

Abstract
Background
Nicotine use increases alcohol drinking, suggesting that the combination of these drugs may produce synergistic effects in activating reward circuitry. Alternatively, use of either of these drugs may facilitate the development of cross-tolerance to the other to promote intake escalation.
Methods
In the current study, adult male Wistar rats were chronically exposed to room air or chronic, intermittent nicotine vapor, which has been shown to produce symptoms of nicotine dependence as evidenced by elevated nicotine self-administration and a host of somatic and motivational withdrawal symptoms during withdrawal. We examined regional neuroadaptations in nicotine-experienced vs. non-experienced animals, focusing on changes in phosphorylation of the AMPA glutamate channel subunit GluA1 in reward-related brain regions since excitatory neuroadaptations are heavily implicated in both alcohol and nicotine addiction.
Results
During withdrawal, nicotine exposure and alcohol challenge (1g/kg) interactively produced neuroadaptations in GluA1 phosphorylation in a brain region-dependent manner. Alcohol robustly increased protein kinase A (PKA)-mediated phosphorylation of GluA1 at serine 845 in multiple regions. However, this neuroadaptation was largely absent in three areas (dorsomedial prefrontal cortex, dorsal striatum, and central amygdala) in nicotine-experienced animals. This interactive effect suggests a molecular tolerance to alcohol-stimulated phosphorylation of GluA1 in the context of nicotine dependence.
Conclusions
Nicotine may modify the rewarding or reinforcing effects of alcohol by altering glutamate signaling in a region-specific manner, thereby leading to increased drinking in heavy smokers.
Keywords: Alcohol, Amygdala, Glutamate, Nicotine, Prefrontal Cortex
Introduction
Nicotine and alcohol are two of the most commonly abused drugs worldwide. Alcohol drinking is strongly correlated with nicotine use and there is a high association of nicotine and alcohol dependence (Falk et al., 2006). In fact, many alcoholics actually die from smoking-related causes (Littleton et al., 2007). Following chronic or excessive use, the transition to nicotine or alcohol dependence produces a constellation of somatic and motivational withdrawal symptoms (Edwards et al., 2012; Cohen et al., 2015). Such motivational disturbances are associated with drug-induced, brain region-specific neuroadaptations that are hypothesized to drive persistent, escalated, and uncontrollable substance use observed in addiction (Edwards and Koob, 2010).
Several preclinical studies have shown that nicotine enhances the acquisition of alcohol drinking (Smith et al., 1999) and that repeated nicotine exposure increases self-administration of alcohol and reinstatement of alcohol seeking (Le et al., 2003; Leao et al., 2015), suggesting that the combination of nicotine and alcohol may produce a synergistic activation of reward circuitry. Alternatively, chronic use of either substance may facilitate the development of a cross-tolerance to the other substance. For example, escalation of drinking following nicotine exposure is associated with a nicotine-driven blunting of alcohol-induced dopamine release in the ventral striatum (Doyon et al., 2013). There is now a growing interest in mechanisms associated with nicotine and alcohol interactions, although information on behavioral and neuronal signaling adaptations that occur in animals exposed to both nicotine and alcohol remains limited compared to the abundance of studies on these substances in isolation (Van Skike et al., 2016).
Virtually all drugs of abuse alter AMPA glutamate receptor channel function, while chronic drug-induced modifications of AMPA channel activity alter excitatory neurotransmission in association with excessive drug use, withdrawal, and relapse (Conrad et al., 2008; Edwards et al., 2011). Most of these effects occur via modification of channel function though direct phosphorylation of GluA1 AMPA subunits, which are located throughout cortical and subcortical brain reward areas and regulated in part by dopaminergic neurotransmission (Wolf et al., 2003; Edwards et al., 2007; Choi et al., 2011). AMPA channel function is increased following phosphorylation by protein kinase A (PKA, at serine 845) and/or protein kinase C and calcium/calmodulin-dependent protein kinase II (PKC/CaMKII, at serine 831), typically via facilitation of channel activity or membrane trafficking (Wang et al., 2005). Such phosphorylation events are thought to be essential for the modulation of synaptic plasticity that may underlie several key aspects of the persistence of drug addiction (Hyman et al., 2006).
In the present study, we employed a rodent model of nicotine dependence that utilizes chronic, intermittent nicotine vapor exposure. This treatment regimen has recently been used to produce several key symptoms of nicotine dependence including escalated nicotine self-administration during withdrawal (Baiamonte et al., 2014; Gilpin et al., 2014). Our current aim was to investigate multiple reward-related brain regions (including the prefrontal cortex, striatum, and amygdala) to determine excitatory neuroadaptations associated with acute, binge-like alcohol administration in the nicotine-experienced vs. nicotine-naïve state. These regions were selected based on their close association with both nicotine and alcohol dependence (for review, see Koob and Volkow, 2010), although few studies have sought to examine interactive neuroadaptations produced via exposure to these two drugs in combination. We employed an acute bolus dose of alcohol (1g/kg) that produces anxiolytic-like effects (Sakharkar et al., 2012) and blood alcohol levels in rats roughly corresponding to those achieved via binge drinking in humans (National Institute on Alcohol Abuse and Alcoholism, 2004).
Materials and Methods
Animals
Adult male Wistar rats (n=24) were obtained from Charles River (Raleigh, NC, USA), and weighed 200–300g at time of arrival. Animals were pair-housed in standard plastic cages with wood chip bedding under a reverse 12-h light-dark cycle (lights off at 8 AM). Animals were fed a standard rat diet with ad libitum access to food and water throughout the experiment. All procedures were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) and NIH guidelines.
Nicotine Vapor Inhalation Protocol
For chronic, intermittent exposure to nicotine vapor, animals were housed in plexiglass vapor delivery chambers (La Jolla Alcohol Research, Inc., La Jolla, CA) and exposed daily to intermittent nicotine vapor (12 hours on/12 hours off) for several weeks, as previously described (George et al., 2010; Baiamonte et al., 2014; Gilpin et al., 2014). Briefly, nicotine vapor was produced by bubbling air at a flow rate of 10 liters/minute/cage through a gas-washing bottle containing a solution of pure nicotine (free base, Sigma-Aldrich, St. Louis, MO). Nicotine vapor was produced by evaporation that was maximized by the bubbling of air with a constant air flow. The highly concentrated nicotine vapor was then passed through a drop-catch bottle and further diluted by the addition of 60 liters/minute of clean air in a 2000 mL Erlenmeyer vacuum flask at room temperature. The final nicotine–air mixture was homogeneously distributed between chambers at a flow rate of 15 liters/minute. Air controls were housed in the room containing the vapor delivery chambers but were exposed to room air only.
Alcohol Challenge and Western Blot Analysis of Phosphoproteins
After 4 weeks of exposure to chronic intermittent nicotine vapor or air, one day prior to sacrifice, rats received a 1 ml/kg (IP) injection of saline to habituate them to the injection procedure. After the final nicotine exposure session, at 8–10 hours of withdrawal (WD), animals were challenged with either saline (1 ml/kg, IP) or ethanol (1 g/kg of 15% w/v ethanol, IP) and euthanized 15 minutes later. Fifteen minutes post-saline or alcohol injection, animals were sacrificed by decapitation under light halothane anesthesia. Brains were rapidly dissected and snap-frozen in isopentane. Regional tissue samples were obtained using 13–16 gauge punches from frozen coronal brain slices (0.5 mm thick) obtained by the use of a cryostat according to anatomical regions depicted in Fig. 1 (Paxinos and Watson, 1998). Tissue samples were homogenized by sonication in lysis buffer (320 mm sucrose, 5 mm HEPES, 1 mm EGTA, 1 mm EDTA and 1% SDS, with protease inhibitor cocktail and phosphatase inhibitor cocktails II and III diluted 1:100; Sigma, St Louis, MO, USA), heated at 100 °C for 5 min and were stored at −80 °C until the determination of protein concentration by a detergent-compatible Lowry method (Bio-Rad, Hercules, CA, USA). Samples of protein (20 μg) were subjected to SDS-polyacrylamide gel electrophoresis on 8% acrylamide gels by using a Tris/Glycine/SDS buffer system (Bio-Rad), followed by electrophoretic transfer to polyvinylidene difluoride membranes (GE Healthcare, Piscataway, NJ, USA). Membranes were blocked overnight in 5% non-fat milk at 4 °C and were then incubated in primary antibody. Primary antibodies include pGluA1S845 (1:2500; Millipore, Temecula, CA, USA; Cat. #04-1073), pGluA1S831 (1:2000; Millipore, Temecula, CA, USA; Cat. #04-823), total GluA1 (1:5000; Cell Signaling, Danvers, MA, USA; Cat. #13185), PKA C-α (1:5000–15000; Cell Signaling, Danvers, MA, USA; Cat. #4782), and β-Tubulin (1:500,000; Santa Cruz Biotechnology, Dallas, TX, USA; Cat. # sc-53140). Membranes were washed and labeled with species-specific peroxidase-conjugated secondary antibody (1:10000; Bio-Rad) for 1 h at room temperature. Following chemiluminescence detection (SuperSignal West Pico; Thermo Scientific, Rockford, IL, USA), blots were stripped for 20 min at room temperature (Restore; Thermo Scientific) and were reprobed for total GluA1 levels. Immunoreactivity was quantified by densitometry (ImageJ 1.45S; Bethesda, MD) under linear exposure conditions. Densitized values were expressed as a percentage of the mean of control values for each gel to normalize data across blots. Individual phosphoprotein levels were normalized to individual total GluA1 protein levels to generate phosphorylation: total ratio values for statistical comparison. No significant changes in total GluA1 levels were observed in any region under any treatment condition.
Figure 1.

(A) Timeline depicting the treatment regimen for chronic, intermittent nicotine vapor exposure and acute alcohol or saline challenges. Animals were exposed to either chronic intermittent nicotine vapor (12h on/12h off) (n=12) or room air (n=12) for four weeks before sacrifice for Western analysis during acute withdrawal. All animals were given habituating intraperitoneal (IP) injections before the day of sacrifice. On the final day of vapor exposure, animals were withdrawn from vapor for 8–10 hours and were injected IP with either 1ml/kg saline or 1g/kg alcohol. Room air-exposed animals received identical treatments. All animals were euthanized 15 minutes post-injection and brains were immediately dissected and snap-frozen in preparation for regional tissue sample collection. (B) Schematic representation of sub-regional brain samples collected (Paxinos and Watson, 1998). DM = dorsomedial prefrontal cortex; VM = ventromedial prefrontal cortex; CC = (posterior) cingulate cortex; DS = dorsal striatum; VS = ventral striatum; HIP = hippocampus; CeA = central amygdala; BLA = basolateral amygdala.
Measurement of Blood Alcohol Levels (BALs)
Tail blood (0.2 ml) was collected and centrifuged, the plasma extracted and injected into an oxygen-rate alcohol analyzer (Analox Instruments, London, UK) for BAL determination. The reaction is based on the oxidation of alcohol by alcohol oxidase in the presence of molecular oxygen (alcohol + O2 → acetaldehyde + H2O2). The rate of oxygen consumption is directly proportional to the alcohol concentration. Single point calibrations were done for each set of samples with reagents provided by Analox Instruments (25–400 mg%).
Statistical Analysis
Western blot data were analyzed using two-way between-subjects ANOVAs with nicotine vapor history and alcohol exposure as factors. Where ANOVAs revealed significant interactions between factors, post hoc analyses were performed via Tukey’s multiple comparison tests.
Results
Phosphorylation of AMPA GluA1 Receptor Subunits in the Prefrontal Cortex
We investigated the interaction of nicotine exposure history and alcohol challenge on excitatory glutamate channel phosphorylation in reward-related brain regions. In the dorsomedial prefrontal cortex (dmPFC), we found that alcohol elicited a robust 87% increase in PKA-mediated GluA1 phosphorylation (q20 = 7.247, p < 0.001; Figure 2A), an effect that was drastically attenuated in animals with a history of nicotine exposure (significant alcohol × nicotine interaction: F1,20 = 4.583, p = 0.0448; significant attenuation of alcohol response in nicotine-exposed animals: q20 = 4.628, p < 0.05) and suggestive of a blunted alcohol-mediated signaling in animals with a history of nicotine exposure. In contrast, alcohol failed to alter PKC-regulated phosphorylation of GluA1 (Figure 2B) in the dmPFC. Within the ventromedial prefrontal cortex (vmPFC), alcohol increased pGluA1S845 to an equal extent (31–33%) in nicotine-exposed and nicotine-naïve groups (main effect of alcohol, F1,20 = 10.74, p = 0.0038; Figure 2C) while again not altering PKC/CaMKII-mediated phosphorylated GluA1 levels (Figure 2D).
Figure 2.

GluA1 phosphorylation in the prefrontal cortex. Alcohol (1 g/kg, 15 minute pretreatment) robustly increases PKA-mediated GluA1 phosphorylation in the dmPFC, although this effect is largely absent in animals with a prior history of nicotine exposure (A). In contrast, alcohol failed to alter PKC/CaMKII-mediated GluA1 phosphorylation in the dmPFC (B). Alcohol modestly increased pGluA1S845 in the vmPFC, and this effect was not modified by prior nicotine exposure (C). This alteration in alcohol-mediated PKA signaling also did not extend to PKC/CaMKII phosphorylation of GluA1 subunits (D). Representative pGluA1S845 and total GluA1 immunoblots are indicated in (E). N=6/group. Pound signs indicate a main group effect of alcohol treatment (##p<0.01, ###p<0.001). Asterisks indicate increased GluA1S845 phosphorylation following alcohol challenge as determined by Tukey’s post hoc test (***p<0.001) or a significant attenuation of this effect in the context of nicotine pre-exposure (*p<0.05).
Phosphorylation of AMPA GluA1 Receptor Subunits in the Striatum
In the dorsal striatum, PKA-mediated GluA1S845 phosphorylation was significantly increased following alcohol challenge in both air- and nicotine-exposed groups (main effect of alcohol, F1,20 = 38.01, p < 0.0001; Figure 3A). However, pGluA1S845 levels were reduced by chronic nicotine vapor exposure in both injection groups (F1,20 = 22.84, p = 0.0001; Figure 3A), reflecting a divergent regulation of excitatory signaling between acute alcohol and chronic nicotine. In comparison, there was no significant regulation of GluA1S831 phosphorylation by either chronic nicotine vapor or following alcohol challenge (Figure 3B). In the ventral striatum, while there were no significant alterations in the phosphorylation levels of GluA1S845 in any groups (Figure 3C), PKC/CaMKII-mediated GluA1S831 phosphorylation was increased by 30–46% following alcohol challenge between air- and nicotine-exposed animals (main effect of alcohol, F1,20 = 5.351, p = 0.0315; Figure 3D).
Figure 3.

GluA1 phosphorylation in the striatum. Alcohol significantly increases PKA-mediated GluA1 phosphorylation in the dorsal striatum in both air- and nicotine-exposed groups, while chronic intermittent nicotine exposure and withdrawal decreases pGluA1S845 (A). Neither of these effects was observed with regard to PKC/CaMKII-mediated protein phosphorylation of GluA1 subunits (B). In contrast to the other regions investigated, alcohol failed to alter PKA-mediated phosphorylation of GluA1 in the ventral striatum (C), although it significantly increased PKC/CaMKII-regulated GluA1 phosphorylation (D). Representative pGluA1S845, pGluA1S831, and total GluA1 immunoblots are indicated in (E). N=6/group. Pound and dollar signs indicate a main group effect of alcohol treatment (#p<0.05, ###p<0.001) or nicotine exposure ($$p<0.01), respectively.
Phosphorylation of AMPA GluA1 Receptor Subunits in the Amygdala
In the central amygdala (CeA), PKA-mediated GluA1S845 phosphorylation was significantly increased by 66% following alcohol challenge in air-exposed animals (q20 = 6.807, p < 0.001; Figure 4A. However, similar to the dmPFC, we observed a significant interaction between alcohol challenge and nicotine exposure (F(1, 20) = 5.601, p = 0.0281) that reflected a substantially diminished ability of alcohol to increase pGluA1S845 in nicotine-exposed animals. Also similar to the dmPFC, these interactive neuroadaptations in PKA-mediated phosphorylation did not generalize to PKC/CaMKII regulation of GluA1 subunits (Figure 4B). As seen in the CeA, PKA-mediated GluA1S845 phosphorylation was increased as a main effect of alcohol in the basolateral amygdala (BLA; F1,20 = 11.53, p = 0.0029), although no evidence of tolerance to alcohol-stimulation of pGluA1S845 was present. Further, there were no significant changes in GluA1S831 phosphorylation following nicotine exposure or alcohol challenge in the BLA.
Figure 4.

GluA1 phosphorylation in the amygdala. Alcohol significantly increases PKA-mediated GluA1 phosphorylation in the central amygdala, although this effect is largely absent in animals with a history of chronic, intermittent nicotine vapor exposure (A). However, no changes were evident in PKC/CaMKII-regulation of GluA1 subunits (B). In comparison to the central amygdala, alcohol also increases GluA1S845 phosphorylation in the basolateral amygdala, but to a similar extent in both air- and nicotine-exposed groups (C), and this effect did not generalize to pGluA1S831 (D). Representative pGluA1S845 and total GluA1 immunoblots are indicated in (E). N=6/group. Pound signs indicate a main group effect of alcohol treatment (##p<0.01, ###p<0.001). Asterisks indicate increased GluA1S845 phosphorylation alcohol as determined by Tukey’s post hoc test (***p<0.001).
Phosphorylation of AMPA GluA1 Receptor Subunits in the Dorsal Hippocampus and Cingulate Cortex
Alcohol produced robust (43–57%, F1,20 = 13.15, p = 0.0017; Figure 5A) increases in pGluA1S845 and modest increases in pGluA1S831 in the dorsal hippocampus (14–23%; F1,20 = 4.880, p = 0.0390; Figure 5B). Prior nicotine exposure did not modify these main effects of acute alcohol challenge. Similarly, alcohol significantly increased PKA-mediated GluA1 phosphorylation in posterior areas of the cingulate cortex (35–49%, F1,20 = 21.36, p = 0.0002; Figure 5C), an effect that did not extend to PKC/CaMKII-regulated GluA1 (Figure 5D).
Figure 5.

GluA1 phosphorylation in the hippocampus and cingulate cortex. Alcohol challenge significantly increases GluA1 phosphorylation in the hippocampus, although this effect is more robust with regard to PKA-mediated phosphorylation (A) in comparison to PKC/CaMKII-mediated regulation (B). In the cingulate cortex, alcohol increases pGluA1S845 (C) without significantly altering pGluA1S831 (D). Nicotine exposure failed to modify alcohol-induced phosphorylation in either of these regions. Representative pGluA1S845, pGluA1S831, and total GluA1 immunoblots are indicated in (E). N=6/group. Pound signs indicate a main effect of alcohol treatment (#p<0.05, ##p<0.01, ###p<0.001).
Protein Kinase A Catalytic Subunit Levels and Blood Alcohol Levels
Finally, given the significant abatement of alcohol-regulated GluA1S845 phosphorylation following withdrawal from chronic nicotine exposure in the dmPFC, dorsal striatum, and CeA (Figures 2–5), we wanted to determine whether this effect was driven by reductions in catalytic protein kinase A (PKA) subunit levels. We observed no changes in PKA levels across groups in the affected regions (Table 1, Figure 6), suggesting alternative signaling or neurocircuitry mechanisms driving nicotine-induced tolerance to the effects of alcohol in these areas. Furthermore, the observed tolerance-like effects were not due to altered metabolism of alcohol in nicotine-exposed rats, as peak blood alcohol levels following a 1 g/kg challenge in separate cohorts of rats were similar in nicotine-naïve (103.7 ± 16.6 mg/dl) and nicotine-exposed (106.3 ± 25.2 mg/dl) groups 15 minutes following injection (commensurate with our molecular studies).
Table 1.
PKA Catalytic Subunit Levels Across Selected Groups
| Brain region | Air/Saline | Air/Alcohol | Nicotine/Saline | Nicotine/Alcohol |
|---|---|---|---|---|
| Dorsomedial PFC | 100 ±8 | 85± 13 | 88 ±7 | 103 ±12 |
| Dorsal striatum | 100 ±20 | 116 ±24 | 108± 16 | 109 ±4 |
| Central amygdala | 100 ±9 | 81 ±7 | 94±18 | 91 ±13 |
Total protein levels are normalized to tubulin and expressed as percentage of drug-naive control.
Figure 6.
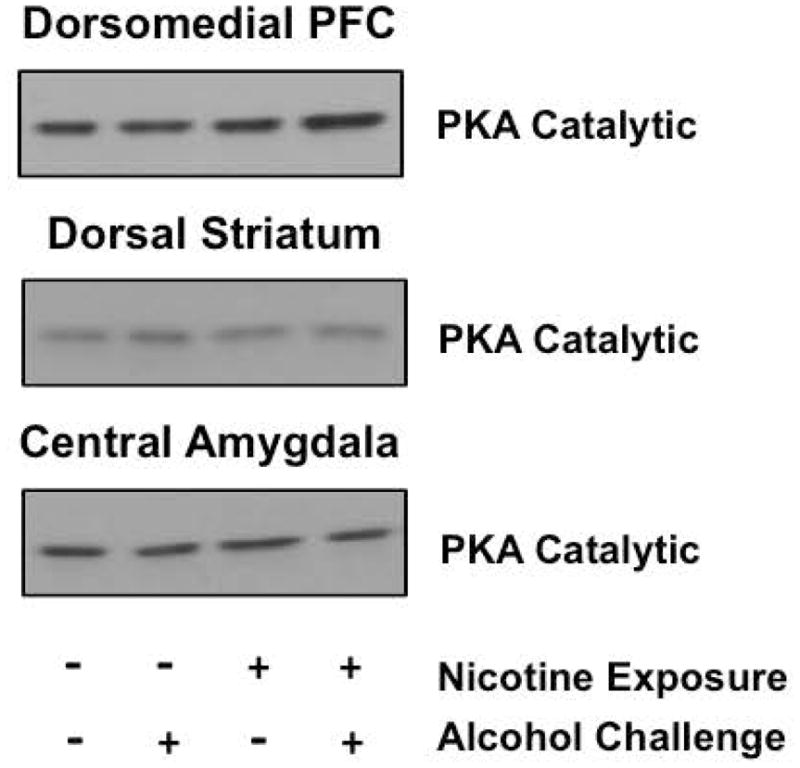
Levels of PKA catalytic subunits. No changes in PKA catalytic subunit immunoreactivity were evident in regions exhibiting tolerance to alcohol-stimulated, PKA-mediated GluA1 phosphorylation in animals with a history of nicotine exposure (see Table 1).
Discussion
Interactive Effects of Nicotine Withdrawal and Alcohol on Reward
The current investigation adds to our understanding of the interactive effects of nicotine and alcohol on glutamatergic signaling within reward-associated brain areas. Whereas alcohol challenge (1 g/kg) increases PKA phosphorylation of GluA1 in a variety of regions, this effect is blunted in the dorsomedial prefrontal cortex (Figure 3), dorsal striatum (Figure 4), and central amygdala (Figure 5) in nicotine-experienced animals. Previous studies have largely focused on brain regional activation one or more hours following acute alcohol administration as indicated by immediate early gene induction (for review, see Vilpoux et al., 2009). In the present study we focused on a much earlier time point and corresponding synaptic substrate (AMPA GluA1 channel phosphorylation) to model the neurobiological effects of acute binge-like intoxication, with or without a history of nicotine exposure, on excitatory signaling across multiple reward-related regions.
A considerable amount of research has described both rewarding and aversive properties associated with nicotine and alcohol, and this balance may change over time or under certain environmental conditions (Verendeev and Riley, 2012). Alcohol reward is mediated by multiple brain regions and likely results from an imbalance in the aversive and appetitive properties of alcohol. Results from our current study (Figures 2–4) lend further support to three brain regions (dorsomedial PFC, dorsal striatum, and central amygdala) as playing critical roles at the intersection of reward and aversion (Hayes et al, 2014). Nicotine-induced attenuation of the ability of alcohol to excite this network (as reflected by reduced GluA1 phosphorylation) may in part explain reports of enhanced alcohol reinforcement in nicotine-dependent animals (Doyon et al., 2013; Leao et al., 2015). Interestingly, activation of nicotinic acetylcholine receptors (nAChRs) in the dorsal hippocampus and basolateral amygdala was previously shown to potentiate alcohol reward (Zarrindast et al., 2010), and both of these areas failed to exhibit tolerance to alcohol-stimulated GluA1 phosphorylation following nicotine exposure in the present study. More definitive mechanisms underlying the magnification of alcohol reward/reinforcement in the context of nicotine dependence remain to be elucidated. For example, it is unknown whether chronic nicotine exposure might enhance alcohol sensitivity or efficacy. Future functional studies employing place conditioning and operant methodologies should shed further light on the specific interactions between alcohol and nicotine reward and reinforcement in relation to excitatory signaling mechanisms.
Altered PKA Signaling in Alcohol-Related Behaviors
As a critical regulator of AMPA function via GluA1 modification, the cyclic AMP-protein kinase A (PKA) signal transduction pathway has been implicated in numerous alcohol and drug dependence-related behaviors (Lee and Messing, 2008). Global disruption of PKA activity increases voluntary alcohol drinking (Thiele et al., 2000). Importantly, cAMP/PKA signaling appears to promote anxiolytic-like behaviors that facilitate excessive drinking (Pandey, 2003). In contrast, inhibition of PKA activity within the central amygdala produces anxiety-like behavior and increases alcohol consumption in non-dependent rodents (Pandey et al. 2003). Pandey and colleagues have demonstrated a role for downstream CREB phosphorylation by PKA and subsequent regulation of neuropeptide Y in these effects (Zhang and Pandey, 2003; Pandey et al., 2005; Zhang et al., 2010), although a similar blunting of alcohol-mediated regulation of PKA activity at the synaptic level following nicotine exposure (Figure 4) may also contribute to an enhanced motivation for alcohol. In the PFC, deficits in PKA signaling were previously shown to promote cognitive deficits associated with alcohol withdrawal, while restoration of cAMP/PKA activity ameliorated these deficits (Dominguez et al., 2016). In the current study, neuroadaptations in alcohol-stimulated PKA phosphorylation of GluA1 produced by nicotine experience were not associated with alterations in total PKA catalytic subunit levels (Figure 6 & Table 1) or alterations in total GluA1 levels, suggesting a more intricate mechanism responsible for this event. Representing one possible mechanism, chronic alcohol vapor exposure is known to increase expression of protein kinase A inhibitor (PKI) alpha in the medial prefrontal cortex and amygdala (Repunte-Canonigo et al., 2007), and a similar change may occur in chronic nicotine vapor-exposed animals to attenuate PKA activity.
Role of Corticostriatal and Corticoamygdalar Circuitry in Nicotine-Alcohol Interactions
A recent study identified discrete neurocircuitry changes associated with nicotine exposure during the transition to alcohol dependence (Leao et al., 2015). As mentioned previously, animals exposed to repeated nicotine injections significantly accelerated escalation of alcohol drinking. Interestingly, alcohol-dependent animals with a history of nicotine exposure displayed increased neuronal activity within the dmPFC and central amygdala as indicated by Fos immunoreactivity. Although the authors did not examine the dorsal striatum, they found similar effects within the more dorsal core subregion of the nucleus accumbens, further implicating dorsal corticostriatal circuitry in alcohol-nicotine interactions in accordance with our results from the dmPFC and dorsal striatum (Figures 2–3). It is possible that enhanced Fos activity in excitatory and/or inhibitory neurons within these brain regions mediates intracellular and/or circuit-level mechanisms of tolerance to excitatory signaling at the synapse following alcohol and nicotine exposure.
Importantly, connections between the PFC and central amygdala have been implicated in binge alcohol drinking (George et al., 2012). Previous work has also shown that nicotine exposure facilitates binge drinking in mice, with associated increases in nAChR levels in frontocortical areas (Locker et al., 2015). Accordingly, nAChR blockade reduces excessive drinking in both binge (Hendrickson et al., 2009) and dependence (Leao et al., 2015) models. The prefrontal cortex regulates a host of executive control functions hypothesized to be dysregulated in drug and alcohol addiction (Crews and Boettiger, 2009), while closely associated mechanisms of impulsivity associate with co-occuring smoking and binge drinking (VanderVeen et al., 2013). The transition to addiction in vulnerable individuals is thought to emerge in association with such cognitive dysfunction, particularly in terms of the various motivational subcomponents regulated by the PFC (George and Koob, 2010).
Over time, and in coordination with regulation of downstream regions (including the dorsal striatum and central amygdala), alterations in PFC function may mediate a progression from impulsive to compulsive drug seeking associated with uncontrollable drug use (Koob, 2009). Neurobiological factors that mediate the balance between alcohol reward and aversion in these or other regions (e.g., the rostromedial tegmental nucleus; Fu et al., 2016, or the lateral habenula; Zuo et al., 2015) may directly contribute to this process. Emerging data that demonstrate highly interactive effects of nicotine and alcohol on the brain support preclinical modeling and a clinical focus aimed at more integrated interventional strategies against problematic use of both drugs (Ames et al., 2014). The present study contributes to our understanding of the neurobiological effects of combined nicotine and alcohol use, and further highlights the potential of restoring disrupted glutamatergic signaling to treat addiction (Knackstedt et al., 2009).
Acknowledgments
This work was generously supported by research and training grants from the National Institute on Alcohol Abuse and Alcoholism (T32AA007577, MAM, AMW, CAI; R01AA023305, NWG; R00AA020839, SE) and by LSUHSC School of Medicine start-up funds. The authors declare no biomedical financial interests or potential conflicts of interest.
References
- Ames SC, Pokorny SB, Schroeder DR, Tan W, Werch CE. Integrated smoking cessation and binge drinking intervention for young adults: a pilot efficacy trial. Addict Behav. 2014;39:848–853. doi: 10.1016/j.addbeh.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Baiamonte BA, Valenza M, Roltsch EA, Whitaker AM, Baynes BB, Sabino V, Gilpin NW. Nicotine dependence produces hyperalgesia: role of corticotropin-releasing factor-1 receptors (CRF1Rs) in the central amygdala (CeA) Neuropharmacology. 2014;77:217–223. doi: 10.1016/j.neuropharm.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Edwards S, Graham DL, Larson EB, Whisler KN, Simmons D, Friedman AK, Walsh JJ, Rahman Z, Monteggia LM, Eisch AJ, Neve RL, Nestler EJ, Han MH, Self DW. Reinforcement-related regulation of AMPA glutamate receptor subunits in the ventral tegmental area enhances motivation for cocaine. J Neurosci. 2011;31:7927–7937. doi: 10.1523/JNEUROSCI.6014-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Treweek J, Edwards S, Leao RM, Schulteis G, Koob GF, George O. Extended access to nicotine leads to a CRF1 receptor dependent increase in anxiety-like behavior and hyperalgesia in rats. Addict Biol. 2015;20:56–68. doi: 10.1111/adb.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Boettiger CA. Impulsivity, frontal lobes and risk for addiction. Pharmacol Biochem Behav. 2009;93:237–247. doi: 10.1016/j.pbb.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez G, Dagnas M, Decorte L, Vandesquille M, Belzung C, Beracochea D, Mons N. Rescuing prefrontal cAMP-CREB pathway reverses working memory deficits during withdrawal from prolonged alcohol exposure. Brain Struct Funct. 2016;221:865–877. doi: 10.1007/s00429-014-0941-3. [DOI] [PubMed] [Google Scholar]
- Doyon WM, Dong Y, Ostroumov A, Thomas AM, Zhang TA, Dani JA. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron. 2013;79:530–540. doi: 10.1016/j.neuron.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Bachtell RK, Guzman D, Whisler KN, Self DW. Emergence of context-associated GluR(1) and ERK phosphorylation in the nucleus accumbens core during withdrawal from cocaine self-administration. Addict Biol. 2011;16:450–457. doi: 10.1111/j.1369-1600.2010.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Graham DL, Bachtell RK, Self DW. Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci. 2007;25:2201–2213. doi: 10.1111/j.1460-9568.2007.05473.x. [DOI] [PubMed] [Google Scholar]
- Edwards S, Koob GF. Neurobiology of dysregulated motivational systems in drug addiction. Future Neurol. 2010;5:393–401. doi: 10.2217/fnl.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Vendruscolo LF, Schlosburg JE, Misra KK, Wee S, Park PE, Schulteis G, Koob GF. Development of mechanical hypersensitivity in rats during heroin and ethanol dependence: alleviation by CRF(1) receptor antagonism. Neuropharmacology. 2012;62:1142–1151. doi: 10.1016/j.neuropharm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Zuo W, Gregor D, Li J, Grech D, Ye JH. Pharmacological Manipulation of the Rostromedial Tegmental Nucleus Changes Voluntary and Operant Ethanol Self-Administration in Rats. Alcohol Clin Exp Res. 2016;40:572–582. doi: 10.1111/acer.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk D, Marinelli PW, Le AD. Biological processes underlying co-use of alcohol and nicotine: neuronal mechanisms, cross-tolerance, and genetic factors. Alcohol Res Health. 2006;29:186–192. [PMC free article] [PubMed] [Google Scholar]
- George O, Grieder TE, Cole M, Koob GF. Exposure to chronic intermittent nicotine vapor induces nicotine dependence. Pharmacol Biochem Behav. 2010;96:104–107. doi: 10.1016/j.pbb.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Koob GF. Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2010;35:232–247. doi: 10.1016/j.neubiorev.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Whitaker AM, Baynes B, Abdel AY, Weil MT, George O. Nicotine vapor inhalation escalates nicotine self-administration. Addict Biol. 2014;19:587–592. doi: 10.1111/adb.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DJ, Duncan NW, Xu J, Northoff G. A comparison of neural responses to appetitive and aversive stimuli in humans and other mammals. Neurosci Biobehav Rev. 2014;45:350–368. doi: 10.1016/j.neubiorev.2014.06.018. [DOI] [PubMed] [Google Scholar]
- Hendrickson LM, Zhao-Shea R, Tapper AR. Modulation of ethanol drinking-in-the-dark by mecamylamine and nicotinic acetylcholine receptor agonists in C57BL/6J mice. Psychopharmacology (Berl) 2009;204:563–572. doi: 10.1007/s00213-009-1488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, Markou A, Kalivas PW. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AD, Wang A, Harding S, Juzytsch W, Shaham Y. Nicotine increases alcohol self-administration and reinstates alcohol seeking in rats. Psychopharmacology (Berl) 2003;168:216–221. doi: 10.1007/s00213-002-1330-9. [DOI] [PubMed] [Google Scholar]
- Leao RM, Cruz FC, Vendruscolo LF, de Guglielmo G, Logrip ML, Planeta CS, Hope BT, Koob GF, George O. Chronic nicotine activates stress/reward-related brain regions and facilitates the transition to compulsive alcohol drinking. J Neurosci. 2015;35:6241–6253. doi: 10.1523/JNEUROSCI.3302-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Messing RO. Protein kinases and addiction. Ann N Y Acad Sci. 2008;1141:22–57. doi: 10.1196/annals.1441.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton J, Barron S, Prendergast M, Nixon SJ. Smoking kills (alcoholics)! shouldn’t we do something about it? Alcohol Alcohol. 2007;42:167–173. doi: 10.1093/alcalc/agm019. [DOI] [PubMed] [Google Scholar]
- Locker AR, Marks MJ, Kamens HM, Klein LC. Exposure to nicotine increases nicotinic acetylcholine receptor density in the reward pathway and binge ethanol consumption in C57BL/6J adolescent female mice. Brain Res Bull. 2015 doi: 10.1016/j.brainresbull.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute on Alcohol Abuse and Alcoholism. NIAAA Newsletter. 3. National Institute on Alcohol Abuse and Alcoholism; Bethesda, MD: 2004. NIAAA Council Approves Definition of Binge Drinking. [Google Scholar]
- Pandey SC. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci. 2003;24:456–460. doi: 10.1016/S0165-6147(03)00226-8. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H. The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol Clin Exp Res. 2003;27:396–409. doi: 10.1097/01.ALC.0000056616.81971.49. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Xu T. Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest. 2005;115:2762–2773. doi: 10.1172/JCI24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Lutjens R, van der Stap LD, Sanna PP. Increased expression of protein kinase A inhibitor alpha (PKI-alpha) and decreased PKA-regulated genes in chronic intermittent alcohol exposure. Brain Res. 2007;1138:48–56. doi: 10.1016/j.brainres.2006.09.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res. 2012;36:61–71. doi: 10.1111/j.1530-0277.2011.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BR, Horan JT, Gaskin S, Amit Z. Exposure to nicotine enhances acquisition of ethanol drinking by laboratory rats in a limited access paradigm. Psychopharmacology (Berl) 1999;142:408–412. doi: 10.1007/s002130050906. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci. 2000;20:RC75. doi: 10.1523/JNEUROSCI.20-10-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Skike CE, Maggio SE, Reynolds AR, Casey EM, Bardo MT, Dwoskin LP, Prendergast MA, Nixon K. Critical needs in drug discovery for cessation of alcohol and nicotine polysubstance abuse. Prog Neuropsychopharmacol Biol Psychiatry. 2016;65:269–287. doi: 10.1016/j.pnpbp.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVeen JW, Cohen LM, Watson NL. Utilizing a multimodal assessment strategy to examine variations of impulsivity among young adults engaged in co-occurring smoking and binge drinking behaviors. Drug Alcohol Depend. 2013;127:150–155. doi: 10.1016/j.drugalcdep.2012.06.026. [DOI] [PubMed] [Google Scholar]
- Verendeev A, Riley AL. Conditioned taste aversion and drugs of abuse: history and interpretation. Neurosci Biobehav Rev. 2012;36:2193–2205. doi: 10.1016/j.neubiorev.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Vilpoux C, Warnault V, Pierrefiche O, Daoust M, Naassila M. Ethanol-sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res. 2009;33:945–969. doi: 10.1111/j.1530-0277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X, Choe ES, Mao L. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32:237–249. doi: 10.1385/MN:32:3:237. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Mangiavacchi S, Sun X. Mechanisms by which dopamine receptors may influence synaptic plasticity. Ann N Y Acad Sci. 2003;1003:241–249. doi: 10.1196/annals.1300.015. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Meshkani J, Rezayof A, Beigzadeh R, Rostami P. Nicotinic acetylcholine receptors of the dorsal hippocampus and the basolateral amygdala are involved in ethanol-induced conditioned place preference. Neuroscience. 2010;168:505–513. doi: 10.1016/j.neuroscience.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Zhang H, Pandey SC. Effects of PKA modulation on the expression of neuropeptide Y in rat amygdaloid structures during ethanol withdrawal. Peptides. 2003;24:1397–1402. doi: 10.1016/j.peptides.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sakharkar AJ, Shi G, Ugale R, Prakash A, Pandey SC. Neuropeptide Y signaling in the central nucleus of amygdala regulates alcohol-drinking and anxiety-like behaviors of alcohol-preferring rats. Alcohol Clin Exp Res. 2010;34:451–461. doi: 10.1111/j.1530-0277.2009.01109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Fu R, Hopf FW, Xie G, Krnjevic K, Li J, Ye JH. Ethanol drives aversive conditioning through dopamine 1 receptor and glutamate receptor-mediated activation of lateral habenula neurons. Addict Biol. 2015 doi: 10.1111/adb.12298. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]