

Abstract
Carbapenem-resistant Enterobacteriaceae (CRE) are resistant to most β-lactam antibiotics due to production of the KPC-2 class A β-lactamase. Here we present the first product complex crystal structures of KPC-2 with β-lactam antibiotics, containing hydrolyzed cefotaxime and faropenem. They provide experimental insights into substrate recognition by KPC-2 and its unique cephalosporinase/carbapenemase activity. These structures also represent the first product complexes for a wild type serine β-lactamase, elucidating the product release mechanism of these enzymes in general.
Graphical Abstract
INTRODUCTION
Each year in the United States, an estimated 2 million people are infected with an antibiotic resistant strain of bacteria, resulting in approximately 23,000 deaths.1 Among all of the bacterial resistance problems, Gram-negative bacterial pathogens pose the greatest threat since they have acquired resistance to many antibiotics, including the β-lactam antibiotics. β-Lactam antibiotics are the most widely used antibiotics in the treatment of a diverse range of bacterial infections.2 These compounds irreversibly inhibit the penicillin-binding proteins (PBPs), which are enzymes that catalyze the transpeptidation reaction that cross-links the bacterial cell wall.3 The predominant mechanism of β-lactam resistance involves the bacterial production of β-lactamases, enzymes that catalyze β-lactam hydrolysis. β-Lactamases are divided into four classes based on sequence homology and mechanism of action. Class A, C, and D β-lactamases use an active site serine residue to carry out β-lactam hydrolysis, whereas class B β-lactamases are metallo-enzymes that require zinc for their activity.4–6
Since the introduction of penicillin G in the early 1940s, and the emergence of resistance a few years later, numerous semisynthetic β-lactam antibiotics, such as cephalosporins, carbapenems, and penems were developed that proved to be more resilient to β-lactamase mediated hydrolysis (Fig. 1).7 However, as previously observed, resistance to these compounds is now extensive.8 CTX-M β-lactamases, which are members of the extended-spectrum β-lactamases (ESBLs), are the most widespread ESBLs and confer resistance to the third-generation cephalosporins.6,9,10 Recently, it has been observed that some class A, B, and D β-lactamases possess carbapenemase activity, allowing them to hydrolyze carbapenems, the current drugs of last resort in the treatment of multi-drug resistant bacterial infections.5
Figure 1.

Structures of cephalosporins (cephalothin, cefotaxime), carbapenem (meropenem), and penem (faropenem) β-lactam antibiotics.
The Klebsiella pneumoniae carbapenemase (KPC) class A β-lactamase poses a serious threat to nearly all β-lactam antibiotics.11 The first member of the KPC family was identified in North Carolina in 1996 and has since spread to many other countries.12 Although initially identified in K. pneumoniae, KPC has also been discovered in other Gram-negative pathogens, mainly belonging to the Enterobacteriaceae family (Carbapenem-resistant Enterobacteriaceae or CRE).13 The blaKPC gene encodes a 293 amino acid enzyme, and there are 23 variants of KPC (KPC-2 through KPC-24) that differ from one another by one or two amino acid changes.14 KPC-2 is the most prevalent carbapenemase in the United States and it has been termed the “versatile β-lactamase” due to its large and shallow active site, allowing it to efficiently hydrolyze virtually all β-lactam antibiotics.15 However, the question still remains as to what specific interactions between β-lactams and the KPC-2 active site allows this enzyme to possess such a broad substrate profile.16,17 The hydrolysis reaction catalyzed by class A β-lactamases proceeds through two steps, acylation and deacylation.13 The attack of Ser70 on the substrate β-lactam carbonyl results in a covalent acyl-enzyme complex. Subsequently, the catalytic water, activated by Glu166, cleaves the acyl-enzyme bond, leading to the formation of the hydrolyzed product. One of the most common ways to capture a β-lactam complex along the reaction pathway of class A β-lactamases is to mutate a key catalytic residue such as Ser70 or Glu166 in order to accommodate the newly generated carboxylate group.18 The complex structures presented herein represent the first product complexes using a wild type (WT) serine β-lactamase, with all native active site residues intact. This provides an accurate picture of the protein microenvironment that is responsible for catalysis, particularly during product release.
RESULTS AND DISCUSSION
We attempted to obtain complex crystal structures with a variety of β-lactam antibiotics which include the cephalosporins ceftazidime, cefotaxime, cefoxitin, and nitrocefin; the penicillins ampicillin, cloxacillin, and penicillin G; the carbapenems biapenem, imipenem, and meropenem; the penem faropenem; and the monobactam aztreonam. Ultimately, we were able to obtain two crystal structures of the third-generation cephalosporin, cefotaxime, and the penem, faropenem, which is structurally similar to the carbapenems, as hydrolyzed products in the active site of KPC-2 (Fig. 1). As discussed therein, the unique features of both KPC-2 and the two substrates may have contributed to the capture of these two complexes.
Product complex with cefotaxime
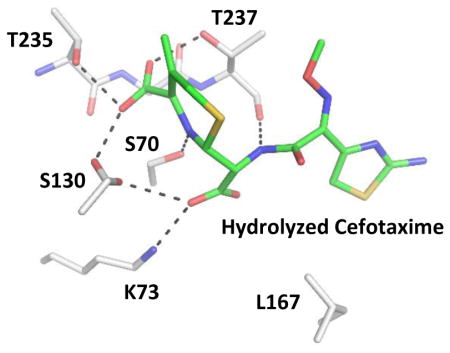
KPC-2 was crystallized in the space group P22121 with one copy of KPC-2 in the asymmetric unit. The crystals routinely diffracted to 1.15–1.5 Å resolution. The apo-structure is similar to previously determined KPC-2 models crystallized in different space groups (Fig. S1).19,20 The complex structure with cefotaxime was determined to 1.45 Å resolution with final Rwork and Rfree values of 16.8% and 21.2%, respectively. The electron density for the product is well defined in the active site as seen in the unbiased Fo-Fc map (Fig. 2A). The occupancy value of the hydrolyzed product was refined to 0.86. There are some differences in the active site when comparing the cefotaxime complex structure to the apo-enzyme, mainly with the residues Ser70, Trp105, and Leu167 (Fig. 2B), due to interactions between the hydrolyzed product and the enzyme. The complex structure illustrates how the bulky oxyimino group of third-generation cephalosporins and the newly generated carboxylate group of the product are accommodated by the KPC-2 active site. It represents the first experimental structure of KPC-2 in complex with a β-lactam antibiotic and sheds new light on the extensive interactions between cefotaxime and the active site residues. The C4 carboxylate group resides in a subpocket formed by Thr235, Gly236, Thr237, and Ser130. This site is highly conserved in serine β-lactamases and PBPs, as shown by previous crystallographic analyses of these enzymes and their complexes.21,22 The six-membered dihydrothiazine group of the hydrolyzed product forms a π–π stacking interaction with Trp105, which has been demonstrated to be important in substrate recognition. 23 The interactions with the substrate also result in a single conformation of Trp105, which is observed to have two conformations in the apo-enzyme (Fig. 2B). Meanwhile, cefotaxime’s acylamide side chain is nestled in a pocket formed by Thr237, Cys238, Gly239, Leu167, and Asn170 (Fig. 2A). The aminothiazole moiety forms extensive non-polar interactions with Leu167, in addition to van der Waals contacts with Asn170, Cys238 and Gly239. Particularly, compared with the apo-structure, the alkyl side chain of Leu167 moves closer to the substrate (Fig. 2B). In comparison, the oxyimino group is largely solvent exposed and establishes relatively few interactions with the protein, mainly with Thr237Cγ2 and Gly239Cα (Fig. 2A).
Figure 2.

KPC-2 cefotaxime product complex. The protein and ligand of the complex are shown in white and yellow, respectively. Hydrogen bonds are shown as black dashed lines. (A) The unbiased Fo-Fc electron density map (gray) of hydrolyzed cefotaxime contoured to 2σ. (B) Superimposition of KPC-2 product complex onto apo-protein (green) with details showing the interactions involving Ser70, Lys73, the cefotaxime ring nitrogen, and the newly formed C8 carboxylate group. PyMOL alignment RMSD = 0.153 Å over 272 residues. (C) Superimposition of KPC-2 product complex and CTX-M-14 E166A penilloate product complex with a ruthenocene-conjugated penicillin (green, with residues unique to CTX-M-14 E166A labeled in green), in stereo view. The ruthenium atom is shown as a gray sphere. PyMOL alignment RMSD = 0.617 Å over 272 residues. (D) Superimposition of KPC-2 product complex with Toho-1 E166A acyl-enzyme complex with cefotaxime (green, with residues unique to Toho-1 E166A labeled in green). PyMOL alignment RMSD = 0.602 Å over 264 residues.
As the first product complex with a WT serine β-lactamase, our KPC-2 structure also captures structural features not seen in previous β-lactamase complexes, particularly concerning the conformations of Ser70, Lys73, Ser130, and the substrate carbonyl group. In class A β-lactamases, Ser70 behaves as a nucleophile that carries out attack on the β-lactam ring of the substrate.24 In the apo-enzyme, Ser70 forms a hydrogen bond (HB) with Lys73, which has been suggested as a potential base in the acylation reaction.25,26 In our structure, the presence of the newly generated C8 carboxylate group, a result of the catalytic water’s attack on the carbon atom of the acyl-enzyme carbonyl group, causes Ser70 to adopt a conformation that places its side chain hydroxyl group in the oxyanion hole formed by the backbone amide groups of Ser70 and Thr237, and previously occupied by the carbonyl oxygen of the acyl-enzyme intermediate (Fig. 2B, 2D). This conformational change abolishes the HB between Ser70 and Lys73 and establishes a new HB between Ser70 and the ring nitrogen, an interaction not observed at any other stage of the reaction (Fig. 2B). Meanwhile, the Lys73 side chain moves to form HB interactions with Ser130. Unlike most previous β-lactamase structures, Ser130 adopts two conformations (Fig. 2A).27 Conformation 1 maintains a weak HB with Lys73, is in close proximity to the ring nitrogen, and is the conformation usually observed in class A β-lactamases;28 conformation 2 swings closer to Lys234 and the substrate C4 carboxylate group, establishing more favorable HBs with the latter two functional groups. Additionally, the amide nitrogen of the cefotaxime product forms a weak HB with the backbone carbonyl group of Thr237 (Fig. 2A). In our previous CTX-M-14 E166A complex structure with a ruthenocene-conjugated penicillin (PDB entry 4XXR)29, Ser130 adopts conformation 1, and serves as a HB acceptor and donor, respectively, in its interactions with a protonated ring nitrogen and a neutral Lys73 (Fig. 2C). In the current structure, Lys73 appears to be protonated and is within HB distance (2.6–2.9 Å) to three HB acceptors, including Ser130O, Asn132Oδ1, and the substrate C8 carboxylate group. In comparison, the distance between Lys73Nζ and Ser130Oγ of conformation 1 is 3.1 Å, suggesting a weak HB contact. Ser130Oγ of conformation 1 is also too far from the ring nitrogen (3.5 Å) for a HB. The weakened interactions between Ser130 and Lys73 or the substrate ring nitrogen are likely part of the reason for Ser130’s adoption of conformation 2, which highlights Ser130’s role in binding the substrate carboxylate group. Taken together, the alternative conformations of Ser70, Lys73, and Ser130 underscore the important non-catalytic roles these residues in the product release process. Previous studies have suggested that steric clash and electrostatic repulsion caused by the newly generated C8 carboxylate group of cefotaxime is responsible for the expulsion of the hydrolyzed product from the enzyme active site.18,30 Our structure supports this hypothesis, while providing important new structural details on potential unfavorable interactions that lead to the release of the product. In our structure, possible clashes between the C8 carboxylate group and Ser70 are alleviated by Ser70’s adoption of an alternative, and likely high energy, conformation with a side chain χ1 angle of 10°, in comparison to the more favorable 60° in Ser70’s usual conformation. In the complex structure of AmpC class C β-lactamase S64G mutant with hydrolyzed cephalothin, the electrostatic repulsion between the C8 and C4 carboxylate groups caused the C4 carboxylate group to move out of the active site, resulting from the rotation of the dihydrothiazine ring and leading to an increase in distance between these two negatively charged groups.31 In our structure, the C4 carboxylate group remains in the active site, albeit in a likely less stable state due to the electrostatic repulsion. Although the C8 carboxylate group can establish new interactions with Lys73, these contacts are accompanied by the loss of HBs between the substrate and the oxyanion hole, between Ser70 and Lys73, and for class A β-lactamases, by possibly additional repulsion between the C8 carboxylate group and Glu166 (Fig. 2B). Another unique feature of the product complex is the lack of a HB between the substrate amide group with Asn132, even though the substrate amide group forms a HB with the backbone O atom of Thr237 (Fig. 2A). The HB with Asn132 is present in nearly all complex structures of serine β-lactamases with β-lactam compounds containing the amide group, including the aforementioned CTX-M-14 E166A product complex and a Toho-1 E166A/cefotaxime complex (PDB entry: 1IYO) structure (Fig. 2D)9,18,29 Instead, in this structure, the carbonyl oxygen is pushed out of the active site and exposed to the solvent (Fig. 2C, 2D). The loss of the HB between the ligand amide group and Asn132 further suggests that the interactions between the product and the enzyme are less favorable compared with the Michaelis substrate complex, which may facilitate substrate turnover during catalysis.
We observed an additional molecule of hydrolyzed cefotaxime near the active site with a refined occupancy value of 0.72 (Fig. S2). This molecule did not make as many interactions in the active site as the other hydrolyzed molecule. There is a HB between Lys270 and the C4 carboxylate group of the second hydrolyzed product, as well as a HB between the aminothiazole group of the first hydrolyzed product and the C4 carboxylate of the second hydrolyzed product. We believe that the presence of this second hydrolyzed product is more of a crystal-packing artifact than it is a species that plays a role in catalysis. It might, however, have partially stabilized the first hydrolyzed product inside the active site in the crystal.
Product complex with faropenem
Faropenem belongs to the penem class of β-lactam antibiotics (Fig. 1). Penems share structural characteristics of both penicillins and cephalosporins.32 Like carbapenems, penems have a hydroxyethyl group attached to the β-lactam ring in the trans-configuration that provides stability against many β-lactamases.5 We captured a single hydrolyzed molecule of faropenem in the active site of KPC-2 (Fig. 3A). The complex structure with faropenem was determined to 1.40 Å resolution with final Rwork and Rfree values of 15.0% and 18.8%, respectively. The occupancy value of the hydrolyzed product was refined to 0.84. Compared to the product complex with cefotaxime, the hydrolyzed faropenem induces similar conformations of Ser70 and Trp105, and makes many similar interactions in the active site (Fig. 3B). For instance, the C7 carboxylate group, which is analogous to the cefotaxime C8 carboxylate group, forms HBs with Ser130, Thr235, and Thr237; conformation 2 of Ser130 forms a HB with the C3 carboxylate group, which is analogous to the cefotaxime C4 carboxylate group (Fig. 3A). Unlike the cefotaxime complex, the ring nitrogen forms a HB with conformation 1 of Ser130, rather than Ser70, likely a result of the five-membered dihydrothiazole ring in faropenem, in comparison to the six-membered dihydrothiazine ring in cefotaxime.
Figure 3.

KPC-2 faropenem product complex. The protein and ligand of the faropenem product complex are shown in white and yellow, respectively. Hydrogen bonds are shown as black dashed lines. (A) The unbiased Fo-Fc electron density map (gray) of hydrolyzed faropenem contoured at 2σ. (B) Superimposition of KPC-2 faropenem complex onto apo-protein (green) with details showing the interactions involving Ser70, Lys73, and the newly formed faropenem C7 carboxylate group. PyMOL alignment RMSD = 0.141 Å over 270 residues. (C) Superimposition of KPC-2 product complex with SED-1 faropenem acyl-enzyme (green) in stereo view. The deacylation water (red sphere) from SED-1 is shown making interactions with Glu166 and the hydroxyethyl group of faropenem. PyMOL alignment RMSD = 0.555 Å over 262 residues. (D) Superimposition of faropenem/cefotaxime product complexes, showing the movement of the Pro104-Trp105 loop and alternative conformations for Val103 and Leu167 (KPC-2/faropenem: white/yellow; KPC-2/cefotaxime: green). PyMOL alignment RMSD = 0.101 Å over 270 residues.
One important observation in this faropenem structure is the conformation of the hydroxyethyl side chain. The faropenem structure shows that the hydroxyethyl side chain has undergone rotation, breaking its interaction with Asn132. Previous studies have shown that in non-carbapenemases, such as SED-1 (PDB entry: 3BFF), the hydroxyethyl group of carbapenems forms a HB with Asn132 and the deacylating water, consequently deactivating the deacylating water molecule and leaving these enzymes unable to hydrolyze the acyl-enzyme linkage.5 However, in carbapenemases like KPC-2, the active site is enlarged, permitting rotation of the hydroxyethyl group, thus abolishing its contact with Asn132 and allowing efficient hydrolysis of carbapenems (Fig. 3C).5,33–35 Importantly, in the current structure, Leu167 is in favorable van der Waal contact with the methyl group of faropenem’s hydroxyethyl moiety, suggesting that Leu167 may play an active role in inducing the rotation of this side chain group to facilitate hydrolysis. Although position 167 is not well conserved among class A β-lactamases, it appears that a leucine is conserved at this position among class A carbapenemases.21 When comparing the cefotaxime and faropenem product complexes, movements are observed in Val103, the Pro104-Trp105 loop, and Leu167 (Fig. 3D). These movements are likely due to the different interactions between the faropenem tetrahydrofuran group and Trp105 and between the cefotaxime aminothiazole group and Leu167. In addition, due to the different chiralities of the C6/C7 atoms linked to the C7/C8 carboxylate group, the newly generated carboxylate group is positioned slightly differently between the two product complexes. Taken together, these findings highlight the flexibility of the KPC-2 active site in accommodating a variety of β-lactam side chains.
Unique KPC-2 structural features for inhibitor discovery
Despite extensive structural analysis of numerous serine β-lactamases, our structures represent the only product complexes with a WT enzyme. All previous studies relied on mutations, such as Ser70Gly to stabilize the interactions between the product and the protein active site.18 The many unfavorable features of the enzyme-product contacts have made it difficult to capture such complexes with WT enzymes, even for KPC-2, as demonstrated by our failures to obtain similar complexes using many other β-lactam antibiotics. The successes with our current two complexes may have been due to particular functional groups of these two substrates and some unique properties of carbapenemases such as KPC-2. In comparison to other β-lactam compounds, the oxyimino side chain of cefotaxime and the tetrahydrofuran ring of faropenem enhance the interactions between the product and KPC-2, whereas avoiding excessive bulkiness that may cause steric clashes with protein residues in a crystal packing environment. More importantly, compared with narrow-spectrum β-lactamases (e.g., TEM-1) and ESBLs (e.g., CTX-M-14), KPC-2 and other class A carbapenemases contain several key features and residues that result in an expanded active site, including a Cys69-Cys238 disulfide bond, Leu167, Pro104, and Trp105. Movements of Ser70 and Asn170 are also observed in the KPC-2 structure. There is a larger distance between Ser70 and Asn170 in KPC-2 as compared to in non-carbapenemases like TEM-1 and CTX-M-14, which may facilitate rotation of the hydroxyethyl group that gives carbapenems their stability.19 Importantly, the KPC-2 active site also appears to be more hydrophobic, with larger non-polar surfaces provided by Pro104, Trp105, Leu167, and Val240, in comparison to their counterparts in TEM-1: Glu104, Tyr105, Pro167, and Asp240 (Fig. 4A); and also Asn104, Tyr105, Pro167, and Asp240 in CTX-M-14 (Fig. 4B). These features may allow KPC-2 to bind to a wide range of β-lactam substrates with a relatively open active site. They are mirrored by similar observations in metallo-β-lactamases that also harbor expanded active sites with a large number of hydrophobic residues.36 Such features may enable carbapenemases to hydrolyze nearly all β-lactam antibiotics, while also exposing a weakness that can facilitate the engineering of high affinity inhibitors against these enzymes.
Figure 4.

Comparison of KPC-2 active site with the narrow-spectrum β-lactamase, TEM-1 and the extended-spectrum beta-lactamase (ESBL), CTX-M-14. (A) KPC-2 complexed with cefotaxime superimposed onto TEM-1 (KPC-2: white, TEM-1: green, cefotaxime: yellow). (B) KPC-2 complexed with faropenem superimposed onto CTX-M-14 (KPC-2: white, CTX-M-14: green, faropenem: yellow).
CONCLUSION
In this work, we present the first crystal structures of the KPC-2 class A β-lactamase in complex with two clinically used β-lactam antibiotics. The structures underscore the role of the expanded and shallow active site in accommodating the bulky cefotaxime oxyimino side chain and the rotation of penem’s 6α-1R-hydroxyethyl group, and particularly Leu167’s critical contribution. The structures demonstrate how alternative conformations of Ser70 and Lys73 facilitate product expulsion, and capture unique substrate conformations at this important milestone of the reaction. Lastly, the structures highlight the increased druggability of the KPC-2 active site, which provides an evolutionary advantage for the enzyme’s catalytic versatility, but also an opportunity for drug discovery.
Supplementary Material
Acknowledgments
We would like to thank Dr. Eric Lewandowski for reading the manuscript. This project was supported by the NIH (AI103158).
ABBREVIATIONS USED
- CRE
Carbapenem-resistant Enterobacteriaceae
- HB
Hydrogen bond
- PBP
penicillin-binding protein
- ESBL
extended-spectrum beta-lactamase
Footnotes
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession codes 5UL8 (apo KPC-2), 5UJ3 (KPC-2/hydrolyzed cefotaxime complex) and 5UJ4 (KPC-2/hydrolyzed faropenem complex).
ORCID
Yu Chen: 0000-0002-5115-3600
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the manuscript.
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Details of the cloning, expression, protein purification, crystallization, X-ray data collection and refinement statistics, superimposition of apo KPC-2 structures, additional copy of hydrolyzed cefo-taxime molecule in the KPC-2 active site, and molecular interactions of the hydrolyzed products.
References
- 1.Sharma VK, Johnson N, Cizmas L, Mcdonald TJ, Kim H. A review of the influence of treatment strategies on antibiotic resistant bacteria and antibiotic resistance genes. Chemosphere. 2016;150:702–714. doi: 10.1016/j.chemosphere.2015.12.084. [DOI] [PubMed] [Google Scholar]
- 2.Cho H, Uehara T, Bernhardt TG. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159(6):1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang YH, Labgold MR, Richards JH. Altering enzymatic activity: Recruitment of carboxypeptidase activity into an RTEM beta-lactamase/penicillin-binding protein 5 chimera. Proc Natl Acad Sci USA. 1990;87(7):2823–2827. doi: 10.1073/pnas.87.7.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Queenan AM, Bush K. Carbapenemases: The versatile β-lactamases. Clin Microbiol Rev. 2007;20(3):440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Carbapenems: Past, present, and future. Antimicrob Agents Chemother. 2011;55(11):4943–4960. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 2009;54(3):969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74(3):417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson KS. Extended-spectrum-β-lactamase, AmpC, and carbapenemase issues. J Clin Microbiol. 2010;48(4):1019–1025. doi: 10.1128/JCM.00219-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimamura T, Ibuka A, Fushinobu S, Wakagi T, Ishiguro M, Ishii Y, Matsuzawa H. Acyl-intermediate structures of the extended-spectrum class A β-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J Biol Chem. 2002;277(48):46601–46608. doi: 10.1074/jbc.M207884200. [DOI] [PubMed] [Google Scholar]
- 10.Adamski CJ, Cardenas AM, Brown NG, Horton LB, Sankaran B, Prasad BVV, Gilbert HF, Palzkill T. Molecular basis for the catalytic specificity of the CTX-M extended-spectrum β-lactamases. Biochemistry. 2015;54(2):447–457. doi: 10.1021/bi501195g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordmann P, Naas T, Poirel L. Global spread of carbapenemase-producing. Enterobacteriaceae Emerging Infect Dis. 2011;17(10):1791–1798. doi: 10.3201/eid1710.110655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown NG, Chow DC, Palzkill T. BLIP-II is a highly potent inhibitor of Klebsiella pneumoniae carbapenemase (KPC-2) Antimicrob Agents Chemother. 2013;57(7):3398–3401. doi: 10.1128/AAC.00215-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walther-Rasmussen J, Hoiby N. Class A carbapenemases. J Antimicrob Chemother. 2007;60(3):470–482. doi: 10.1093/jac/dkm226. [DOI] [PubMed] [Google Scholar]
- 14.Mehta SC, Rice K, Palzkill T. Natural variants of the KPC-2 carbapenemase have evolved increased catalytic efficiency for ceftazidime hydrolysis at the cost of enzyme stability. PLoS Pathog. 2015;11(6):e1004949. doi: 10.1371/journal.ppat.1004949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papp-Wallace KM, Winkler ML, Taracila MA, Bonomo RA. Variants of β lactamase KPC-2 that are resistant to inhibition by avibactam. Antimicrob Agents Chemother. 2015;59(7):3710–3717. doi: 10.1128/AAC.04406-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta N, Limbago BM, Patel JB, Kallen AJ. Carbapenem-resistant Enterobacteriaceae: Epidemiology and prevention. Clin Infect Dis. 2011;53(1):60–67. doi: 10.1093/cid/cir202. [DOI] [PubMed] [Google Scholar]
- 17.Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA. Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob Agents Chemother. 2009;54(2):890–897. doi: 10.1128/AAC.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delmas J, Leyssene D, Dubois D, Birck C, Vazeille E, Robin F, Bonnet R. Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J Mol Biol. 2010;400(1):108–120. doi: 10.1016/j.jmb.2010.04.062. [DOI] [PubMed] [Google Scholar]
- 19.Ke W, Bethel CR, Thomson JM, Bonomo RA, Akker FVD. Crystal structure of KPC-2: Insights into carbapenemase activity in class A β-lactamases. Biochemistry. 2007;46(19):5732–5740. doi: 10.1021/bi700300u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrella S, Ziental-Gelus N, Mayer C, Renard M, Jarlier V, Sougakoff W. Genetic and structural insights into the dissemination potential of the extremely broad-spectrum class A β-lactamase KPC-2 identified in an Escherichia coli strain and an Enterobacter cloacae strain isolated from the same patient in France. Antimicrob Agents Chemother. 2008;52(10):3725–3736. doi: 10.1128/AAC.00163-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majiduddin FK, Palzkill T. Amino acid residues that contribute to substrate specificity of class A β-lactamase SME-1. Antimicrob Agents Chemother. 2005;49(8):3421–3427. doi: 10.1128/AAC.49.8.3421-3427.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massova I, Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papp-Wallace KM, Taracila M, Wallace CJ, Hujer KM, Bethel CR, Hornick JM, Bonomo RA. Elucidating the role of Trp105 in the KPC-2 β-lactamase. Protein Sci. 2010;19(9):1714–1727. doi: 10.1002/pro.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minasov G, Wang X, Shoichet BK. An ultrahigh resolution structure of TEM-1 β-lactamase suggests a role for Glu166 as the general base in acylation. J Am Chem Soc. 2002;124(19):5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 25.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. Ab initio QM/MM study of class A β-lactamase acylation: Dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc. 2005;127(44):15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 26.Nichols DA, Hargis JC, Sanishvili R, Jaishankar P, Defrees K, Smith EW, Wang KK, Prati F, Renslo AR, Woodcock HL, Chen Y. Ligand-induced proton transfer and low-barrier hydrogen bond revealed by X-ray crystallography. J Am Chem Soc. 2015;137(25):8086–8095. doi: 10.1021/jacs.5b00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun T, Bethel CR, Bonomo RA, Knox JR. Inhibitor-resistant class A β-lactamases: Consequences of the Ser130-to-Gly mutation seen in apo and tazobactam structures of the SHV-1 variant. Biochemistry. 2004;43(44):14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 28.Helfand MS, Bethel CR, Hujer AM, Hujer KM, Anderson VE, Bonomo RA. Understanding resistance to β-lactams and β-lactamase inhibitors in the SHV β-lactamase: Lessons from the mutagenesis of SER-130. J Biol Chem. 2003;278(52):52724–52729. doi: 10.1074/jbc.M306059200. [DOI] [PubMed] [Google Scholar]
- 29.Lewandowski EM, Skiba J, Torelli NJ, Rajnisz A, Solecka J, Kowalski K, Chen Y. Antibacterial properties and atomic resolution X-ray complex crystal structure of a ruthenocene conjugated β-lactam antibiotic. Chem Commun. 2015;51(28):6186–6189. doi: 10.1039/c5cc00904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leyssene D, Delmas J, Robin F, Cougnoux A, Gibold L, Bonnet R. Noncovalent complexes of an inactive mutant of CTX-M-9 with the substrate piperacillin and the corresponding product. Antimicrob Agents Chemother. 2011;55(12):5660–5665. doi: 10.1128/AAC.00245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beadle BM, Trehan I, Focia PJ, Shoichet BK. Structural milestones in the reaction pathway of an amide hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC beta-lactamase. Structure. 2002;10(3):413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]
- 32.Milazzo I. Faropenem, a new oral penem: Antibacterial activity against selected anaerobic and fastidious periodontal isolates. J Antimicrob Chemother. 2003;51(3):721–725. doi: 10.1093/jac/dkg120. [DOI] [PubMed] [Google Scholar]
- 33.Fonseca F, Chudyk EI, Kamp MWVD, Correia A, Mulholland AJ, Spencer J. The basis for carbapenem hydrolysis by class A β-lactamases: A combined investigation using crystallography and simulations. J Am Chem Soc. 2012;134(44):18275–18285. doi: 10.1021/ja304460j. [DOI] [PubMed] [Google Scholar]
- 34.Stewart NK, Smith CA, Frase H, Black DJ, Vaku-lenko SB. Kinetic and structural requirements for carbapenemase activity in GES-type β-lactamases. Biochemistry. 2015;54(2):588–597. doi: 10.1021/bi501052t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nukaga M, Bethel CR, Thomson JM, Hujer AM, Distler A, Anderson VE, Knox JR, Bonomo RA. Inhibition of class A β-lactamases by carbapenems: Crystallographic observation of two conformations of meropenem in SHV-1. J Am Chem Soc. 2008;130(38):12656–12662. doi: 10.1021/ja7111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiou J, Leung TYC, Chen S. Molecular mechanisms of substrate recognition and specificity of New Delhi Metallo-β-Lactamase. Antimicrob Agents Chemother. 2014;58(9):5372–5378. doi: 10.1128/AAC.01977-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.