A) | 5.04 | 3.28 | E4 | |
| i02035Gh | At07 | 72200974 | C/T | 0.07(C) | 5.06 | 3.30 | E4 | |
| i02037Gh | At07 | 72204773 | C/T | 0.07(C) | 5.04 | 3.28 | E4 | |
| i49171Gh | At07 | 72213592 | T/C | 0.11(T) | 4.09 | 2.66 | E4 | |
| i37604Gh | At07 | 72249786 | C/T | 0.11(C) | 4.09 | 2.66 | E4 | |
| i12279Gh | At10 | 98329643 | A/G | 0.38(A) | 4.57 | 2.98 | E3 | |
| i24710Gh | At10 | 98399227 | A/G | 0.30(A) | 4.55 | 2.99 | E3 | |
| i23677Gh | At10 | 98423067 | T/C | 0.30(T) | 4.55 | 2.99 | E3 | |
| i03207Gh | Dt03 | 3409179 | G/A | 0.05(G) | 3.98 | 2.60 | E8 | |
| i09441Gh | Dt05 | 12903672 | A/G | 0.34(A) | 3.97–4.33 | 2.58–2.83 | E2,E4,E8 | |
| i34936Gh | Dt05 | 13762535 | T/G | 0.31(T) | 4.69 | 3.07 | E5 | |
| i49170Gh | Dt06 | 174910 | G/A | 0.37(G) | 4.66 | 3.03 | E4 | |
| i44154Gh | Dt07 | 42826098 | C/A | 0.13(C) | 3.99 | 2.59 | E4 | |
| i44994Gh | Dt07 | 42837391 | G/A | 0.13(G) | 3.99 | 2.59 | E4 | |
| i07326Gh | Dt11 | 23906867 | C/T | 0.27(C) | 4.41 | 2.91 | E1 | |
| i22531Gh | Dt11 | 23959318 | A/G | 0.27(A) | 4.41 | 2.91 | E1 | |
| i07327Gh | Dt11 | 24008823 | T/C | 0.27(T) | 4.41 | 2.91 | E1 | |
| i60962Gt | Dt11 | 24030081 | A/G | 0.18(A) | 4.32–7.26 | 2.12–4.78 | E1,E2,E4‐E6,E8 | |
| FS | i65738Gm | At01 | 96310264 | A/C | 0.07(A) | 4.11 | 2.69 | E1 |
| i18340Gh | At07 | 71993462 | C/A | 0.12(C) | 5.17 | 3.38 | E4 | |
| i44206Gh | At07 | 72008085 | G/A | 0.12(G) | 5.17 | 3.38 | E4 | |
| i39753Gh | At07 | 72067994 | G/A | 0.09(G) | 4.27–7.56 | 3.22–5.38 | E1‐E4 | |
| i02033Gh | At07 | 72193182 | G/T | 0.12(G) | 5.29 | 3.53 | E4 | |
| i02034Gh | At07 | 72198802 | A/G | 0.07(A) | 4.04–9.16 | 2.81–6.07 | E1‐E4 | |
| i02035Gh | At07 | 72200974 | C/T | 0.07(C) | 4.03–9.15 | 2.80–6.07 | E1‐E4 | |
| i02037Gh | At07 | 72204773 | C/T | 0.07(C) | 4.04–9.16 | 2.81–6.07 | E1‐E4 | |
| i49171Gh | At07 | 72213592 | T/C | 0.11(T) | 5.32 | 3.48 | E4 | |
| i37604Gh | At07 | 72249786 | C/T | 0.11(C) | 5.32 | 3.48 | E4 | |
| i30934Gh | At13 | 5168143 | T/C | 0.30(T) | 4.59 | 2.97 | E7 | |
| i38606Gh | Dt06 | 60561982 | T/C | 0.08(T) | 4.16 | 2.72 | E5 | |
| i22089Gh | Dt06 | 60565551 | A/G | 0.08(A) | 4.16 | 2.72 | E5 | |
| i20058Gh | Dt10 | 23109967 | T/C | 0.10(T) | 4.32 | 2.79 | E7 | |
| i60962Gt | Dt11 | 24030081 | A/G | 0.18(A) | 4.50 | 2.94 | E8 | |
| i41872Gh | Dt13 | 52825264 | G/T | 0.27(G) | 4.26 | 2.96 | E2 | |
| i09756Gh | Dt13 | 52919060 | T/C | 0.34(T) | 4.18 | 2.73 | E4 | |
| i20350Gh | Dt13 | 52934633 | C/T | 0.35(C) | 4.32 | 2.93 | E6 | |
| FM | i49257Gh | At13 | 13057186 | G/A | 0.45(G) | 4.02 | 2.57 | E7 |
| i45804Gh | At13 | 13494588 | A/G | 0.45(A) | 4.38 | 2.81 | E7 | |
| i25107Gh | Dt05 | 60697515 | C/T | 0.49(C) | 4.33 | 2.90 | E5 | |
| i46147Gh | Dt06 | 2045744 | C/T | 0.47(C) | 4.61 | 2.96 | E7 | |
| FU | i04566Gh | At08 | 97378358 | G/A | 0.47(G) | 4.90 | 3.19 | E3 |
| i15200Gh | At08 | 97451057 | T/C | 0.49(T) | 4.39 | 2.85 | E3 | |
| i04572Gh | At08 | 97452659 | G/A | 0.49(G) | 4.27 | 2.77 | E3 | |
| i54149 Gb | At08 | 97482110 | G/A | 0.49(G) | 4.48 | 2.91 | E3 | |
| FE | i24710Gh | At10 | 98399227 | A/G | 0.30(A) | 4.18 | 2.67 | E3 |
| i23677Gh | At10 | 98423067 | T/C | 0.30(T) | 4.18 | 2.67 | E3 | |
| i27874Gh | At12 | 2806482 | C/T | 0.20(C) | 4.28 | 2.80 | E5 | |
| i24077Gh | At12 | 2849310 | G/T | 0.20(G) | 4.33 | 2.83 | E5 | |
| i16244Gh | At12 | 2885883 | C/T | 0.21(C) | 4.09 | 2.67 | E5 | |
| i24987Gh | Dt01 | 39191073 | A/G | 0.05(A) | 5.04 | 3.28 | E8 | |
| i21001Gh | Dt01 | 39365919 | A/G | 0.05(A) | 5.04 | 3.28 | E8 | |
| i51597 Gb | Dt03 | 425957 | C/T | 0.37(C) | 4.12 | 2.59 | E6 | |
| i12839Gh | Dt04 | 47872770 | A/C | 0.32(A) | 8.23 | 5.45 | E5 | |
| i12840Gh | Dt04 | 47872954 | T/G | 0.32(T) | 7.83 | 5.18 | E5 | |
| i09441Gh | Dt05 | 12903672 | A/G | 0.34(A) | 4.16 | 2.68 | E2 |
FL, fibre length; FS, fibre strength; FM, fibre micronaire; FU, fibre uniformity; FE, fibre elongation; ‐Log10(P) value indicates the significance levels and R2 (%) indicates the percentage of phenotypic variation explained by each SNP; Chr., chromosome; MAF, minor allele frequency.
For FL, 20 significant SNPs were identified on chromosomes At07, At10, Dt03, Dt05, Dt06, Dt07 and Dt11 (Table 3; Figure S2), explaining approximately 57.72 % of the total phenotypic variation (Table 3). Seven loci in At07 and four loci in Dt11 were close in their respective chromosomes. Moreover, the locus i60962Gt in Dt11 was detected across six environments (Table 3).
For FS, 18 significant SNPs on chromosomes At01, At07, At13, Dt06, Dt10, Dt11 and Dt13 were detected (Table 3; Figure S3), contributing 2.69 to 6.07 % of the total phenotypic variation (Table 3). Among these SNPs, four loci were detected repeatedly in four environments (Table 3; Figure S4). Seven SNPs in At07 were also obtained from loci related to FL (Table 3).
For the other three traits, FM, FU and FE, 4, 4 and 11 loci were detected, respectively (Table 3). There were four SNPs located in three chromosomes significantly associated with FM (Table 3; Figure S4), and all of four loci for FU were located close together in At08 (Table 3; Figure S5). The 11 SNPs for FE on six chromosomes contributed 2.67 to 5.45 % of the phenotypic variation, but three (i24710Gh, i23677Gh and i09441Gh) were also present among significant SNPs associated with FL (Table 3; Figure S6).
Identification of candidate genes associated with SNPs
We confirmed potential candidate genes near 46 significant SNP loci based on genes annotated in the G. hirsutum TM‐1 genome (Zhang et al., 2015b). Within 200 kb of significant SNPs, a total of 612 candidate genes were identified (Table S3). We scrutinized the distribution of these candidate genes among each chromosome according to the distance to the nearest associated SNP (Figure 3a; Table S3). The results showed that genes detected in the Dt subgenome overall doubled those in the At subgenome (Figure 3a). The genes on Dt03, Dt05 and Dt06 increased as the distance increased, and genes on At07, At10 and Dt11 showed no obvious change (Figure 3a).
Figure 3.
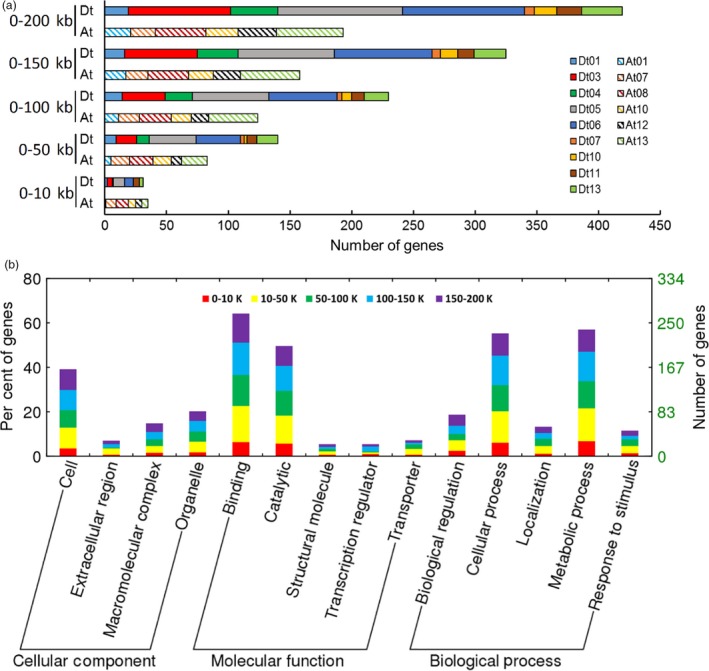
Chromosome distribution and gene ontology (GO) analysis of 612 candidate genes in five consecutive intervals and their distance to the nearest associated SNP. (a) Gene numbers in different chromosome of the At subgenome and Dt subgenome. (b) Functional classification of all genes in three levels: cellular component, molecular function and biological process. The five colours represent five different intervals.
Gene ontology (GO) analysis indicated that the functions of most genes were binding and catalytic reactions, regardless of the intervals (Figure 3b). We also conducted KEGG pathway enrichment of all candidate genes, and found 284 genes enriched in 72 pathways, with approximately 40 genes involved in important fibre development pathways (Table S4). The top three concrete pathways containing more than ten genes were plant hormone signal transduction, carbon metabolism and ribosome pathways. For the nucleotide sugar metabolism and starch and sucrose metabolism pathways clearly related to fibre development, two genes, Gh_A13G0573 and Gh_D07G1799, code for galacturonosyltransferase (GAUT), and Gh_A07G1759 and Gh_D06G1953 code for ADP glucose pyrophosphorylase and UDP‐arabinose 4‐epimerase, respectively (Tables S3, S4). We analyse these genes and their related transcriptome data below.
Analysis of major SNP loci relevant to fibre length and strength
We analysed major SNP loci relevant to the two critical quality traits, fibre length and strength, that breeders most often consider in selection. For SNPs associated with FL, a significant peak appeared in chromosome Dt11 (Figure 4a, b), and there were 20 candidate genes associated with the significant SNPs (Figure 4b). Haplotype analysis showed a high level of LD between the associated SNPs in Dt11 (Figure 4c). The four SNPs resulted in four haplotypes in our association panel (Figure 4d). The average FL of Hap4 was 29.24 mm, greater than those of the other three (Figure 4e). Based on the polymorphism of the SNP markers, each SNP locus of Hap4 could be classified into three genotypes (Figure S7). The genotypes of the four SNP loci containing in Hap4 displayed a higher average FL than the other nonincluded genotypes (Figure S7).
Figure 4.
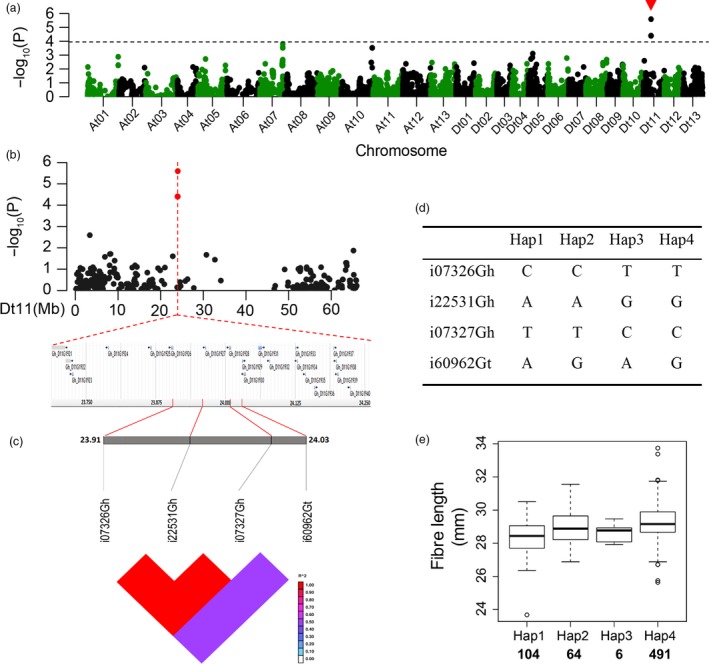
GWAS results for fibre length and analysis of the peak on chromosome Dt11. (a) Manhattan plots for fibre length. The dashed line represents the significance threshold (P < 10−3.97). The arrowhead indicates the position of the strong peak investigated in this study. (b) Manhattan plot (top) and genes surrounding the peak (bottom) on chromosome Dt11. (c) Genomic location of four SNP loci and LD based on pairwise R2 values between the SNPs estimated in Dt11. The R2 values are indicated using the colour bar. (d) Haplotypes observed in 719 accessions using the four SNPs. (e) Phenotypic differences of fibre length among four haplotypes.
Seven significant SNPs on chromosome At07 were simultaneously analysed for fibre length and strength across multiple environments (Table 3; Figure 5a, b). We selected seven SNPs to further investigate the allelic variation. There were also 20 candidate genes surrounding them (Figure 5b). Haplotype analysis showed a high level of LD between these SNP markers (Figure 5c). Among the 719 accessions, there were three haplotypes with the seven SNPs (Figure 5d). An overwhelming majority of the accessions belonged to Hap1, while Hap3 was composed of 47 accessions (Table S5), some of which were known high‐quality upland cotton cultivars. The average fibre length and strength of Hap3 were 29.85 mm and 30.81 cN/tex, respectively, significantly greater than those of other two (Figure 5e, f).
Figure 5.
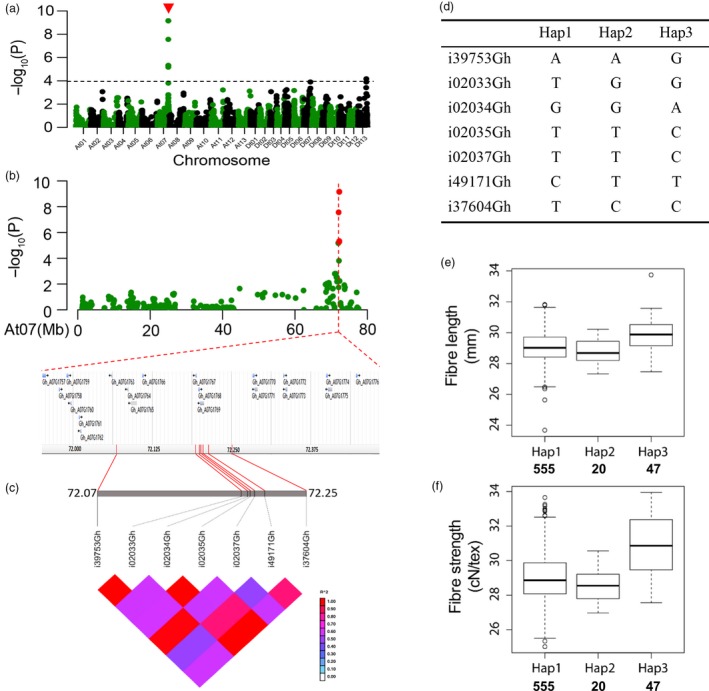
GWAS results for fibre strength and analysis of the peak on chromosome At07. (a) Manhattan plots for fibre strength. The dashed line represents the significance threshold (P < 10−3.97). The arrowhead indicates the position of the strong peak investigated in this study. (b) Manhattan plot (top) and genes surrounding the peak on chromosome At07 (bottom). (c) Genomic location of seven SNP loci and LD based on the pairwise R2 values between the SNPs estimated in At07. The R2 values are indicated using the colour bar. (d) Haplotypes observed in 719 accessions using the seven SNPs. (e‐f) Phenotypic differences of fibre length and strength among three haplotypes.
For the genetic effect of each SNP in Hap3, taking locus i39753Gh for example, the average fibre length and strength in the accessions with the GG genotype were 29.90 mm and 31.37 cN/tex, respectively (Figure S8). The three SNPs i02034Gh, i02035Gh and i02037Gh had the same effect on the phenotype as the locus i39753Gh (Figure S8). Moreover, the genotype of the other loci in Hap3 had a higher average fibre length and strength than those of the other haplotypes (Figure S8).
Verification of candidate genes by transcriptome analysis
We further validated these candidate genes using transcriptome sequencing data. We investigated the expression level (log2(1+RPKM) > 1) of all 212 and 161 genes related to FL and FS, respectively (Table S3), and found that approximately two‐thirds of genes (163 and 120) were significantly expressed during some time points (0–30 days post anthesis, DPA) of fibre development, including initiation, cell elongation and cell wall thickening (Figure S9). Among them, there were 107 and 95 genes (including 30 and 27 hypothetical proteins) not reported in cotton (Table S3). This means the genes are novel candidates for fibre development. The genes that had been reported in cotton (Deng et al., 2016; Guo et al., 2009; Huang et al., 2013; Samuel et al., 2006; Wang et al., 2004; Zhou et al., 2015a) were mainly (63 % in FL and 68 % in FS) involved in fibre development, which confirmed the accuracy of the fibre‐associated candidate genes described in this study. The differences in fibre length and strength between cultivars could be determined by gene expression in different stages of fibre development. For genes in the fibre elongation stage (0–15 DPA), those with significantly increased expression levels in at least two of the three time points (5, 10 and 15 DPA) compared with 0 DPA (predominately expressed) were screened in four upland cotton cultivars. There were nine candidate genes that matched this condition (Figure 6a). In the fibre thickening stage, spanning 20–30 DPA, we identified eight differentially expressed genes (Figure 6b).
Figure 6.
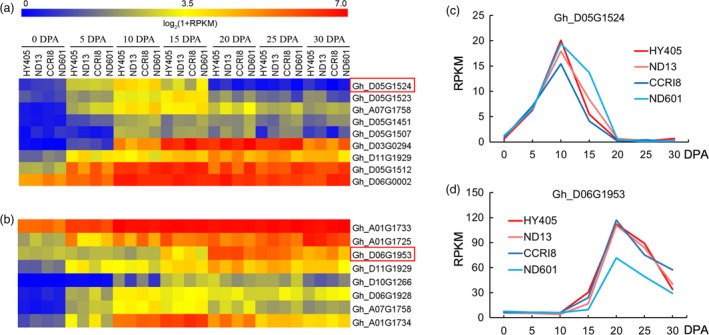
Expression pattern of the promising genes involved in fibre development in four upland cotton cultivars (HY405, ND13, CCRI8 and ND601). (a‐b) Heat map of the expression of genes related to fibre length and strength. (c‐d) Expression levels of two representative genes associated with fibre length and strength.
All 17 selected genes (nine in FL and eight in FS) were related to diverse functions (Table 4) and were classified into three expression patterns (Figure 6). First, some genes, for example Gh_D05G1524, encoding a lipid transfer protein, showed steeply increasing expression from 0 DPA of fibre elongation stage and sharply decreased after 10 DPA among four upland cotton cultivars (Figure 6c). We designated this type of gene ‘elongation pattern’. Second, another type of gene, for example Gh_D06G1953, associated with fibre length, coding for UDP‐arabinose 4‐epimerase in the nucleotide sugar metabolism pathway (Table S4) and catalysing the epimerization between UDP‐D‐Xyl and UDP‐L‐Ara in the cell wall polysaccharides (Burget et al., 2003), showed a striking increase in expression from 15 DPA, peaking at 20 DPA (Figure 6d). We designated this type of gene ‘thickening pattern’. Third, several of the 17 genes, such as Gh_D06G0002 and Gh_A01G1733, were highly expressed from 5 to 30 DPA (Figure 6a, b). We designated this type of gene ‘elong‐ckening pattern’. Other than the five known genes (Gh_D03G0294, Gh_D05G1451, Gh_D05G1507, Gh_D05G1523 and Gh_D07G1799), ten (excluding two genes found in both FL and FS) of these 17 predominately expressed genes were not previously reported in cotton (Table S3). Additionally, there were three expression patterns of fibre development for the 163 and 120 screened genes of FL and FS (Figure S9).
Table 4.
Promising genes associated with fibre length and strength identified by GWAS combined with RNA‐seq
| Trait | Gene ID | Homologue | Gene annotation | Distance (kb) |
|---|---|---|---|---|
| FL | Gh_A07G1758 | AT4G17170.1 | RAB GTPase homologue B1C | 88.4 |
| Gh_D03G0294 | AT5G65730.1 | Xyloglucan endotransglucosylase/hydrolase 6 | 129.8 | |
| Gh_D05G1451 | AT1G78580.1 | trehalose‐6‐phosphate synthase | 21.1 | |
| Gh_D05G1507 | AT1G28480.1 | Thioredoxin superfamily protein | 190.5 | |
| Gh_D05G1512 | AT2G20560.1 | DNAJ heat‐shock family protein | 132.2 | |
| Gh_D05G1523 | AT5G49760.1 | Leucine‐rich repeat protein kinase family protein | 10.6 | |
| Gh_D05G1524 | AT5G49800.1 | Polyketide cyclase/dehydrase and lipid transport superfamily protein | 1.2 | |
| Gh_D06G0002 | AT1G47830.1 | SNARE‐like superfamily protein | 167.8 | |
| Gh_D11G1929 | AT3G19150.1 | KIP‐related protein 6 | 123.2 | |
| Gh_D07G1799 | AT3G02350.1 | galacturonosyltransferase 9 | 32.4 | |
| FS | Gh_A01G1725 | AT3G11660.1 | NDR1/HIN1‐like 1 | 98.5 |
| Gh_A01G1733 | AT3G52560.1 | ubiquitin E2 variant 1D‐4 | 26.9 | |
| Gh_A01G1734 | AT5G06270.1 | unknown protein | 52.7 | |
| Gh_A07G1758 | AT4G17170.1 | RAB GTPase homologue B1C | 88.4 | |
| Gh_D06G1928 | AT5G58380.1 | CBL‐interacting protein kinase | 153.2 | |
| Gh_D06G1953 | AT1G30620.2 | UDP‐arabinose 4‐epimerase 1 | 155.0 | |
| Gh_D10G1266 | AT3G57880.1 | CaLB domain plant phosphoribosyltransferase family protein | 124.6 | |
| Gh_D11G1929 | AT3G19150.1 | KIP‐related protein 6 | 123.2 | |
| Gh_D13G1792 | AT3G04470.1 | Ankyrin repeat family protein | 24.9 |
FL, fibre length; FS, fibre strength; FM, fibre micronaire; FU, fibre uniformity; FE, fibre elongation; Distance indicates the distance to the nearest SNP.
We found two genes with significant differences in expression between high‐quality and normal‐quality cultivars (Figure 7; Table 4). Gh_D07G1799, coding for galacturonosyltransferase (GAUT), is involved in pectin biosynthesis, which is part of glycan metabolism (Sterling et al., 2006). In the KEGG analysis, we found that it was from the nucleotide sugar metabolism and sucrose metabolism pathways (Table S4). In the previous study of our team, we cloned a novel gene, GbGAUT1, from G. barbadense and proposed that GbGAUT1 plays a key role in fibre development (Chi et al., 2009). The expression level of Gh_D07G1799 showed an obvious increase in fibre elongation stage and was expressed at much higher levels in normal‐quality cultivars than in high‐quality cultivars (Figure 7a, b). Another gene Gh_D13G1792 belonged to the Arabidopsis ankyrin repeat family protein, and this protein served as a molecular chaperone that plays many important roles in plant cellular metabolism (Shen et al., 2010). It is associated with membrane‐enclosed organelles and is required for pollen tube growth in lilies (Huang et al., 2006). However, it was not previously reported in cotton (Table S3). This gene had a much higher expression level in high‐quality cultivars than in normal‐quality cultivars (Figure 7c, d).
Figure 7.
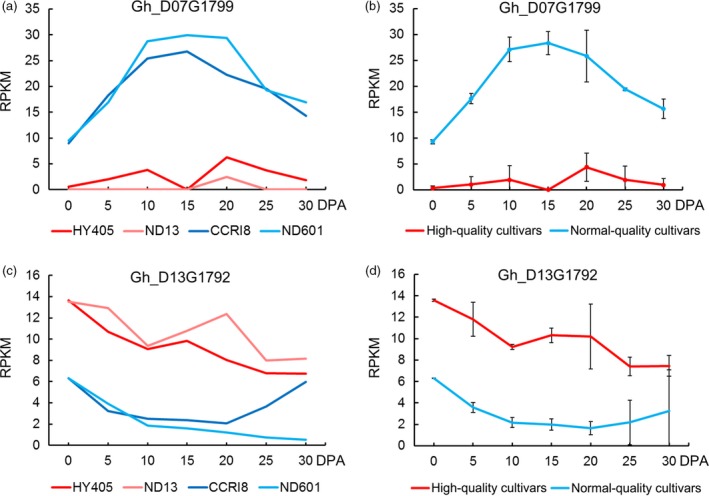
Expression levels of genes significantly differentially expressed between high‐quality cultivars (HY405 and ND13) and normal‐quality cultivars (CCRI8 and ND601). (a) Expression of the gene Gh_D07G1799, associated with fibre length, in the four cultivars. (b) Expression of the gene Gh_D07G1799 between high‐quality cultivars and normal‐quality cultivars. (c) Expression of the gene Gh_D13G1792, associated with fibre strength, in the four cultivars. (d) Expression of the gene Gh_D13G1792 between high‐quality cultivars and normal‐quality cultivars.
In addition, we verified 20 associated genes in FL and 20 genes in FS surrounding the peak SNPs in Dt11 and At07 based on expression (Figure 8a, b). Three genes (Gh_D11G1926, Gh_D11G1928 and Gh_D11G1929) in Dt11 for FL contained a significant SNP locus (Figure 4b) and had higher expression level, especially Gh_D11G1929 (Figure 8a). This gene was highly expressed at all time points of fibre development (Figure 6a, b; Figure 8a). There were seven genes not expressed during fibre development. Most of the genes (except the five genes that were not expressed) associated with the seven SNPs in At07 for FS showed high expression levels (Figure 8b). The two loci i02034Gh and i02037Gh were located inside gene Gh_A07G1768 and Gh_A07G1769, respectively (Figure 5b). Moreover, there were three SNPs near the two genes (Figure 5b) that were steadily expressed from 0 to 30 DPA (Figure 8b). These significant SNPs had an effect on the neighbouring genes. One of these genes, Gh_A07G1768, annotated as an unknown protein, may be a novel gene for fibre development (Table S3). In addition, the expression of gene Gh_A07G1758 near locus i39753Gh remained high (Figure 6a, b; Figure 8b). These data suggested that the above candidate genes play important roles in different stages of fibre development. Additionally, among the 26 significantly expressed genes in the haplotypes, there were three known and 23 not reported genes in cotton (Table S3).
Figure 8.
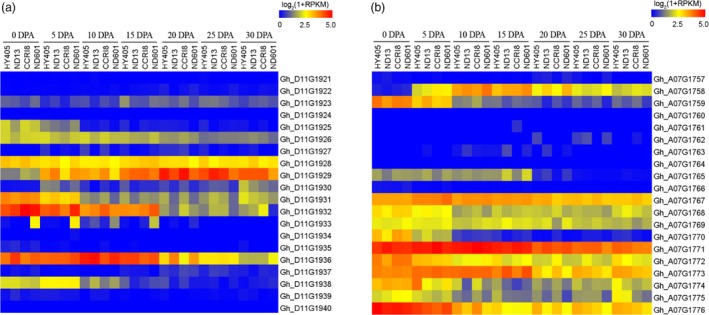
Expression pattern of candidate genes in the peak regions of Dt11 and At07. (a) Heat map of the expression of 20 genes associated with fibre length in Dt11 among four upland cotton cultivars. (b) Heat map of the expression of 20 genes in At07 among four upland cotton cultivars.
Discussion
In this study, we first performed a genome‐wide association analysis of fibre quality traits based on the CottonSNP63K array with a large number of natural accessions of G. hirsutum. This study uncovered numerous loci underlying variation in fibre quality traits and identified a set of candidate genes that could be exploited to alter fibre development to improve upland cotton cultivars.
By phenotyping the 719 cotton accessions in various environments, relatively abundant variation and significant differences among genotypes and environments were observed for the fibre quality traits (Table 1; Table S2), which showed higher diversity compared with previous reports in cotton (Jamshed et al., 2016; Shen et al., 2005; Yang et al., 2014b). The panel was classified into two groups based on molecular analyses, with 68 unique SNPs in G1 and 360 unique SNPs in G2 (Figure 1c, d); however, they were not completely consistent with geographical sources due to the wide adaptation of cotton, as in previous studies (Tyagi et al., 2014; Wang et al., 2016). The differences in polymorphic SNPs likely reflected the history of extensively interspecific crosses between diverse accessions of the two groups, resulting in introgression among germplasms. Therefore, it was difficult to fully classify the cotton accessions according to geographical source (Li et al., 2014; Xu et al., 2016). In addition, LD decay limits the mapping resolution of GWAS. Our study found that the LD decay was 0.82 Mb, which provided a more accurate reference for selecting candidate genes than previous studies using SSRs (Abdurakhmonov et al., 2008; Nie et al., 2016). Taking account of the greater continuous phenotypic variation among the tested fibre quality traits, our not highly structured association panel is applicable for performing GWAS.
Historically, QTL mapping has been utilized to identify and map causative genomic locations controlling fibre quality using biparental populations. Hundreds of QTLs have been located in intraspecific and interspecific populations of cotton (Said et al., 2015), but this method has rarely led to candidate gene isolation because it only captures limited allelic diversity existing in two parental lines. In contrast to traditional linkage mapping, GWA mapping does not require biparental crosses between individuals and provides greater precision in the localization of QTLs. This approach has been widely used for various traits in other crops, including morphological traits (Crowell et al., 2016; Li et al., 2014; Meijon et al., 2014; Porth et al., 2013), disease resistance (Dimkpa et al., 2015; Wang et al., 2012; Wen et al., 2014), environmental adaptation (Famoso et al., 2011; Nagel et al., 2015; Wu et al., 2015; Yang et al., 2014a; Zhang et al., 2015a) and flowering time (Harsh et al., 2015; Stracke et al., 2009; Xu et al., 2016). In cotton, efforts have been made to detect QTLs using GWAS with SSR markers in recent years (Abdurakhmonov et al., 2008; Cai et al., 2014; Nie et al., 2016; Zeng et al., 2009). These GWAS results, although conducted with few markers and samples, are important for fibre improvement. However, limitations of mapping resolution and genome coverage are inevitable. Therefore, in this study, the genetic factors controlling fibre quality discovered by GWA mapping with more SNP markers and cotton accessions could provide more precise insight into fibre development.
A combination analysis of our GWAS and transcriptome sequencing data revealed 163 and 120 significantly expressed candidate genes in FL and FS, respectively. Two of these genes, Gh_A07G1758 and Gh_D11G1929, were associated with fibre length and strength. Gh_A07G1758 on At07 is homologous to AT4G17170 (RAB GTPase B1C) and Gh_D11G1929 on Dt11 codes for KIP‐related protein 6 (KRP6) in Arabidopsis. The Rab family encodes key factors of vesicle‐targeting specificity proteins in A. thaliana (Rutherford and Moore, 2002). A plant Rab GTPase, RabA4b, was proposed to regulate membrane trafficking in cells (Preuss et al., 2004). Another plant‐unique RAB5 protein, ARA6, was demonstrated to have a functional link between a specific RAB acting in the endosomal trafficking pathway and a specific SNARE complex in Arabidopsis thaliana (Ebine et al., 2011). Complexes of SNARE proteins mediate intracellular membrane fusion between vesicles and organelles to facilitate transport cargo proteins in plant cells (Baker and Hughson, 2016). These results indicated that Gh_A07G1758 could play a key role in the formation of cotton fibre, as indicated by a QTL (Zhang et al., 2011) near Gh_A07G1758 found in previous research. The detailed mechanism of this gene should be further investigated. In addition, we identified a gene, Gh_D06G0002, encoding a SNARE‐like superfamily protein homologue, which was not previously reported in cotton. Gh_D11G1929 is a homologue of A. thaliana KRP6, which is a cyclin‐dependent kinase inhibitor. In Arabidopsis thaliana, KRP6 overexpression accelerated entry into mitosis, but delayed mitotic progression (Vieira et al., 2014). The regulator KRP6 partially repressed GA‐dependent activation of the cell cycle during germination (Nieuwland et al., 2016). However, analysis of this gene in cotton fibre development has not been reported.
Many genes participating in nucleotide sugar metabolism are important in fibre cells. An identified gene, Gh_D03G0294, homologous to Arabidopsis xyloglucan endotransglucosylase/hydrolase (XTH), made plant cells undergo cell expansion, acting as a cell wall‐loosening enzyme (Van Sandt et al., 2007). GhXTH1 was the predominant XTH in elongating fibres, and its expression limited cotton fibre elongation (Lee et al., 2010). Another gene, Gh_D05G1451, homologous to Arabidopsis trehalose‐6‐phosphate synthase (TPS), was implicated in the regulation of sugar metabolism/embryo development (Eastmond et al., 2002). Arabidopsis TPS1 may play a major role in coordinating cell wall biosynthesis and cell division in cellular metabolism (Gomez et al., 2006). These two genes play different roles during fibre development based on their expression patterns in cotton (Figure 6); however, the functions of these genes in cotton remain to be elucidated.
In conclusion, we genotyped 719 upland cotton accessions using the CottonSNP63K array for the first time and identified 46 SNPs significantly associated with fibre quality traits across eight environments and a number of novel genes, including 19 promising genes, of which ten were not reported in cotton, for FL and FS by GWA mapping. The identified genetic variation and candidate genes deepen our understanding of the molecular mechanisms underlying cotton fibre development. The validated accessions with excellent haplotypes are valuable breeding materials to improve cultivars. Our study provides a new resource for the improvement of cotton fibre quality through biotechnology‐assisted selection in future breeding efforts.
Experimental procedures
Cotton accessions and field experiments
A collection of 719 upland cotton germplasms (Gossypium hirsutum L.) was used for the association analysis in this study. These accessions were from different countries, 588 were collected from China and 131 were from other countries (Table S1). The 719 accessions were grown in eight natural environments in a randomized complete block design at Baoding (115°47′N, 38°87′E), Hejian (116°13′N, 38°42′E), Xinji (115°12′N, 37°54′E) and Qingxian (116°91′N, 38°65′E) in Hebei Province and Yacheng (109°20′N, 18°38′E) in Hainan Province in 2014, denoted 14BD (E1), 14HJ (E2), 14XJ (E3), 14QX (E4) and 14HN (E5), respectively, and Xinji and Qingxian in Hebei Province and Yacheng in Hainan Province in 2015, denoted 15XJ (E6), 15QX (E7) and 15HN (E8), respectively. Two replicates were performed for each accession in five locations in 2014, and three replicates were performed for the three locations in 2015. Briefly, one row of each accession was planted for each replicate, with 20–22 plants per row, 30–35 cm between plants within rows and 80 cm between rows.
Phenotypic evaluation and statistical analyses
When mature, 20 naturally open bolls from the central part of the plants from each accession were hand harvested at each location and ginned. Fibre samples were sent to the Supervision and Testing Center of Cotton Quality, Ministry of Agriculture of China in Anyang, Henan Province for fibre property determination. Fibre quality traits, including the upper half mean fibre length (FL, mm), fibre strength (FS, cN/tex), fibre micronaire (FM), fibre uniformity (FU, %) and fibre elongation (FE, %), were measured using a high volume instrument (HVI). Statistical analyses, Pearson correlation between traits and significance analyses were conducted using SPSS 22.0 software. Differences were tested for significance at the 1% probability level.
Genotyping and SNP marker screening
Genomic DNA of each accession was extracted from young leaf tissues for genotyping using a modified CTAB method (Zhang and Stewart, 2000). A CottonSNP63K array containing 63 058 SNPs (Hulse‐Kemp et al., 2015), which was recently developed by an international cotton SNP consortium, was applied to genotype the 719 accessions using the Illumina Infinium platform according to the manufacturer's protocol. All the SNP data were clustered and selectively analysed by Illumina GenomeStudio genotyping software. The SNP data set was further filtered with a calling rate < 0.85 and MAF < 0.05. For the physical localization of SNP markers, the probe sequences of the SNPs were used to perform a local BLAST (Altschul et al., 1990) search against the G. hirsutum TM‐1 reference genome (Zhang et al., 2015b). SNPs that could not be assigned to a G. hirsutum chromosome were excluded from further analysis.
Population structure and association mapping analysis
STRUCTURE 2.3.4 software (Evanno et al., 2005) was used to estimate the genetic structure of the population consisting of 719 accessions based on polymorphic SNPs. The numbers of hypothetical groups ranged from K = 1 to 10, using an admixture model with ten independent runs of 10 000 burn‐in time and 10 000 MCMC (Markov chain Monte Carlo) replication number. The output from STRUCTURE was analysed for the delta K value (ΔK) in STRUCTURE HARVESTER (Earl and vonHoldt, 2011). The optimal K value was determined by the log probability of LnP(K) and delta K based on the rate of change of LnP(K) between successive K. The Q matrix was derived for the subsequent association mapping which was the result of the integration of the cluster membership coefficient of replicate runs from STRUCTURE using CLUMPP software (Jakobsson and Rosenberg, 2007). Principal component analysis (PCA) using GCTA software (Yang et al., 2011) was used to assess the population structure. PowerMarker version 3.25 (Liu and Muse, 2005) was used to construct a NJ phylogenetic tree by calculating Nei's genetic distance among individuals.
For the genome‐wide association analysis, TASSEL 3.0 software (Bradbury et al., 2007) was used to determine the association between SNPs and phenotypic traits using a mixed linear model (MLM) (Zhang et al., 2010). The Q matrix from STRUCTURE and the kinship calculated with TASSEL 3.0 were included as fixed and random effects, respectively. The LD parameter (r2) between pairwise SNPs (MAF > 0.05) was estimated using TASSEL 3.0 software. The significance of the associations between SNPs and traits was based on the threshold of the Bonferroni correction for multiple tests (1/n), where n was the total number of SNPs used in the association analysis.
Transcriptome sequencing
Two high‐quality upland cotton cultivars (HY405 and ND13) and two normal‐quality upland cotton cultivars (CCRI8 and ND601) were grown in the field of Baoding, China, in 2014. For the cotton fibre samples, bolls were collected at 0, 5, 10, 15, 20, 25 and 30 DPA. Samples from different plants were pooled. Total RNA was extracted from these samples using the EASY spin Plant RNA kit (Aidlab, Beijing, China). The qualified RNA was used for sequencing analysis using TopHat v2.0 on a HiSeq 2500 platform at the Novogene Bioinformatics Institute, Beijing, China.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Correlation analysis between five traits related to fibre quality.
Figure S2. Manhattan plots showing the GWAS for FL in eight environments.
Figure S3. Manhattan plots showing the GWAS for FS in eight environments.
Figure S4. Manhattan plots showing the GWAS for FM in eight environments.
Figure S5. Manhattan plots showing the GWAS for FU in eight environments.
Figure S6. Manhattan plots showing the GWAS for FE in eight environments.
Figure S7. Boxplots depicting the genetic effects of SNPs with significant associations with fibre length in Dt11.
Figure S8. Boxplots depicting the genetic effects of SNPs with significant associations with fibre length and strength in At07.
Figure S9. Expression of all candidate genes related to fibre length and strength.
Table S1. List of 719 upland cotton accessions used for association mapping.
Table S2. Analysis of variance (ANOVA) results of the fibre quality traits.
Table S3. List of 612 candidate genes with fibre quality traits.
Table S4. KEGG analysis of all candidate genes.
Table S5. Phenotypes of the fibre length and strength of 47 upland cotton accessions belonging to Hap3.
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2016YFD0101006 and 2016YFD0101405) and the Top Talent Fund of Hebei Province. The authors are grateful to Dr. Meng Kuang, Dr. Nan Wang and Miss Yaru An for their assistance in data analysis.
References
- Abdurakhmonov, I.Y. , Kohel, R.J. , Yu, J.Z. , Pepper, A.E. , Abdullaev, A.A. , Kushanov, F.N. , Salakhutdinov, I.B. et al (2008) Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics, 92, 478–487. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Baker, R.W. and Hughson, F.M. (2016) Chaperoning SNARE assembly and disassembly. Nat. Rev. Mol. Cell Biol. 17, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL, software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Burget, E.G. , Verma, R. , Mølhøj, M. and Reiter, W.D. (2003) The biosynthesis of L‐arabinose in plants, molecular cloning and characterization of a golgi‐localized UDP‐D‐xylose 4‐epimerase encoded by the MUR4 gene of Arabidopsis. Plant Cell, 15, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, C.P. , Ye, W.X. , Zhang, T.Z. and Guo, W.Z. (2014) Association analysis of fiber quality traits and exploration of elite alleles in Upland cotton cultivars/accessions (Gossypium hirsutum L.). J. Integr. Plant Biol. 56, 51–62. [DOI] [PubMed] [Google Scholar]
- Chen, Z.J. , Scheffler, B.E. , Dennis, E. , Triplett, B.A. , Zhang, T.Z. , Guo, W.Z. , Chen, X.Y. et al (2007) Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 145, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, J.N. , Han, G. , Wang, X.F. , Zhang, G.Y. , Sun, Y.X. and Ma, Z.Y. (2009) Isolation and molecular characterization of a novel homogalacturonan galacturonosyl‐ transferase gene (GbGAUT1) from Gossypium barbadense . Afr. J. Biotechnol. 8, 4755–4764. [Google Scholar]
- Coart, E. , Lamote, V. , Loose, M.D. , Bockstaele, E.V. , Lootens, P. and Roldán‐Ruiz, I. (2002) AFLP markers demonstrate local genetic differentiation between two indigenous oak species [Quercus robur L. and Quercus petraea (Matt.) Liebl.] in flemish populations. Theor. Appl. Genet. 105, 431–439. [DOI] [PubMed] [Google Scholar]
- Crowell, S. , Korniliev, P. , Falcao, A. , Ismail, A. , Gregorio, G. , Mezey, J. and McCouch, S. (2016) Genome‐wide association and high‐resolution phenotyping link Oryza sativa panicle traits to numerous trait‐specific QTL clusters. Nat. Commun. 7, 10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, T. , Yao, H.Y. , Wang, J. , Wang, J. , Xue, H.W. and Zuo, K.J. (2016) GhLTPG1, a cotton GPI‐anchored lipid transfer protein, regulates the transport of phosphatidylinositol monophosphates and cotton fiber elongation. Sci. Rep. 6, 26829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimkpa, S.O. , Lahari, Z. , Shrestha, R. , Douglas, A. , Gheysen, G. and Price, A.H. (2015) A genome‐wide association study of a global rice panel reveals resistance in Oryza sativa to root‐knot nematodes. J. Exp. Bot. 22, 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl, D.A. and vonHoldt, B.M. (2011) STRUCTURE HARVESTER, a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. [Google Scholar]
- Eastmond, P.J. , Dijken, A.J.H.V. , Spielman, M. , Kerr, A. , Tissier, A.F. , Dickinson, H.G. , Jones, J.D.G. et al (2002) Trehalose‐6‐phosphate synthase 1, which catalyses the first step in trehalose synthesis, is essential for Arabidopsis embryo maturation. Plant J. 29, 225–235. [DOI] [PubMed] [Google Scholar]
- Ebine, K. , Fujimoto, M. , Okatani, Y. , Nishiyama, T. , Goh, T. , Ito, E. , Dainobu, T. et al (2011) A membrane trafficking pathway regulated by the plant‐specific RAB GTPase ARA6. Nat. Cell Biol. 13, 853–859. [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. and Goudet, J. (2005) Detecting the number of clusters of individuals using the software STRUCTURE, a simulation study. Mol. Ecol. 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Famoso, A.N. , Zhao, K.Y. , Clark, R.T. , Tung, C.W. , Wright, M.H. , Bustamante, C. , Kochian, L.V. et al (2011) Genetic architecture of aluminum tolerance in rice (Oryza sativa) determined through genome‐wide association analysis and QTL mapping. PLoS Genet. 7, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint‐Garcia, S.A. , Thornsberry, J.M. and Buckler, E.St. . (2003) Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54, 357–374. [DOI] [PubMed] [Google Scholar]
- Gomez, L.D. , Baud, S.A. , Li, Y. and Graham, I.A. (2006) Delayed embryo development in the ARABIDOPSIS TREHALOSE‐6‐PHOSPHATE SYNTHASE 1 mutant is associated with altered cell wall structure, decreased cell division and starch accumulation. Plant J. 46, 69–84. [DOI] [PubMed] [Google Scholar]
- Gore, M.A. , Chia, J.M. , Elshire, R.J. , Sun, Q. , Ersoz, E.S. , Hurwitz, B.L. , Peiffer, J.A. et al (2009) A first‐generation haplotype map of maize. Science 326, 1115–1117. [DOI] [PubMed] [Google Scholar]
- Grover, C.E. , Zhu, X. , Grupp, K.K. , Jareczek, J.J. , Gallagher, J.P. , Szadkowski, E. , Seijo, J.G. et al (2014) Molecular confirmation of species status for the allopolyploid cotton species, Gossypium ekmanianum Wittmack. Genet. Resour. Crop Ev. 62, 103–114. [Google Scholar]
- Guo, Y.H. , Yu, Y.P. , Wang, D. , Wu, C.A. , Yang, G.D. , Huang, J.G. and Zheng, C.C. (2009) GhZFP1, a novel CCCH‐type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol. 183, 62–75. [DOI] [PubMed] [Google Scholar]
- Gupta, P.K. , Rustgi, S. and Kulwal, P.L. (2005) Linkage disequilibrium and association studies in higher plants, present status and future prospects. Plant Mol. Biol. 57, 461–485. [DOI] [PubMed] [Google Scholar]
- Han, Y.P. , Zhao, X. , Liu, D.Y. , Li, Y.H. , Lightfoot, D.A. , Yang, Z.J. , Zhao, L. et al (2015) Domestication footprints anchor genomic regions of agronomic importance in soybeans. New Phytol. 209, 871–884. [DOI] [PubMed] [Google Scholar]
- Harsh, R. , Rosy, R. , Neil, C. , Song, J. , Roslyn, P. , Champa, B. , Tahira, R. et al (2015) Genome‐wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola. Plant Cell Environ. 94, 1125–1127. [DOI] [PubMed] [Google Scholar]
- Huang, X.H. and Han, B. (2014) Natural variations and genome‐wide association studies in crop plants. Annu. Rev. Plant Biol. 65, 531–551. [DOI] [PubMed] [Google Scholar]
- Huang, J. , Chen, F. , Casino, C.D. , Autino, A. , Shen, M. , Yuan, S. , Peng, J. et al (2006) An ankyrin repeat‐containing protein, characterized as a ubiquitin ligase, is closely associated with membrane‐enclosed organelles and required for pollen germination and pollen tube growth in Lily. Plant Physiol. 140, 1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X.H. , Wei, X.H. , Sang, T. , Zhao, Q. , Feng, Q. , Zhao, Y. , Li, C.Y. et al (2010) Genome‐wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967. [DOI] [PubMed] [Google Scholar]
- Huang, G.Q. , Gong, S.Y. , Xu, W.L. , Li, W. , Li, P. , Zhang, C.J. , Li, D.D. et al (2013) A fasciclin‐like arabinogalactan protein, GhFLA1, is involved in fiber initiation and elongation of cotton. Plant Physiol. 161, 1278–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse‐Kemp, A.M. , Lemm, J. , Plieske, J. , Ashrafi, H. , Buyyarapu, R. , Fang, D.D. , Frelichowski, J. et al (2015) Development of a 63k snp array for cotton and high‐density mapping of intraspecific and interspecific populations of Gossypium spp. G3: Genes ‐ Genomes ‐ Genetics 5, 1187–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, M.S. , Zeng, L. , Thyssen, G.N. , Delhom, C.D. , Kim, H.J. , Li, P. and Fang, D.D. (2016) Mapping by sequencing in cotton (Gossypium hirsutum) line MD52ne identified candidate genes for fiber strength and its related quality attributes. Theor. Appl. Genet. 129, 1–16. [DOI] [PubMed] [Google Scholar]
- Jakobsson, M. and Rosenberg, N.A. (2007) CLUMPP, a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jamshed, M. , Jia, F. , Gong, J.W. , Palanga, K.K. , Shi, Y.Z. , Li, J.W. , Shang, H.H. et al (2016) Identification of stable quantitative trait loci (QTLs) for fiber quality traits across multiple environments in Gossypium hirsutum recombinant inbred line population. BMC Genom. 17, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, G.Q. , Huang, X.H. , Zhi, H. , Zhao, Y. , Zhao, Q. , Li, W.J. , Chai, Y. et al (2013) A haplotype map of genomic variations and genome‐wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 45, 957–961. [DOI] [PubMed] [Google Scholar]
- Lacape, J.M. , Llewellyn, D. , Jacobs, J. , Arioli, T. , Becker, D. , Calhoun, S. , Al‐Ghazi, Y. et al (2010) Meta‐analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol. 10, 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. , Burns, T.H. , Light, G. , Sun, Y. , Fokar, M. , Kasukabe, Y. , Fujisawa, K. et al (2010) Xyloglucan endotransglycosylase/hydrolase genes in cotton and their role in fiber elongation. Planta 232, 1191–1205. [DOI] [PubMed] [Google Scholar]
- Li, H. , Peng, Z.Y. , Yang, X.H. , Wang, W.D. , Fu, J.J. , Wang, J.H. , Han, Y.J. et al (2013) Genome‐wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50. [DOI] [PubMed] [Google Scholar]
- Li, F. , Chen, B.Y. , Xu, K. , Wu, J.F. , Song, W.L. , Bancroft, I. , Harper, A.L. et al (2014) Genome‐wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res. 21, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F.G. , Fan, G.Y. , Lu, C.R. , Xiao, G.H. , Zou, C.S. , Kohel, R.J. , Ma, Z.Y. et al (2015) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM‐1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. [DOI] [PubMed] [Google Scholar]
- Liu, K. and Muse, S.V. (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129. [DOI] [PubMed] [Google Scholar]
- Mace, E.S. , Tai, S. , Gilding, E.K. , Li, Y.H. , Prentis, P.J. , Bian, L. , Campbell, B.C. et al (2013) Whole‐genome sequencing reveals untapped genetic potential in Africa's indigenous cereal crop sorghum. Nat. Commun. 4, 2320.1–2320.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijon, M. , Satbhai, S.B. , Tsuchimatsu, T. and Busch, W. (2014) Genome‐wide association study using cellular traits identifies a new regulator of root development in Arabidopsis . Nat. Genet. 46, 77–81. [DOI] [PubMed] [Google Scholar]
- Morris, G.P. , Ramu, P. , Deshpande, S.P. , Hash, C.T. , Shah, T. , Upadhyaya, H.D. , Riera‐Lizarazu, O. et al (2013) Population genomic and genome‐wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. USA 110, 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel, M. , Kranner, I. , Neumann, K. , Rolletschek, H. , Seal, C.E. , Colville, L. , Fernández‐Marín, B. et al (2015) Genome‐wide association mapping and biochemical markers reveal that seed ageing and longevity are intricately affected by genetic background and developmental and environmental conditions in barley. Plant Cell Environ. 38, 1011–1022. [DOI] [PubMed] [Google Scholar]
- Nie, X.H. , Huang, C. , You, C.Y. , Li, W. , Zhao, W.X. , Shen, C. , Zhang, B.B. et al (2016) Genome‐wide SSR‐based association mapping for fiber quality in nation‐wide upland cotton inbreed cultivars in China. BMC Genom. 17, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwland, J. , Stamm, P. , Wen, B. , Randall, R.S. , Murray, J.A.H. and Bassel, G.W. (2016) Re‐induction of the cell cycle in the Arabidopsis post‐embryonic root meristem is ABA‐insensitive, GA‐dependent and repressed by KRP6 . Sci. Rep. 6, 23586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M. , Borevitz, J.O. , Bergelson, J. , Berry, C.C. , Chory, J. , Hagenblad, J. , Kreitman, M. et al (2002) The extent of linkage disequilibrium in Arabidopsis thaliana . Nat. Genet. 30, 190–193. [DOI] [PubMed] [Google Scholar]
- Nuzhdin, S.V. , Friesen, M.L. and McIntyre, L.M. (2012) Genotype‐phenotype mapping in a post‐GWAS world. Trends Genet. 28, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A.H. , Saranga, Y. , Menz, M. , Jiang, C. and Wright, R.J. (2003) QTL analysis of genotype × environment interactions affecting cotton fiber quality. Theor. Appl. Genet. 106, 384–396. [DOI] [PubMed] [Google Scholar]
- Porth, I. , Klapste, J. , Skyba, O. , Hannemann, J. , McKown, A.D. , Guy, R.D. , DiFazio, S.P. et al (2013) Genome‐wide association mapping for wood characteristics in Populus identifies an array of candidate single nucleotide polymorphisms. New Phytol. 200, 710–726. [DOI] [PubMed] [Google Scholar]
- Preuss, M.L. , Serna, J. , Falbel, T.G. , Bednarek, S.Y. and Nielsen, E. (2004) The Arabidopsis Rab GTPase RabA4b localizes to the tips of growing root hair cells. Plant Cell 16, 1589–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington, D.L. , Thornsberry, J.M. , Matsuoka, Y. , Wilson, L.M. , Whitt, S.R. , Doebley, J. , Kresovich, S. et al (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98, 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, S. and Moore, I. (2002) The Arabidopsis, Rab GTPase family: another enigma variation. Curr. Opin. Plant Biol. 5, 518–528. [DOI] [PubMed] [Google Scholar]
- Said, J.I. , Lin, Z.X. , Zhang, X.L. , Song, M.Z. and Zhang, J.F. (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 14, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said, J.I. , Song, M.Z. , Wang, H.T. , Lin, Z.X. , Zhang, X.J. , Fang, D.D. and Zhang, J.F. (2015) A comparative meta‐analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 290, 1003–1025. [DOI] [PubMed] [Google Scholar]
- Samuel, Y.S. , Cheung, F. , Lee, J.J. , Ha, M. , Wei, N.E. , Sze, S.H. , Stelly, D.M. et al (2006) Accumulation of genome‐specific transcripts, transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton. Plant J. 47, 761–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, X.L. , Guo, W.Z. , Zhu, X.F. , Yuan, Y.L. , Yu, J.Z. , Kohel, R.J. and Zhang, T.Z. (2005) Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol. Breeding 15, 169–181. [Google Scholar]
- Shen, G.X. , Kuppu, S. , Venkataramani, S. , Wang, J. , Yan, J.Q. , Qiu, X.Y. and Zhang, H. (2010) ANKYRIN REPEAT‐CONTAINING PROTEIN 2A is an essential molecular chaperone for peroxisomal membrane‐bound ASCORBATE PEROXIDASE3 in Arabidopsis . Plant Cell 22, 811–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling, J.D. , Atmodjo, M.A. , Inwood, S.E. , Kumar, K.V.S. , Quigley, H.F. , Hahn, M.G. and Mohnen, D. (2006) Functional identification of an Arabidopsis pectin biosynthetic homogalacturonan galacturonosyltransferase. Proc. Natl. Acad. Sci. USA 103, 5236–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracke, S. , Haseneyer, G. , Veyrieras, J.B. , Geiger, H.H. , Sauer, S. , Graner, A. and Piepho, H.P. (2009) Association mapping reveals gene action and interactions in the determination of flowering time in barley. Theor. Appl. Genet. 118, 259–273. [DOI] [PubMed] [Google Scholar]
- Thornsberry, J.M. , Goodman, M.M. , Doebley, J. , Kresovich, S. , Nielsen, D. and Buckler, E.S. (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28, 286–289. [DOI] [PubMed] [Google Scholar]
- Tyagi, P. , Gore, M.A. , Bowman, D.T. , Campbell, B.T. , Udall, J.A. and Kuraparthy, V. (2014) Genetic diversity and population structure in the US Upland cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 127, 283–295. [DOI] [PubMed] [Google Scholar]
- Van Sandt, V.S. , Suslov, D. , Verbelen, J.P. and Vissenberg, K. (2007) Xyloglucan endotransglucosylase activity loosens a plant cell wall. Ann. Bot. 100, 1467–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, P. , De Clercq, A. , Stals, H. , Van Leene, J. , Van De Slijke, E. , Van Isterdael, G. , Eeckhout, D. et al (2014) The cyclin‐dependent kinase inhibitor KRP6 induces mitosis and impairs cytokinesis in giant cells induced by plant‐parasitic nematodes in Arabidopsis . Plant Cell 26, 2633–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Wang, J.X. , Yu, N. , Li, C.H. , Luo, B. , Gou, J.Y. , Wang, L.J. et al (2004) Control of plant trichome development by a cotton fiber MYB gene. Plant Cell 16, 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Yan, J.B. , Zhao, J.R. , Song, W. , Zhang, X.B. , Xiao, Y.N. and Zheng, Y.L. (2012) Genome‐wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 196, 125–131. [DOI] [PubMed] [Google Scholar]
- Wang, Y.Y. , Zhou, Z.L. , Wang, X.X. , Cai, X.Y. , Li, X.N. , Wang, C.Y. , Wang, Y.H. et al (2016) Genome‐wide association mapping of glyphosate‐resistance in Gossypium hirsutum races. Euphytica 209, 209–221. [Google Scholar]
- Wen, Z.X. , Tan, R.J. , Yuan, J.Z. , Bales, C. , Du, W.Y. , Zhang, S.C. , Chilvers, M. et al (2014) Genome‐wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom. 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J.F. (1989) New World tetraploid cottons contain Old World cytoplasm. Proc. Natl. Acad. Sci. USA 86, 4132–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J.F. and Cronn, R.C. (2003) Polyploidy and the evolutionary history of cotton. Adv. Agron. 78, 139–186. [Google Scholar]
- Wu, D. , Sato, K. and Ma, J.F. (2015) Genome‐wide association mapping of cadmium accumulation in different organs of barley. New Phytol. 208, 817–829. [DOI] [PubMed] [Google Scholar]
- Xu, L.P. , Hu, K.N. , Zhang, Z.Q. , Guan, C.Y. , Chen, S. , Hua, W. , Li, J.N. et al (2016) Genome‐wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res. 23, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Lee, S.H. , Goddard, M.E. and Visscher, P.M. (2011) GCTA, a tool for genome‐wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Lu, Y.L. , Yang, X.H. , Huang, J. , Zhou, Y. , Ali, F. , Wen, W.W. et al (2014a) Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 10, 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X.L. , Zhou, X.D. , Wang, X.F. , Li, Z.K. , Zhang, Y. , Liu, H.W. , Wu, L.Q. et al (2014b) Mapping QTL for cotton fiber quality traits using simple sequence repeat markers, conserved intron‐scanning primers, and transcript‐derived fragments. Euphytica 201, 215–230. [Google Scholar]
- Yano, K. , Yamamoto, E. , Aya, K. , Takeuchi, H. , Lo, P.C. , Hu, L. , Yamasaki, M. et al (2016) Genome‐wide association study using whole‐genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927–934. [DOI] [PubMed] [Google Scholar]
- Yu, J.M. , Pressoir, G. , Briggs, W.H. , Vroh Bi, I. , Yamasaki, M. , Doebley, J.F. , McMullen, M.D. et al (2006) A unified mixed‐model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. [DOI] [PubMed] [Google Scholar]
- Zeng, L.H. , Meredith, W.R. , Gutierrez, O.A. and Boykin, D.L. (2009) Identification of associations between SSR markers and fiber traits in an exotic germplasm derived from multiple crosses among Gossypium tetraploid species. Theor. Appl. Genet. 119, 93–103. [DOI] [PubMed] [Google Scholar]
- Zhang, J.F. and Stewart, J.M. (2000) Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci. 4, 193–201. [Google Scholar]
- Zhang, Z. , Ersoz, E. , Lai, C.Q. , Todhunter, R.J. , Tiwari, H.K. , Gore, M.A. , Bradbury, P.J. et al (2010) Mixed linear model approach adapted for genome‐wide association studies. Nat. Genet. 42, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , Zhang, J. , Ma, J. , Tang, S.Y. , Liu, D.J. , Teng, Z.H. , Liu, D.X. et al (2011) Genetic mapping and quantitative trait locus analysis of fiber quality traits using a three‐parent composite population in upland cotton (Gossypium hirsutum L.). Mol. Breeding 29, 335–348. [Google Scholar]
- Zhang, J. , Singh, A. , Mueller, D.S. and Singh, A.K. (2015a) Genome‐wide association and epistasis studies unravel the genetic architecture of sudden death syndrome resistance in soybean. Plant J. 31, 1–12. [DOI] [PubMed] [Google Scholar]
- Zhang, T.Z. , Hu, Y. , Jiang, W.K. , Fang, L. , Guan, X.Y. , Chen, J.D. , Zhang, J.B. et al (2015b) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM‐1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. [DOI] [PubMed] [Google Scholar]
- Zhao, K.Y. , Tung, C.W. , Eizenga, G.C. , Wright, M.H. , Ali, M.L. , Price, A.H. , Norton, J.G. et al (2011) Genome‐wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa . Nat. Commun. 2, 1020–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Zhang, Z.T. , Li, M. , Wei, X.Z. , Li, X.J. , Li, B.Y. and Li, X.B. (2015a) Cotton (Gossypium hirsutum) 14‐3‐3 proteins participate in regulation of fiber initiation and elongation by modulating brassinosteroid signalling. Plant Biotechnol. J. 13, 269–280. [DOI] [PubMed] [Google Scholar]
- Zhou, Z.K. , Jiang, Y. , Wang, Z. , Gou, Z.H. , Lyu, J. , Li, W.Y. and Yu, Y.J. (2015b) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33, 408–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation analysis between five traits related to fibre quality.
Figure S2. Manhattan plots showing the GWAS for FL in eight environments.
Figure S3. Manhattan plots showing the GWAS for FS in eight environments.
Figure S4. Manhattan plots showing the GWAS for FM in eight environments.
Figure S5. Manhattan plots showing the GWAS for FU in eight environments.
Figure S6. Manhattan plots showing the GWAS for FE in eight environments.
Figure S7. Boxplots depicting the genetic effects of SNPs with significant associations with fibre length in Dt11.
Figure S8. Boxplots depicting the genetic effects of SNPs with significant associations with fibre length and strength in At07.
Figure S9. Expression of all candidate genes related to fibre length and strength.
Table S1. List of 719 upland cotton accessions used for association mapping.
Table S2. Analysis of variance (ANOVA) results of the fibre quality traits.
Table S3. List of 612 candidate genes with fibre quality traits.
Table S4. KEGG analysis of all candidate genes.
Table S5. Phenotypes of the fibre length and strength of 47 upland cotton accessions belonging to Hap3.