

Abstract
A new NaCl based method for preparation of gallium-68 labeled radiopharmaceuticals has been adapted for use with an automated gallium-68 generator system. The method was evaluated based on 56 preparations of [68Ga]DOTATOC and compared to a similar acetone-based approach. Advantages of the new NaCl approach include reduced preparation time (< 15 min) and removal of organic solvents. The method produces high peptide-bound % (> 97%), and specific activity (> 40 MBq nmole−1 [68Ga]DOTATOC) and is well-suited for clinical production of radiopharmaceuticals.
Keywords: Gallium-68, Ion Exchange, NaCl, Radiopharmaceuticals, Positron emission tomography, DOTA peptides
1. Introduction
The use of 68Ga labeled compounds for PET imaging is of increasing interest to Nuclear Medicine investigators and physicians around the world (Maecke et al., 2005; Baum et al., 2008; Baum and Prasad, 2008; Roesch and Riss, 2010; Roesch and Baum, 2011; Prata, 2012). As the potential of 68Ga labeled compounds for molecular imaging is increasingly recognized, advances in software-controlled automated systems to streamline the routine production process have been developed and introduced commercially (Breeman et al., 2005; De Decker and Turner, 2012; Zhernosekov et al., 2007). While manual systems and methods had previously been the mainstay for most production, increased demand for routine clinical production in our facilities has prompted investigations into new methods that are adaptable to automation and have the potential to streamline processes and reduce costs. In this light, new automated systems hold the promise to reduce radiation exposure to technical staff and potentially reduce operator errors, thereby improving overall production process control (Dwivedi et al., 2011). In addition, automation can have added benefit to organizations as manufacturers realize economies of scale for production of “kits”. Radiopharmaceutical production kits have the potential to reduce costs and time consuming documentation for receipt of materials by providing complete-packaged assemblage of reagents and materials, accompanied by documentation attesting to adherence to Good Manufacturing Practices (GMP). Recent investigations also suggest that 68Ga labeled compounds may result in additional cost savings to organizations compared to the use of mainstream 111In labeled radiopharmaceuticals (Schreiter et al., 2012). On-demand production and operation in the absence of cyclotron facilities provides further rationale for the use of 68Ga and 68Ga labeled compounds for molecular imaging. However, start-up costs for procurement and installation of automated systems are not insignificant and quality control instrumentation costs are potentially burdensome. Thus, evaluations of the performance of such systems for routine production are of interest to an increasing number of investigators and clinical operations as the potential of 68Ga labeled compounds for molecular imaging is recognized.
As an alternative technique to the widely used acetone based labeling procedure (Zhernosekov et al., 2007) we investigated the suitability of the combined cationic/anionic concentration method (Müller et al. 2011) as initial step for an automated synthesis of 68Ga labeled DOTA conjugated peptides. This process takes advantage of the occurrence of different gallium species (Ga3+ cations and anionic [GaCl4]−) depending on varying pH and chloride concentration. In addition this concentration method is particularly well suited for the subsequent labeling of fragile peptides such as DOTA conjugated Affibody molecules (e.g., DOTA-ZHer2:342-pep2; Mueller et al., 2009; Müller et al. 2011) and organic solvents are not required during the labeling. In this procedure 68Ga is trapped by a silica based strong cation exchange cartridge (SCX) and then converted into [68GaCl4]− and eluted with a small volume of 5.5 M HCl. In step two, the [68GaCl4]− is adsorbed to a strong anion exchange cartridge (SAX) and 68Ga3+ is subsequently eluted with water (Fig. 1; Route 1). Because of the high purity of the labeled peptide after the radiolabeling reaction step, a final purification step is usually not required. However, the method requires two cartridges and corrosive 5.5 M HCl during the synthesis. During the course of these investigations, it was found that because of the high chloride concentration, a 5 M NaCl solution (containing a very low amount of HCl) can also be used to convert cationic bound 68Ga (adsorbed to a silica based strong cation exchange cartridge, SCX) into the [68GaCl4]− species (Fig. 1 route 2). The transformed [68GaCl4]− moiety is eluted with a minimal loss of activity from the SCX cartridge. As shown in Fig. 2, this concentration step is highly efficient and can be used to consistently obtain 98–99% of the total 68Ga activity of the generator. Furthermore, the replacement of 5.5 M HCl in the initial step for 68Ga eluate concentration allows direct use of this SCX eluate for labeling reactions by subsequent reconversion of [68GaCl4]− to 68Ga3+ using a suitable buffer system.
Fig. 1.
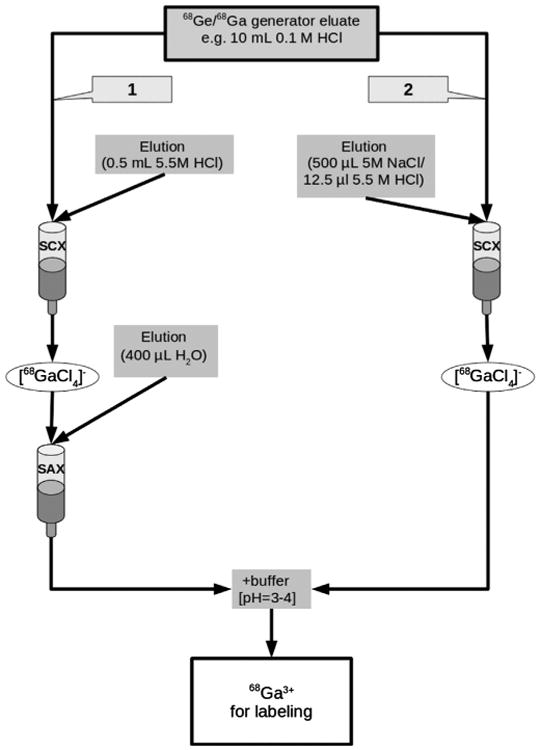
Schematic drawing of the combined cationic/anionic (1)—and the NaCl (2) based 68Ga generator eluate concentration procedure.
Fig. 2.
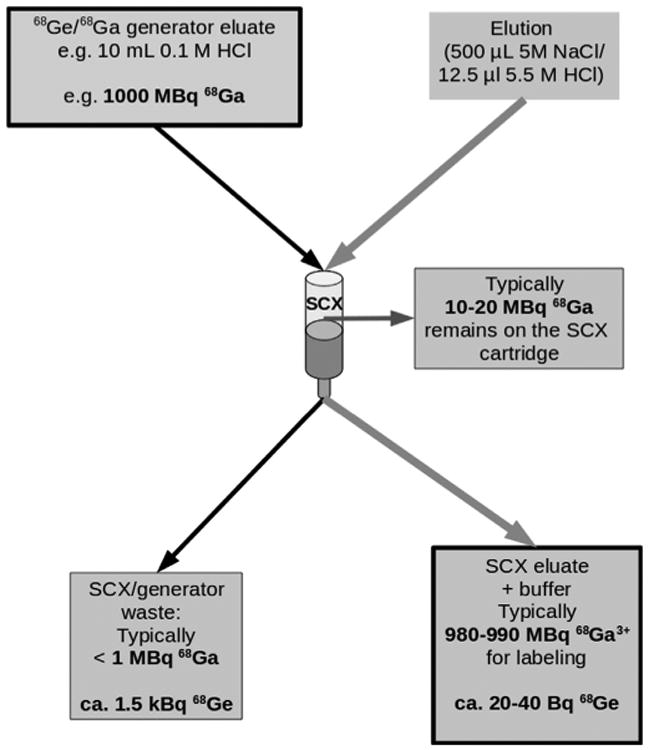
Schematic drawing of the efficiency of the NaCl based 68Ga generator eluate concentration procedure, representatively shown for 1000 MBq 68Ga start activity.
In this paper, we present the adaptation and use of an automated 68Ga generator and fluid handling system (Modular-Lab PharmTracer), applied for production of [68Ga]DOTA-peptides by the new NaCl-based 68Ga pre-purification and radiolabeling approach (further referred to as the Mueller Method). DOTATOC and DOTATATE were used as models. The method is based on founding studies using manual methods presented elsewhere ((Mueller et al., 2012). For reference, the automated NaCl method is compared to an acetone-based method adapted for use with the same automated generator and fluid handling instrumentation. Details of the acetone-based method are provided to facilitate this comparison/evaluation.
2. Experimental
All reagents were the highest grade available from commercial sources and used as received unless otherwise stated. Precise methods for preparation of specific reagents are presented in detail where deemed appropriate (e.g., pH sensitive buffers). Decay corrections are based on half lives of 68Ge (270.95 days) and 68Ga (67.71 min) obtained from the National Nuclear Data Center, Brookhaven National Laboratory (Brookhaven, NY USA). Radiochemical yields are expressed as a percentage of total activity of 68Ga of the generator eluates and are not decay corrected. Unless otherwise stated explicitly, all uncertainties are standard uncertainties, corresponding to a one uncertainty interval and are estimated based on experimental standard deviations or approximations thereof.
Generators
For all Mueller Method experiments, a 68Ga generator (IGG100 68Ga generator; Eckert & Ziegler Europe) was used. The total activity for the labeling reactions was between 2.4 and 1.6 GBq over a period of one year. For automated preparation of [68Ga]DOTATOC by the acetone method, radiolabeling with 68Ga was carried out through the use of a 68Ga/68Ge generator system IGG100 (Eckert Ziegler, GmBH, Berlin, Germany) with a total 68Ga activity of between approximately 1.8 to 1.3 GBq at the time of experiments presented here. The system was aged between 3–6 months for experiments conducted for this investigation.
2.1. Reagents and quality control methods
NaCl (Mueller) Method
The silica based SCX cartridge (Strong cation exchange, further referred as SCX cartridge, purchased from Varian, Bond Elut-SCX, 100 mg, 1 mL) was preconditioned with 1 mL 5.5 M HCl and 10 mL water prior to the generator elution step. Peptide-bound % was determined by standard radio-thin layer chromatography (radioTLC) using ITLC-SG strips (Varian); mobile phase 50% acetonitrile/0.1% trifluoroacetic acid (TFA) and validated by radio-High Performance Liquid Chromatography (radioHPLC) under the following conditions: HPLC pump: Jasco PU-1580; quaternary gradient unit: Jasco LG-1580-04; radio detector: Biostep IsoScan LC; Multiwavelength Detector: Jasco MD 1510; column: RP-18, LiChroCART 250-4, LiChrosphere 100, RP-18 (5 μm). As 1 M sodium acetate buffer a solution of 4.1 g sodium acetate in 50 mL water and 1 mL concentrated HCl was used and the pH of this buffer was adjusted with glacial acetic acid to 4.5. Adaptations were made for automated preparation of [68Ga]DOTATOC as described below.
NaCl (Mueller) Method Reagents
Reagents were trace-metal grade or highest available purity including: Water (Ultrapur, VWR) was used for preparation of 1 M sodium acetate buffer, 5.5 M HCl and 5 M NaCl solution; water for injection (Fresenius-Kabi) was used for the labeling reaction and subsequent dilution;0.1 M HCl (TitriPUR, VWR); 5.5 M HCl prepared by dilution of hydrochloric acid 37% (Emprove, VWR) 23 mL and water (Ultrapur) 27 mL; sodium chloride 99.99% (VWR); sodium acetate (Suprapur, VWR); acetic acid glacial (100%, for analysis; VWR); l- ascorbic acid (ACS reagent ≥ 99%, Sigma-Aldrich).
Acetone Method
Initial development experiments were conducted using a manual system, and subsequently adapted for automated production. For manual preparations, 68Ga was eluted from the generator (IGG100 Eckert & Ziegler) with 10 mL of 0.1 M HCl at a flow rate of approximately 2 mL per minute directly to a cation exchange (Strata™-XC, 33 μm, Strong Cation, 30 mg/mL #8B-S029-TAK, Phenomenex) column mounted by supports fabricated in house for manual preparations. Following generator elution, the generator outflow line is disconnected and the Strata-XC column is air dried using a 20 mL plastic syringe (2 ×). Once dry, pure 68Ga was eluted from the Strata-XC column with 400 μL of 98% acetone/0.05 M HCl (prepared weekly and stored at 4 °C) directly to a glass vial containing 5–10 nmoles DOTATOC dissolved in 5 mL of pure water (preheated to approximately 80 °C). This solution was then heated (open vessel) to 100 °C and 68Ga (with the DOTATOC) were incubated for 15 min. The manual method is not used for human studies and acetone is expected to be evaporated to acceptable levels (∼300 ppm) in the final purified drug product by this method, which is well below acceptable US Pharmacopeia limits (5000 ppm). These values are in agreement with previously reported values (Zhernosekov et al., 2007). Following the radiolabeling incubation period, the reaction mixture containing the [68Ga]-peptide and a small amount of non-incorporated 68Ga (further referred to as “free” 68Ga) was drawn up through a Strata™-X cartridge (33 μm Polymeric Reversed Phase C-18, 30 mg/1 mL, #8B-S100-TAK, Phenomenex® Inc., Torrance, CA USA) that had been preconditioned with 1 mL of 95% ethanol (USP for Injection) and 2.5 mL of pure water. The cartridge was then rinsed with 2 mL of water to remove any residual free 68Ga and finally the purified 68Ga-peptide conjugate was eluted in 500 μL of 1:1 ethanol (95%):isotonic saline solution. Peptide-bound %, and identity were determined by standard radioHPLC methods. The presence of parent radionuclide 68Ge was assessed by radiometric assay of an aliquot of the final drug product vial after at least seven days of decay by gamma ray spectrometry. No presence of 68Ge-supported 68Ga could be detected with overnight counts of these QC aliquots using a high purity germanium gamma ray detection system after allowing approximately one week for decay. Adaptation of the method for automation is presented below.
Acetone Method Reagents
Reagents were trace-metal grade or highest available purity including: 0.1 M HCl prepared by diluting 1 mL of concentrated HCl (trace metal grade, Aristar® Ultra, 32%-35%, VWR Intl. 87003-218) with to 100 mL with trace metal grade water (Aristar® Ultra, VWR, 87003-236); 98% acetone:0.02 M HCl solution was prepared by adding 0.23 mL HCl (Aristar) and 2.23 mL water (Aristar) to 98 mL > 99% acetone; Citric acid solution was prepared by dissolving 21 g citric acid monohydrate (Sigma-Aldrich, ACS grade, C1909-500G) to 1 L with 18 MΩ deionized-distilled water prepared in house; 0.2 M acetic acid (HOAc) was prepared by diluting 1.145 mL concentrated acetic acid (99.99 + %, Sigma-Aldrich, 338826-500ML) to 100 mL with Aristar water; 0.2 M sodium acetate (NaOAc) was prepared by diluting and dissolving 2.72 g ultra-pure sodium acetate reagent (sodium acetate trihydrate, Sigma-Aldrich, 71188 or equivalent) to 100 mL Aristar water; radiolabeling buffer solution was then prepared by mixing 82 mL 0.2 M HOAc with 18 mL 0.2 M NaOAc to achieve the desired pH 4 buffer. The pH of buffer solutions was checked by common pH paper and confirmed using a glass electrode electronic pH meter, calibrated daily using standard buffers. Solution for final purification of radiolabeled peptide by disposable C-18 catridge (included with cassette kit (47.5% ethanol in water) was prepared by diluting 9.5 mL absolute ethanol (USP grade) with 11.5 mL USP water for injection.
2.2. Automated 68Ga concentration and labeling procedures and quality control
Automated Mueller Method
DOTATOC was labeled on a Modular-Lab PharmTracer module (Eckert & Ziegler). The 68Ga generator was eluted with a total of 10 mL of 0.1 M HCl and the eluate was collected into a 15 mL plastic vial. 99.9% of 68Ga of the generator eluate was then trapped on a SCX cartridge by using a syringe pump and the activity was then eluted, with minimal loss (1–2%), using a mixture of 12.5 μL of 5.5 M HCl and 500 μL of 5 M NaCl. This eluate was slowly added to a solution of 350 μL sodium acetate buffer, 5 mg ascorbic acid and 50 μg of the DOTA conjugated peptide (DOTATOC) (25 μg/GBq 68Ga) in 3.0 mL of water. During the labeling, the pH of the reaction mixture inside the reactor was determined to be 3.6 ± 0.3. After heating the solution for 400 s at 90 °C, the 68Ga chloride is quantitatively coupled to the DOTA moiety of the peptide conjugate. The reaction mixture was diluted with 2 mL of water and sterile filtered. Peptide-bound % of > 95% is routinely achieved as determined by radio TLC. The radiochemical yield after sterile filtration is about 65%. The final product was neutralized with 2 mL of sterile sodium phosphate buffer (1 mmol/L Na+, 0.6 mmol/L ; B. Braun, Melsungen AG, Germany). The automated synthesis was finished within about 14 min (Fig. 3).
Fig. 3.
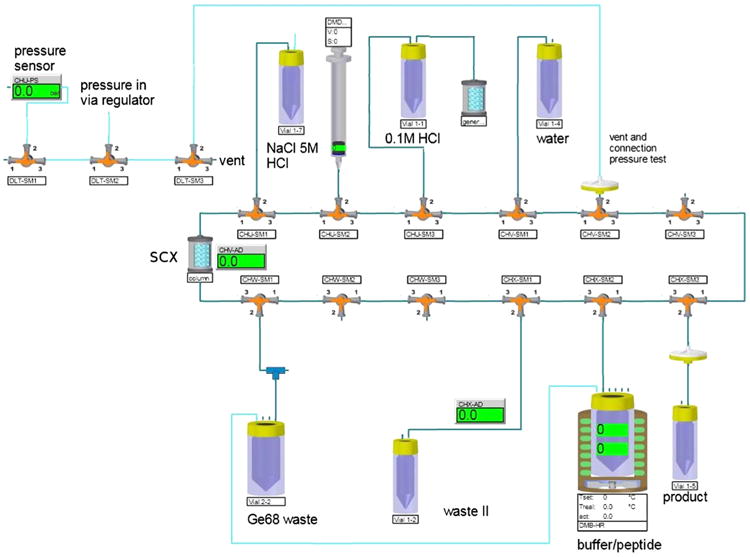
Software interface module diagram of the automated ModularLab PharmTracer (Eckert & Ziegler) radiosynthesis system used to prepare 68Ga DOTATOC by the NaCl based method.
Automated Acetone Method
For these experiments, a 68Ga generator (IGG100 68Ga generator; Eckert & Ziegler Europe) was used. In this case, the total activity for the labeling reactions was between 1.8 and 1.3 GBq over a few months. Recently, we conveyed summarized performance data for the use of the automated method. Here, we report a detailed description of the procedure for this method for comparison to the NaCl approach of 13 clinical runs. The manual acetone method described above was adapted for use with an automated system (Modular Lab PharmTracer, Eckert Ziegler). The system is housed in a sterile laminar flow hood for routine radiopharmaceutical production. Single-use, sterile pyrogen-free cassette-based “kits”, which had been adapted for this method by the manufacturer were used as received and were accompanied by documentation certifying adherence to GMP standards. For results presented here, preparations and quality control were conducted according to validated protocols and procedures previously documented. The complete liquid flow-path is self-contained in the cassette-based kit and requires only a single connection to the generator outflow connected by standard Luer-type fitting, which is disconnected and capped at the conclusion of each radiolabeled peptide preparation. For experiments presented here, transfers of liquid reagents required to elute 68Ga, to radiolabel DOTATOC, and purify/sterilize final [68Ga]DOTATOC were controlled by integrated software-controlled syringe pump. The glass reaction vessel, final drug product vial, sterilizing filter, acetone/HCl cation-exchange eluant vial, and ethanol/saline final purification solution vial (as well as pipet tips, syringes, and all other disposables required for the complete procedure) were included in the kit purchased from the manufacturer. GMP-grade DOTATOC (Bachem, Germany) was dissolved in USP water for injection and was dispensed in 30 μg portions and stored at −20 °C for use.
Prior to elution of the generator, DOTATOC aliquots were removed from the −20 °C freezer, dissolved in 2 mL of sodium acetate buffer solution and transferred to the reaction vessel (included with cassette system) via plastic syringe (with a stainless steel needle) included with the cassette-based kit, according to manufacturer instructions. Transfer is expected to be nearly quantitative, but residual DOTATOC will remain in the DOTATOC vial. No attempt was made to quantify this residual DOTATOC material. Acetone-based Strata-XC 68Ga eluant was prepared as described above and 3 mL was transferred via electronic-calibrated pipet with plastic tip to the appropriate vial contained in the cassette-based kit according to manufacturer instructions. The 47.5% ethanol solution septum-vial and a 50 mL septum vial containing common USP isotonic saline (as received) were connected to the cassette-based kit according to manufacturer instructions. The product line leading from the cassette was equipped with sterilizing filter and needle (as received in the cassette-based kit), and connected appropriately to the product vial. Finally, waste lines leading from the cassette were attached to the waste vial according to instructions and the system was ready for preparation of [68Ga]DOTATOC.
The automated preparation begins with elution of the generator with 6 mL 0.1 M HCl directly to the cation exchange cartridge included with the disposable single-use kit. Following syringe-pump driven expelling of excess HCl, the cation exchange cartridge is eluted of 68Ga using 1 mL 98% acetone/0.02 M HCl eluant directly to the reaction vessel, containing DOTATOC dissolved in pH 4 NaOAc buffer solution (pH measured as above). The solution is heated to 95 °C for 7 min, at which time the heat source is turned off automatically, the solution is cooled further and diluted with saline, and transferred via automated fluid control to the C-18 cartridge, where radiolabeled [68Ga]DOTATOC is retained and any remaining free 68Ga is removed by rinsing with saline. Finally, purified [68Ga]DOTATOC is eluted from the C-18 cartridge with 47.5% ethanol solution and filter-sterilized before entering the final product vial. The final product vial is vortexed briefly to ensure homogeneity, the radioactivity measured by dose calibrator, at the manufacturer dial setting, and a small aliquot is removed for required quality control. Quality control for clinical application includes endotoxin, peptide-bound %, pH, and half life, as well as gas chromatography for acetone and ethanol assay of the final radiopharmaceutical product. The process from elution of the generator to sterilization of the [68Ga]DOTATOC and dilution in saline requires 32 min according to the manufacturer instructions. The 32-minute process can be left unattended and requires no operator intervention. Breakthrough for automated clinical studies is performed by gamma ray measurement using a well-type sodium iodide detection system (ORTEC-Ametek, USA). To assess the potential for 68Ge breakthrough and incorporation in the final DOTATOC drug product, after 7 days of decay time, the QC reference vial is counted overnight in the detector well and the presence of 68Ge in the drug product is assessed by supported 68Ga, identified by the presence of a 511 keV gamma ray peak.
3. Results
NaCl (Mueller) Method
A total of 56 clinical production runs of [68Ga]DOTATOC were prepared by a NaCl based method for pre-purification of 68Ga eluted from a well-characterized 68Ga generator. Elution of the generator was by manufacturers instructions (10 mL 0.1 M HCl) directly to an SCX cartridge, which was adapted to integrate into a modified cassette-based single use kit, provided by the manufacturer for the Modular Lab PharmTracer system. The absence of organic solvents during the synthesis allows an inclusion of an easy to handle cassette-cleaning step after the labeling and therefore, multiple use of the cassettes in compliance with GMP regulations. Only the reaction vessel should be changed daily in order to ensure a high peptide-bound. The final [68Ga]DOTATOC drug product was consistently ready for delivery to the nuclear medicine clinic within 14 min from the start of generator elution, owing at least in part, to the removal of a C-18 final purification step required by most methods to remove small, but significant, amounts of residual unlabeled free 68Ga. Quality control parameters were all within specifications (Table 1); including high peptide-bound > 97% (Fig. 4); and specific activity > 40 MBq n mole−1. The average radiochemical yield for the 56 production runs is about 65% (not decay corrected; 1.6–1.0 GBq), which is similar to the manual synthesis using the acetone based method (Table 1). Gas chromatography assays for organic solvents were not required for this method. All other quality control parameters were within specification for all runs (Table 1). The automated system has reduced operator extremity dose (fingers) to negligible levels (Fig. 5). It was found that the 68Ga-DOTATOC labeled using the NaCl method is stable for more than two half lives. As shown in Fig. 6A, radiolysis is not detectable by radio HPLC, even two hours after the labeling reaction. In contrast, even after a final purification step the 68Ga labeled DOTATOC produced by the acetone/HCl concentration procedure, side products can be detected by radio HPLC (Fig. 6B). For the labeling of DOTATATE using the HCl/acetone procedure, larger concentrations of side products are frequently visible in the final solution (Fig. 7B). A subsequent purification with the help of a solid phase extraction would only separate the free 68Ga. In these cases, the radiochemical purity of the final product can be lower than 90% and therefore may not be sufficient for use as clinical radiopharmaceutical. On the other hand, the NaCl method delivers the 68Ga-DOTATOC in high radiochemical purity (with no apparent side products) as detected by radio HPLC (Fig. 7A). After complete decay of 68Ga using a high purity germanium detector, the content of parent radionuclide 68Ge could be determined and is approximately 2–5 Bq/ml (per GBq 68Ga) in the final products. The measurement method was similar to that, which has described in Breeman et al., 2005. The main 68Ge breakthrough from the generator was not adsorbed on the SCX cartridge during the initial adsorption of 68Ga and is therefore found in the SCX waste. A small amount of 68Ge activity is retained on the cartridge (Fig. 2). Routine production has resulted in consistent high-quality PET images of neuroendocrine tumor patient studies.
Table 1.
Averaged results of 56 automated runs using the NaCl based labeling procedure.
| Parameter | Test method | Specification | Average (min–max) |
|---|---|---|---|
| Peptide bound 68Ga (%) | ITLC | > 90 | 99.4 (97–100) |
| Specific activity (MBqnmole−1) | Dose calibrator | > 20 | 47.5 (30–62) |
| pH | pH-meter | 4.5–8.5 | 6.4 (6.0–6.8) |
Fig. 4.
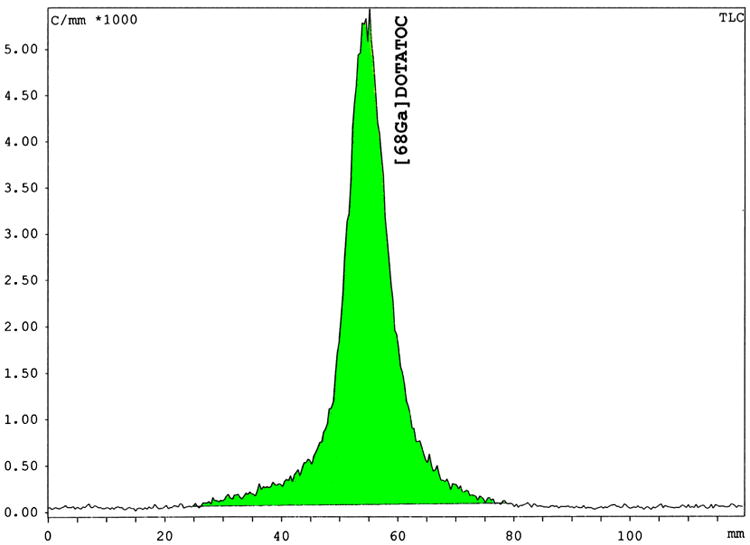
ITLC-SG of the 68Ga labeled DOTATOC: 50% acetonitrile, 0.1% TFA; Rf (free 68Ga)=0, Rf(68Ga-DOTATOC)=0.4–0.7 demonstrating the peptide-bound % of the radiolabeled peptide prepared by the NaCl based method using the automated system.
Fig. 5.

Synthesis module for the radiopharmaceutical production of 68Ga-DOTATOC.
Fig. 6.
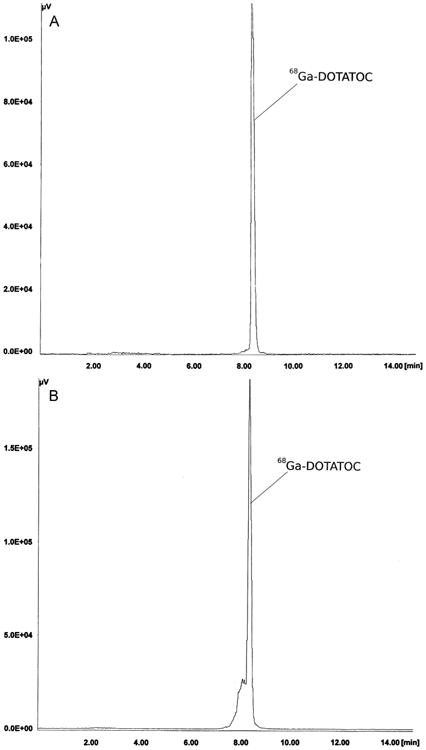
(A) Typical HPLC chromatogram of 68Ga-DOTATOC using the NaCl based labeling procedure, 2 h after synthesis. (B) Typical HPLC chromatogram of purified 68Ga-DOTATOC using the acetone/HCl method immediately after synthesis; HPLC: column: RP-18, LiChroCART 250-4, LiChrospher 100, RP-18 (5 mm); solvent A: acetonitrile solution in water (5%), 0.1% TFA; solvent B: 95% acetonitrile solution in water, 0.1% TFA; flow rate: 1.2 mL/min; gradient: from 0–2 min 100% A, 2–15 min to 100% B.
Fig. 7.
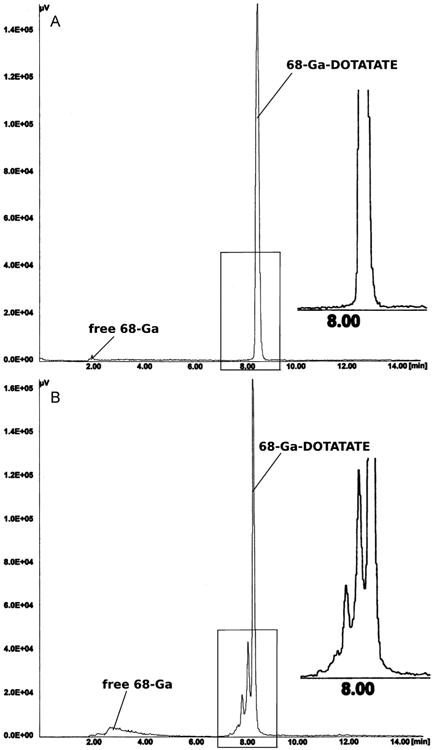
(A) Typical HPLC chromatogram of the final reaction mixture of 68Ga-DOTATATE synthesis using the NaCl based labeling procedure; (B) Typical HPLC chromatogram of the final reaction mixture of 68Ga-DOTATATE synthesis using the acetone/HCl method; HPLC conditions similar as described in Fig. 6.
Acetone Method
A total of 13 automated production runs of [68Ga]DOTATOC were prepared by an acetone-based method for pre-purification of 68Ga eluted from a well-characterized 68Ga generator (same model as used for the Mueller Method evaluation described above). Elution of the generator was by manufacturers instructions (6 mL 0.1 M HCl) directly to a cation exchange cartridge (SCX), which was used as received in a cassette-based single-use kit, provided by the manufacturer for the Modular Lab PharmTracer system. Using this method, 68Ga is preconcentrated on the SCX cartridge (adapted to the cassette) and eluted with 1 mL of 98% acetone/0.02 M HCl directly to a pH 4 buffer solution containing 30 μg (21 nmole) DOTATOC. Radiolabeling is carried out for 7 min at 95 °C; 2 mL of isotonic saline is added to cool the solution and the diluted mixture containing [68Ga]DOTATOC and a small, but not insignificant amount of free 68Ga is passed through a C-18 reverse phase disposable cartridge (also included with the kit). Free 68Ga is washed from the cartridge with saline, and [68Ga]DOTATOC is subsequently eluted with a 47.5% ethanol solution in saline directly through a sterilizing filter to the final product vial. The final radiopharmaceutical product was consistently ready for delivery to the nuclear medicine clinic within 60 min from the start of generator elution (including time for quality control). Quality control parameters were all within specifications and are reported elsewhere for this method (Mueller et al., 2012) including high peptide-bound % 68Ga (> 98%) and specific activity > 30 MBq nmole-1. The average radiochemical yield for the 13 production runs presented here is about 43% (not decay corrected), which is similar to the manual acetone based method used routinely in our research laboratory (data not shown). The automated system has reduced operator extremity dose (fingers) to negligible levels because the system is unattended through the radiolabeling steps (Fig. 3). No presence of supported 68Ga (indicative of 68Ge breakthrough) could be detected in any final radiopharmaceutical product solution by the methods described here.
4. Discussion
Methods employed for the automated comparison shown here were developed based on pioneering approaches to 68Ga generator elution, purification, and radiolabeling developed previously (Breeman et al., 2005; De Decker and Turner, 2012; Zhernosekov et al., 2007). While generators and methods for 68Ga labeling have been known for more than half a century, discovery of new peptides for targeted molecular imaging in recent years has stimulated a renaissance in 68Ga methods research (Fani and Maecke, 2012; Ginj and Maecke, 2004; Maecke et al., 2005). As we describe in detail elsewhere, while these new methods have been used successfully for research and clinical studies previously, (Breeman et al., 2005; Zhernosekov et al., 2007) each possesses potential disadvantages that we sought to avoid in optimizing our routine methods for clinical radiopharmaceutical production (Mueller et al., 2012). Importantly, for the purposes of this report, direct comparisons of the presented methods are not necessarily meant to be quantitative, but rather point to the acceptability of each method, and perhaps demonstrate the potential advantages of the new NaCl method for routine production, which is the subject of further investigation. Thus, our intention here is not to quantitatively demonstrate the superior performance for recommendation, but rather to inform on the acceptability of these methods for those researchers or institutions involved in 68Ga research or clinical implementation of new 68Ga labeled compounds. There are therefore several important differences in the systems and methods applied that should be pointed out within this context. Firstly, our research in this area represents collaboration between institutions. Thus, NaCl and acetone methods presented were conducted at separate radiopharmacy facilities (University of Iowa, Iowa City, IA USA and Zentralklinik Bad Berka, Bad Berka, Germany). Secondly, the total activity of the generators used at each site were different over the lifetime of the data collection (Iowa 1.8–1.3 GBq; Bad Berka 2.4–1.6 GBq). Thirdly, due to differences in regulatory procedural documentation and the total activity of the available generators, the total amount of peptide included at the radiolabeleing step is different at the individual sites (Iowa 30 μg; Bad Berka 50 μg). Other obvious differences include the operators who performed the daily radiolabeling, as well as preparation and specific manufacturers of reagents and apparatus employed. On the other hand, the results do represent routine clinical preparations of [68Ga]DOTATOC within the setting of clinical radiopharmacies and our results can thus be used as a gauge of the potential for automated systems to produce 68Ga peptides on a routine basis for clinical applications, while pointing out potential advantages that require additional quantitative assessments for final decisions to be reached.
It was found that the automated system could be readily adapted for routine production of [68Ga]DOTATOC by the new NaCl method. Through 56 clinical preparations of [68Ga]DOTATOC presented here, the system and approach consistently delivered high peptide-bound % and specific activity. All other quality control parameters (e.g., pH, endotoxins, sterility) were within specifications. Total preparation time for [68Ga]DOTATOC of less than 15 min was consistently realized by use of the automated NaCl method, which compares favorably to previously reported methods, as well as to an acetone-based method described here for comparison (preparation time 32 min). A major advantage of the new NaCl based method is removal of organic solvents from the radiolabeling procedure, which eliminates gas chromatography (GC) quality control requirements compulsory to other methods, reducing instrumentation costs (GC is not required) and improving timely delivery of 68Ga labeled compounds for clinical PET imaging. Based on our results, the acetone method in its current form, results in a lengthened preparation and delivery time due to the requirement to include the C-18 final purification step to remove free 68Ga and additional time needed to complete quality control assays, which includes tests for acetone and ethanol by gas chromatography.
5. Conclusion
A NaCl based method for preparation of 68Ga labeled radio-pharmaceuticals has been adapted for use with an automated 68Ga generator system. The method was evaluated based on 56 preparations of [68Ga]DOTATOC and compared to a similar acetone-based approach. Advantages of the new NaCl approach include reduced preparation time (< 15 min) and removal of need for organic solvents in the preparation process, which can improve delivery time and reduce costs for radiopharmacies interested in clinical use of 68Ga radiopharmaceuticals. Importantly, no cleavage products of the peptide (potentially caused by radiolysis) could be detected by radio HPLC for as long as two hours post synthesis. The method produced high peptide-bound (97–100%), and specific activity (> 40 MBq nmole−1) [68Ga] DOTATOC and is well-suited for clinical production of other radiopharmaceuticals.
Highlights.
A NaCl based automated production of Ga-68-radiopharmaceuticals is described.
Using 5 M NaCl for pre-purification of 68Ga eliminates the need for organic solvents.
The method provides for high efficiency, specific activity, and radiochemical purity.
The new method eliminates the need for the quality control by gas chromatography.
Acknowledgments
The authors kindly acknowledge support of the Dance Marathon (Iowa City, IA USA), the Holden Comprehensive Cancer Center at the University of Iowa, and the Neuroendocrine Tumor Fund. Support and discussions with M. Sue O'Dorisio and technical assistance from Kathy Thedes-Reynolds, University of Iowa, are kindly acknowledged.
References
- Baum RP, Prasad V, Hommann M, Hörsch D. Receptor PET/CT imaging of neuroendocrine tumors. Recent Results Cancer Res. 2008;170:225–242. doi: 10.1007/978-3-540-31203-1_18. [DOI] [PubMed] [Google Scholar]
- Baum RP, Prasad VPET. and PET/CT Imaging of Neuroendocrine Tumors. In: Wahl RL, editor. Principles and Practice of PET and PET/CT. 2nd. Wolters Kluwer/Lippincott Williams and Wilkins; Philadelphia: 2008. pp. 411–437. [Google Scholar]
- Breeman WA, de Jong M, de Blois E, Bernard BF, Konijnenberg M, Krenning EP. Radiolabelling DOTA-peptides with 68Ga. Eur J Nucl Med Mol Imaging. 2005;32:478–485. doi: 10.1007/s00259-004-1702-y. [DOI] [PubMed] [Google Scholar]
- De Decker M, Turner JH. Automated module radiolabeling of peptides and antibodies with gallium-68, lutetium-177 and iodine-131. Cancer Biother Radiopharm. 2012;27:72–76. doi: 10.1089/cbr.2011.1073. [DOI] [PubMed] [Google Scholar]
- Dwivedi DK, Snehlata, Dwivedi AK, Lochab SP, Kumar R, Naswa N, Sharma P, Malhotra A, Bandopadhayaya GP, Bal C, Pant GS. Radiation exposure to nuclear medicine personnel handling positron emitters from Ge-68/Ga-68 generator. Indian J Nucl Med. 2011;26:86–90. doi: 10.4103/0972-3919.90258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fani M, Maecke HR. Radiopharmaceutical development of radiolabeled peptides. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):11–30. doi: 10.1007/s00259-011-2001-z. [DOI] [PubMed] [Google Scholar]
- Ginj M, Maecke HR. Radiometallo-labeled peptides in tumor diagnosis and therapy. Met Ions Biol Syst. 2004;42:109–142. [PubMed] [Google Scholar]
- Maecke HR, Hofmann M, Haberkorn U. (68)Ga-labeled peptides in tumor imaging. J Nuc Med: Off Publ, Soc Nucl Med. 2005;46(Suppl 1):172S–178S. [PubMed] [Google Scholar]
- Mueller D, Klette I, Gottschaldt M, Baum RP. Radiolabeling of fragile macroligands with Ga-68. J Labelled Compd Radiopharm. 2009;52:477. [Google Scholar]
- Muller D, Klette I, Baum RP. The combined cationic–anionic purification of the 68Ge/68Ga generator eluate for the labelling of fragile peptides. Abstracts of Poster Presentations (Chemistry) World J Nucl Med. 2011;10(P-008):73–89. [Google Scholar]
- Mueller D, Klette I, Baum RP, Gottschaldt M, Schultz MK, Breeman WAP. Simplified NaCl based (68)Ga concentration and labeling procedure for rapid synthesis of (68)Ga radiopharmaceuticals in high radiochemical purity. Bioconjug Chem. 2012;23(8):1712–1717. doi: 10.1021/bc300103t. [DOI] [PubMed] [Google Scholar]
- Prata MI. Gallium-68: A New Trend in PET Radiopharmacy. Curr Radiopharm. 2012;5:142–149. doi: 10.2174/1874471011205020142. [DOI] [PubMed] [Google Scholar]
- Roesch F, Baum RP. Generator-based PET radiopharmaceuticals for molecular imaging of tumours: On the way to THERANOSTICS. Dalton Trans. 2011;40:6104–6111. doi: 10.1039/c0dt01504k. [DOI] [PubMed] [Google Scholar]
- Roesch F, Riss PJ. The Renaissance of the 68Ge/68Ga radionuclide generator initiates new developments in 68 Ga radiopharmaceutical chemistry. Curr Top Med Chem. 2010;10:1633–1668. doi: 10.2174/156802610793176738. [DOI] [PubMed] [Google Scholar]
- Schreiter NF, Brenner W, Nogami M, Buchert R, Huppertz A, Pape UF, Prasad V, Hamm B, Maurer MH. Cost comparison of 111In-DTPA-octreotide scintigraphy and 68Ga-DOTATOC PET/CT for staging enteropancreatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2012;39:72–82. doi: 10.1007/s00259-011-1935-5. [DOI] [PubMed] [Google Scholar]
- Zhernosekov KP, Filosofov DV, Baum RP, Aschoff P, Bihl H, Razbash AA, Jahn M, Jennewein M, Rosch F. Processing of generator-produced 68Ga for medical application. J Nucl Med. 2007;48:1741–1748. doi: 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]