

Abstract
The present study explored the molecular mechanisms by which instant cell membrane resealing (CMR) controls the activation of NOD-like receptor pyrin domain containing 3 (Nlrp3) inflammasomes. Using wavelength-switching fluorescent microscopy with PI and fura-2 as indicators, we monitored instant CMR simultaneously with (Ca2+)i in mouse microvascular endothelial cell (MVECs). LCWE or saponin wad found to produce membrane injury, which was resealed in a Ca2+-dependent manner, but abolished by FasL, a membrane raft (MR) clustering stimulator. Even in the presence of Ca2+, FasL prolonged the CMR time as shown by an earlier onset of PI influx (48±12 sec vs. 17±3 min. of control). These effects of FasL were substantially blocked by an MR disruptor, methyl-beta-cyclodextrin (MCD). The failure of CMR upon FasL activated Nlrp3 inflammasomes, which was blocked by MCD, a membrane resealing compound, VA64 or siRNA of an MR-facilitating enzyme, acid sphingomyelinase. This inflammasome activation was due to increased lysosomal permeability and cathepsin B release. It is concluded that an MR-associated CMR protects ECs from Nlrp3 inflammasome activation induced by membrane injury.
Keywords: Endothelial cells, Nlrp3 inflammasome, Membrane repair, Membrane rafts, Inflammatory machinery, Cathepsins B
2. Introduction
Plasma membrane disruption is a common form of cell injury under physiological and pathological conditions (1). It has been known that rapid membrane resealing or repairing during injury is an important adaptive mechanism, which is essential for cell survival and cell function (2). If the cell membrane disruption be not quickly repaired, the cells may function abnormally and even die due to the loss of cytoplasm and to the entry of extracellular molecules (3). It has been reported that membrane rafts (MRs) and an important MR sphingolipid, ceramide play a critical role in cell membrane repair during cell injury (4) and that acid sphingomyelinase (ASM), the ceramide-producing enzyme that promotes MR clustering participates in the control of cell membrane resealing (5). However, it remains poorly understood whether this MR and ceramide-associated cell membrane resealing is implicated in the regulation of vascular function or related diseases to endothelial dysfunction and injury.
Recently, work in our laboratory has demonstrated that MR clustering is importantly involved in the formation and activation of membrane redox signaling platforms in ECs, which contributes to the regulation of arterial endothelial function such as endothelium-dependent vasodilation (6). In particular, this MR redox signaling platform is implicated in endothelial injury in response to death receptor activation and other danger factors, ultimately leading to vascular diseases (7, 8). It has also been reported that the deficiency of ceramide producing enzyme, ASMase is involved in the development of vascular inflammation and atherosclerosis (9). It is now imperative to know whether this vascular inflammation or atherogenesis associated with ASM activity or ceramide production is attributed to endothelial membrane injury and dysfunction at the early stage of cell exposure to different pathological stimuli or danger factors. In particular, it is interesting to know whether MR clustering and associated instant membrane repairing is important in the control of inflammasome activation, which ultimately results in vascular inflammation or injury via its instigation of inflammation or non-canonical action on cell metabolism, surviving and organellar stabilization (10-13).
Among different inflammasomes, NOD-like receptor pyrin domain containing 3 (Nlrp3) inflammasome has been best characterized in mammalian cells, and this Nlrp3 inflammasome may be involved in the vascular inflammation and atherosclerosis (14-16). Most recently, we have reported that Nlrp3 inflammasome activation importantly contributes to endothelial dysfunction or injury induced by different danger factors under pathological conditions (13, 17). It has been known that the Nlrp3 inflammasome is usually formed as a complex consisting of three main components, including Nlrp3 that functions as a pattern recognition receptor, the adaptor protein, ASC (apoptotic speck-containing protein with a CARD) and pro-caspase-1 (18). As an intracellular inflammatory machinery, the activation of this inflammasome mainly produces various pro-inflammatory cytokines such as interleukin-1beta (IL-1beta) and interleukin-18 (IL-18) (19, 20), thereby initiating inflammatory response. In addition, recent studies have also indicated that Nlrp3 inflammasome activation may produce a number of uncanonical actions to alter cell function or cellular activities producing cell injury and ultimate disease, which are beyond its canonical inflammation instigating effects. (21-24). However, it remains poorly understood how this inflammasome is activated in ECs in response to different stimuli or danger factors. In particular, given that the bacteria infection or bacterial toxin usually activate inflammasomes due to their early effects on the membrane of host cells, altered cell membrane integrity or membrane injury may be importantly involved in the activation of inflammasome as a host cell response. In this regard, previous studies have indeed shown that MRs importantly participate in the host response during infection of different pathogens including bacteria, viruses and parasites (25-27).
In the present study, we first determined whether the MR clustering promotes the Nlrp3 inflammasome formation and activation in ECs and whether this MR clustering-enhanced Nlrp3 activation is due to membrane injury or resealing. Then, we explored the molecular mechanisms by which the deficient membrane resealing or repair activates Nlrp3 inflammasomes in ECs. In these experiments, the Lactobacillus casei cell wall fragments (LCWE) as a commonly used bacterial extract to produce arteritis and a classical cell membrane hole producing reagent, saponin were used as a plasma membrane damaging agent, and the membrane injury and instant resealing response observed by dynamic monitoring of fluorescent dye influx and removal. It has been demonstrated that a MR-associated instant cell membrane resealing mechanism functions instantly in ECs when they are exposed to membrane injury factors and that the resealing mechanism determines whether Nlrp3 inflammasomes are activated through enhanced lysosomal release of lysosomal cathepsin B.
3. Materials and Methods
3.1. Cell culture and stimulation
The mouse microvascular endothelial cell (MVEC) line EOMA was purchased from ATCC. The cell line was originally isolated from mouse hemangioendothelioma. MVECs were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (FBS). The cells were cultured in a humidified incubator a mixture at 37°C with 5% CO2 and 95% air. Cells were passaged by trypsinization (Tripsin/EDTA, Sigma, St. Louis, MO, USA), followed by stopping the reaction in the DMEM medium containing 10% fetal bovine serum. According to previous studies, a short time period was applied for FasL stimulation (10 ng/ml) to induce MR clustering and to avoid other action such as apoptosis (6). To test whether the MR clustering plays a role for plasma membrane resealing during injury, MVECs were pretreated with membrane damage reagents, different siRNAs or chemicals as described in individual experiments described below.
3.2. Simultaneous measurement of Ca2+ and PI production in MVECs
Plasmic membrane damage and intracellular Ca2+ response to different stimulants were determined by using Propidium iodide (PI) as an indicator of membrane open and resealing and by fura-2, a Ca2+-sensitive fluorescent dye, respectively. A fluorescent microscopic imaging system with high speed wave-length switching was used as described previously by using an inverted microscope (Diaphot 200, Nikon, Tokyo, Japan) and a digital camera (SPOT RT Monochrome, Diagnostic Instruments, Sterling Heights, MI,USA) (28-30), which dynamically monitored the PI signals when cell membrane is damaged or resealed in MVECs. Briefly, the cells were cultured in the recording chamber, which was filled with Hanks' buffer. After a 10 min equilibration, the bath solution was exchanged with 1 ml of Hanks' buffer containing fura 2-AM (10 μM; Molecular probes) and the cells was incubated in this loading solution at 37°C for 30 min. The cells were then rinsed three times with Ca2+ (1.5. mM CaCl2) or Ca2+-free Hanks' buffer to remove extracellular fura 2-AM and incubated in Hanks' buffer containing PI (2 μg/ml) for an additional 5 min. During this 5 min incubation time, intracellular deesterification of fura 2-AM was allowed. After that, the PI fluorescence was recorded at an emission wavelength of 636 nm after excitation of 493 nm. The ratio of Fura-2 emissions at 340 and 380 nm (F340/F380) were monitored as we described in our previous studies (31). Meta-fluor imaging and analysis software was used to acquire, digitize and store the images for off-line processing and statistical analysis.
3.3. Differential conversion of PI fluorescence intensity
PI is commonly used as an indicator of cytoplasmic membrane damage. However, PI binds to DNA to produce red fluorescence which cannot be dissociated. An obvious disadvantage of this method for plasma membrane injury measurements is that a plateau area of the PI fluorescence curve does not represent the actual degree of plasma membrane injury because it is possible that PI is saturated without recovery. To address the problem, we performed a differential conversion of time-dependent PI fluorescence curve to calculate df/dt, which represents the flow velocity of PI into ECs. This velocity of PI flow may primarily represent the degree of plasma membrane injury. By calculating the area under the df/dt curve, we can estimate the extent of the impairment or repair on the plasma membrane. We first performed regression analysis of the PI fluorescence curve recorded during plasma membrane injury/repair to obtain a regression equation. A differential conversion was then undertaken based on the regression equation. For example, the PI curve recorded during LCWE stimulation was fitted with a sigmoidal four-parameter Gompertz growth model with SigmaPlot 5.0. An equation for LCWE-induced time-dependent change in fluorescence (F) was described as follows:
Where t is time, t0 is time when LCWE is added and a, b, and c are constants, which define the shape for a specific-fitting curve of flow velocity of PI entry into cells. These constant parameters, were calculated from recorded curve with the curve fitting program of SigmaPlot 5.0. To obtain the rate of plasma membrane injury level, the differential conversion of Eq. 1 (df/dt) was calculated with the equation below:
3.4. Flow-cytometric analysis
MVECs were harvested and washed with PBS and then PI (final concentration, 2μg/ml) was added during or after LCWE or saponin incubation. After 30 min, the cells were incubated with SYTOX (Life Technologies, Grand Island, NY, USA) at 37 °C for 4 hours in the presence of PI. Stained cells were run through a Guava Easycyte Mini Flow Cytometry System (Guava Technologies, Hayward, CA, USA) according to the manufacturer's instructions (32) and analyzed with Guava acquisition and analysis software (Guava Technologies). Cell populations with strong PI staining indicate cell injury.
3.5. Immunofluorescence microscopy
Indirect immunofluorescent staining was used to determine colocalization of inflammasome molecules in MVECs exposed to cell membrane damaging reagents. Anti-Nlrp3 antibody (1:200, Abcam, Cambridge, MA, USA) and anti-caspase-1 antibody or anti-ASC antibody (1:100, Santa Cruz, Dallas, TX, USA) were used for these experiments. In brief, MVECs grown on 8-well chamber with or without pretreatment of FasL (10 ng/ml) were incubated with LCWE or saponin to stimulate cell membrane injury. To explore the mechanisms for cell membrane injury or repairing, membrane sealer, VA64 (33, 34), MR disruptor, MCD, and ASM inhibitor or siRNA were separately or together added to the cells medium. All treatments were terminated by fixation of the cells in 4% PFA for 15 minutes. These fixed cells were washed in phosphate-buffer saline (PBS) and incubated for overnight at 4°C with goat anti-Nltp3 and rabbit anti-ASC, or rabbit anti-caspase-1 antibodies. Then, the cell slides were stained for 1 hour with fluorescence-conjugated secondary antibodies (1:500, Invitrogen) according to the manufacturer's instructions (13). Then, the cells were visualized through sequentially scanning protocol on an Olympus laser scanning confocal microscope (Fluoview FV1000, Olympus, Japan). Co-localization was analyzed by Image Pro Plus software, and the co-localization coefficient was represented by Pearson's correlation coefficient as we described previously (13).
3.6. Western blot analysis
After different treatment, MVECs were washed twice with PBS and scraped in sucrose buffer (20 mM HEPES, 1 mM EDTA, 255 mM sucrose, and a protease inhibitor cocktail tablet (Roche, Nutley, NJ, USA), pH7.4.). Then, 20 μg protein samples were denatured with reducing 5 × Laemmli SDS-sample buffer and heated at 95°C for 5 min. Samples were run into SDS-PAGE gel, transferred into PVDF membrane and blocked with skimmed milk. The membranes were probed with primary antibody of anti-caspase-1 (1:500, Santa Cruz, Dallas, TX, USA) overnight at 4°C followed by incubation with secondary antibody conjugated to horseradish peroxidase (1:5000, Santa Cruz, Dallas, TX, USA). The immunoreactive bands were enhanced by chemiluminescence reagents (Pierce, Rockford, IL, USA) and imaged on Kodak Omat film. beta-actin reporting was used as a loading control. The intensity of the bands was quantified by densitometry.
3.7. RNA interference of ASM
Small interference RNAs (siRNAs) against ASM and scrambled small RNA were obtained from Qiagen (Qiagen Valencia, CA, USA). The sequence for ASM siRNA was confirmed to be effective in silencing ASM gene in ECs in our previous studies (35). The scrambled small RNA has been also confirmed as non-silencing double-stranded RNA and was used as control in the present study. Transfection of siRNA was performed using the siLentFect Lipid Reagent (Bio-Rad, CA, USA) according to the manufacturer's instructions.
3.8. IL-1beta quantitation
IL-1beta was quantitated in the supernatants of EC cultures. After treatments, the cell supernatants were collected and IL-1beta concentrations measured by an ELISA kit (R&D System, Minneapolis, MN, USA) according to the protocol described by the manufacturer.
3.9. Analysis of lysosome membrane permeability and cathepsin B activation
MVECs were cultured in 8-well chamber plates. After treatments, the cells were incubated with CV-(RR)2 reagent and 1 μM acridine orange (AO) for 20 min at 37°C and rinsed with PBS. In addition, a fluorescent substrate, z-Arg-Arg-cresyl violet was loaded into MVECs to determine cathespin B activity in lysosomes, which was shown by red fluorescence. These cells were immediately analyzed and photographed using an inverted fluorescence microscope (LSM 710, Zeiss, Bonn, Germany) according to the manufacturer's protocol of a commercial kit (Enzo, Ann Arbor, MI, USA).
3.10. Statistics
Data are presented as means ± SE. Significant differences between and within multiple groups were examined using ANOVA for repeated measures, followed by Duncan's multiple-range test. Student's t-test was used to evaluate the significance of differences between two groups of observations. P < 0.0.5 was considered statistically significant.
4. Results
4.1. Dynamic monitoring of cell membrane injury and resealing simultaneously with recording of intracellular Ca2+ transient using PI and Fura-2
Figure 1 shows representative images and recordings of simultaneously measured (Ca2+)i and PI fluorescence intensity in MVECs in response to LCWE. Saponin was used as a positive control reagent for cell membrane permeabilization in these studies (36). As shown in panel A and D, the F340-to-F380 ratio dramatically increased as shown by changes in fluorescent intensity from green to red upon LCWE or saponin stimulation, indicating intracellular (Ca2+) increase when these cells were exposed to LCWE, which was similar to the cell response to cell permeabilizing reagent. Under control condition, there was no red PI fluorescence observed. However, when LCWE or saponin was added into the bath solution, the red fluorescence appeared indicating the entry of PI into cells through damaged plasma membrane. In addition to these recorded images, the PI fluorescence intensity and (Ca2+)i in MVECs were also digitized and monitored simultaneously throughout the experiment, which are shown in Figure 1B and 1E. Plateau of PI fluorescence was observed, which was formed because PI binding to produce fluorescence was not associated with a constant association and dissociation. Therefore, a differential conversion was performed as we described previously in the use of similar dyes (37). As shown in Figure 1C and 1F, df/dt represents the speed of intracellular PI fluorescence increase, indicating the level of cell membrane injury. A larger increase in df/dt reflects more severe injury of cell membrane and less resealing of injured area. It was found that a peak increase in (Ca2+)i upon LCWE or saponin stimulation occurred within 15 min or 20 min, which was followed by a largest rise of df/dt at 20 min and 25 min for LCWE and saponin, respectively. This maximal increase in df/dt followed by a peak (Ca2+) increase reflects an balance of cell membrane injury by LCWE or saponin and Ca2+-dependent membrane resealing.
Figure 1.
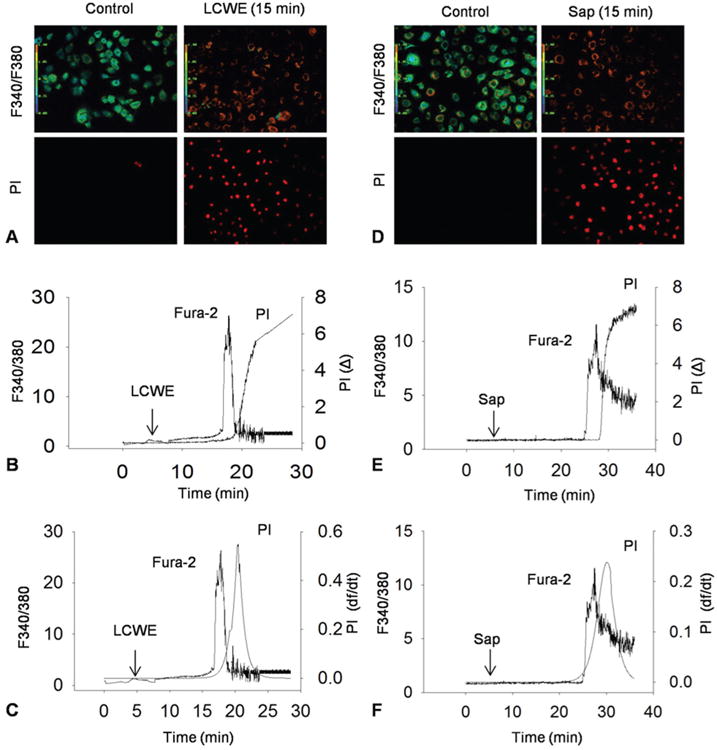
Dynamic monitoring of cell membrane injury and resealing simultaneously with recording of intracellular Ca2+ transient using PI and Fura-2. A and B) Representative fura 2 fluorescence ratio images taken at excitation wavelengths of 340 and 380 nm (F340/F380) and an emission wavelength of 510 nm and representative PI fluorescence images taken at a excitation wavelength of 535 nm and an emission wavelength of 617 nm under control conditions and during incubation with LCWE (20 μg/ml) or Saponin (Sap, 40 μg/ml). C and D) Typical simultaneous recordings of LCWE-induced or Sap-induced increase in (Ca2+)i and PI fluorescence in the MVECs. E and F) Relationship of LCWE-induced or Sap-induced increase in (Ca2+)i and flow velocity of PI shown by change in relative fluorescence with time (df/dt) of PI fluorescence.
4.2. Ca2+-dependent plasma membrane resealing in response to LCWE or saponin in MVECs
Using the simultaneous recording system to record intracellular (Ca2+) and membrane injury by PI fluorescence, we found that a Ca2+-dependent membrane resealing or repair mechanism is working when LCWE or saponin was used to damage cell membrane of MEVCs. This cell membrane resealing occurs instantly due to Ca2+ influx as soon as membrane lesions are triggered (38). As shown in Figure 2A and D, in the presence of Ca2+ both LCWE and saponin produced much less membrane injury, as shown by minimal df/dt seen in MVECs exposed to LCWE and much smaller df/dt to saponin compared to cells bathed with normal Hank's solution containing Ca2+. The maximal df/dt was summarized in Figure 2B and 2E, LCWE or saponin-induced cell membrane injury as shown by maximal df/dt was significantly larger in MVECs bathed in Ca2+-free Hank's solution compared to the cells in normal Hank's solution, indicating the counteraction of Ca2+-dependent repair on the membrane injury during the process. By calculation of a time needed to reach maximal df/dt, we found that MVECs in Ca2+-free Hank's solution needed much shorter time to reach maximal df/dt compared to MVECs with normal Hank's solution (Figure 2C and 2F). This further showed that the presence of Ca2+ could protect cell membrane from LCWE or saponin-induced membrane injury.
Figure 2.
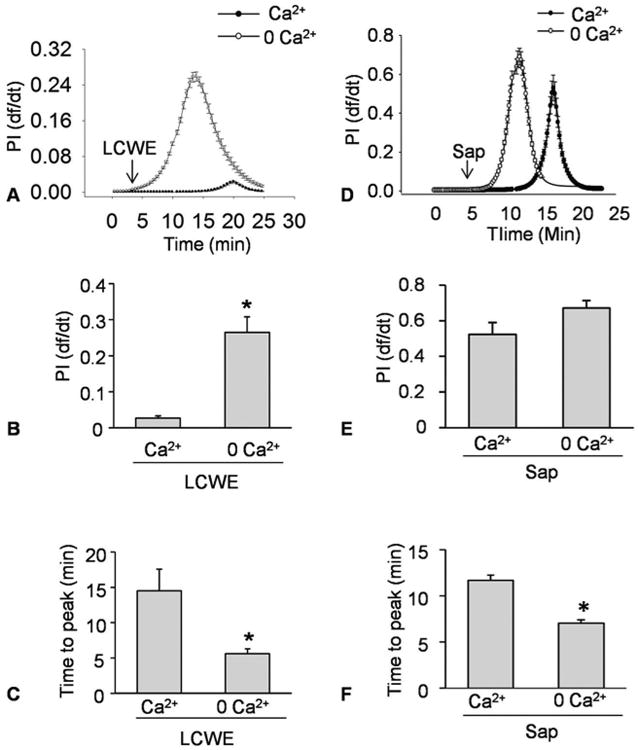
Ca2+-dependent plasma membrane resealing in response to LCWE or saponin in MVECs. MVECs were incubated with or without Ca2+ Hanks' buffer and then stimulated with LCWE (15 μg/ml) or Saponin (Sap, 40 μg/ml). A and B) Representative curves showing the relationship of LCWE-induced or Saponin-induced increase in plasma membrane injury to the flow velocity of PI increase by change in relative fluorescence with time (df/dt). The df/dt represents the flow velocity of PI into MVECs. C and D) Representative histograms show the maximal df/dt (N=6). E and F) Representative histograms showing the time to reach maximal df/dt (N=6). *P < 0.0.5 vs. Ca2+ Hanks' buffer.
4.3. Enhanced MR clustering blocked membrane resealing during LCWE-induced injury
Given that MRs and its main lipid component ceramide are importantly implicated in cell membrane repair (4, 5), we tested whether MRs clustering alters membrane resealing during LCWE-induce plasma membrane injury in MVECs. In these experiments, FasL was used to stimulate endothelial cell MR clustering and MCD used for blockade of MR clustering as we demonstrated previously (39, 40). As shown in Figure 3A, in the presence of Ca2+ MVECs almost had no increases in df/dt under control condition and FasL pretreatment led to occurrence of a large increase in df/dt. This FasL-enhanced df/dt increase was largely reduced and delayed when these cells were treated with MCD (Figure 3D). As summarized in Figure 3B-3E, FasL significantly increased the maximal df/dt, which was blocked by MCD. In addition, the time to reach maximal df/dt was significantly reduced when MVECs were pre-treated with FasL. In the presence of MCD, however, the time to reach maximal df/dt was recovered to a level comparable to the control cells with Ca2+ in the bath solution.
Figure 3.
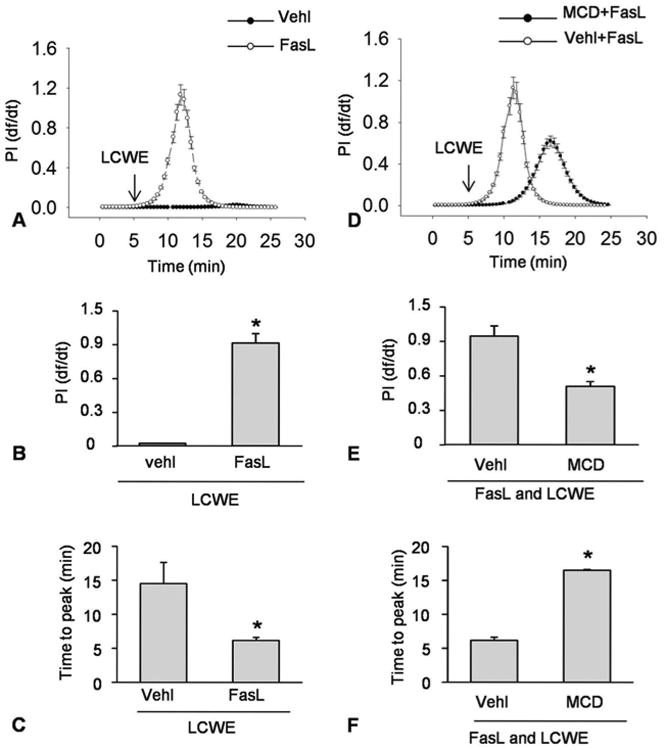
Enhanced MR clustering blocked membrane resealing during LCWE-induced injury. MVECs were stimulated without (control: ctrl) or with LCWE (15 μg/ml) in the absence and presence of MR clustering inducer (FasL) and MR clustering inhibitor (MCD). A and B) Representative curves depicting the relationship of FasL enhanced LCWE-induced the degree of plasma membrane injury to the flow velocity of PI by change in relative fluorescence with time (df/dt) of PI fluorescence. C and D) Representative histograms showing the maximal df/dt (N=6). E and F) Representative histograms presenting the time to reach maximal df/dt (N=6). *P < 0.0.5 vs. LCWE or FasL + LCWE.
4.4. LCWE-induced plasma membrane injury and repair in MVECs analyzed by flow cytometry
In addition to dynamic observation of cell membrane injury and resealing, we also performed cytometric analysis to confirm LCWE-induced plasma membrane injury and repair and to test whether ASM-mediated formation of MR platforms is involved in this LCWE-induced plasma membrane injury. In these experiments, MVECs were first stained by the SYTOX dye to determine cell viability and then these cells were treated using LCWE. After 5 minutes treatment, the cells were stained by PI dye and then immediately counted using flow cytometry (Figure 4A and 4C). If plasma membrane was injured, the PI would enter cells and be stained. Increases of the cell numbers in Q4 area indicate enhanced cell damage. The cell numbers in Q1 and Q2 areas are normal and membrane repaired cells. As summarized in Figure 4B, LCWE-induced cell injury was significantly enhanced by FasL as shown largely increased cell number in the area of Q4. In the presence of MRs disruptor, MCD, damaged cells induced by LCWE and enhanced by FasL were substantially reduced. Similar results were obtained when the Asm gene in the cells was silenced, as showed by significant decrease in damaged cells induced by FasL/LCWE treatments (Figure 4D).
Figure 4.
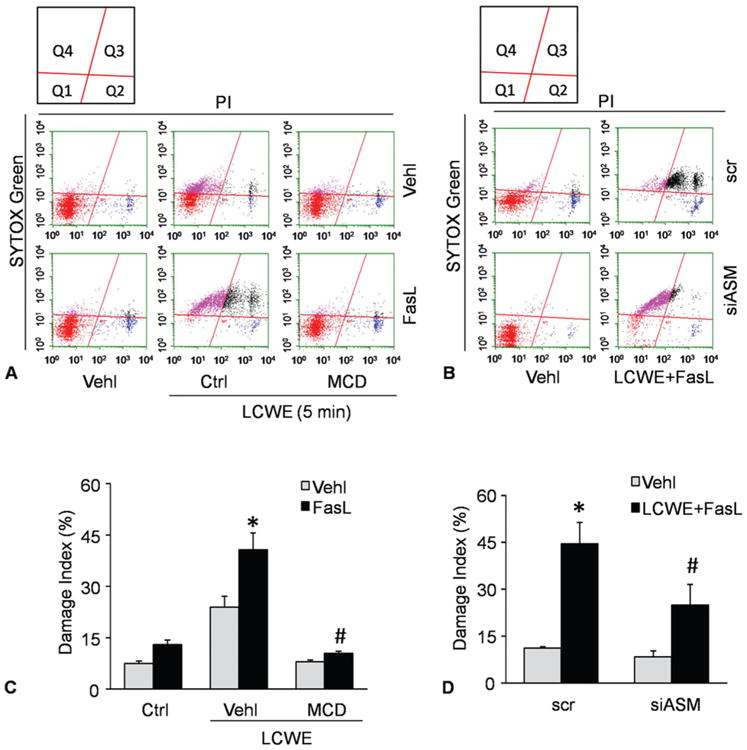
LCWE-induced plasma membrane injury and repair in MVECs analyzed by flow cytometry. MVECs were stimulated without (control: ctrl) or with LCWE (15 μg/ml, 4 h) in the absence and presence of MR clustering inducer (FasL) and MR clustering inhibitor (MCD). A and B) Representative dot plots of sorted cells by flow cytometry, where MVECs were first stained by the SYTOX dye and the by PI to determine cell viability. Summarized data in panels C and D showing the cell number of Q4 area. *P < 0.0.5 vs. vehicle control; #P < 0.0.5 vs. vehicle + FasL + LCWE.
4.5. Role of decreased cell plasma membrane resealing in activation of Nlrp3 inflammasome by LCWE
In previous studies, Nlrp3 inflammasome has been demonstrated to trigger the endothelial dysfunction induced by LCWE in MVECs (13). The present study determined whether cell membrane injury due to failed membrane repairing importantly contributes the formation and activation of Nlrp3 inflammasome induced by LCWE. By treatment of MVECs with an artificial cell membrane resealing compound, VA64, we observed Nlrp3 inflammasome activation by determination of the colocalization of Nlrp3 inflammasome components, the cleavage of pro-caspase-1 to active caspase-1, and the production of IL-1beta in response to LCWE stimulation. As shown in Figure 5A, LCWE increased the co-localization between Nlrp3 (green) and ASC (red) as shown by increased yellow spots in overlaid images. In the presence of VA64, however, LCWE failed to increase colocalization of these Nlrp3 inflammasome molecules. The co-localization coefficient of Nlrp3 vs. ASC or caspase-1 was summarized in Figure 5B and 5C, showing that enhanced membrane resealing during cell injury by LCWE significantly inhibited the co-localization of Nlrp3 vs. ACS or caspase-1, which indicates the blockade of the formation of Nlrp3 inflammasomes in these ECs. By Western blot analysis, we further demonstrated that LCWE increased Nlrp3 inflammasome activity in MVECs as shown by increased level of active caspase-1 level (cle-casp-1 in Figure 5D), which was blocked by VA64. In addition, biochemical analyses demonstrated that LCWE-induced increases in IL-1beta production and caspase-1 activity were significantly attenuated by VA64, further suggesting that membrane resealing enhancement prevents MVECs from Nlrp3 inflammasome formation and activation.
Figure 5.
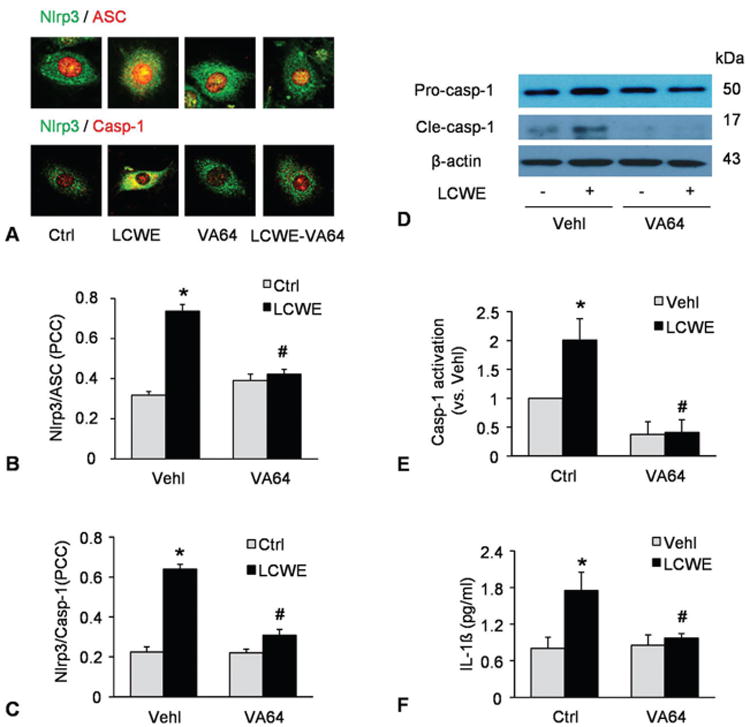
Role of decreased cell plasma membrane resealing in activation of Nlrp3 inflammasome by LCWE. MVECs were stimulated without (control: ctrl) or with LCWE (15 μg/ml, 8 h) in the absence and presence of artificial cell membrane resealing reagent (VA64). A) Confocal microscopic images of MVECs stained with Alexa488-conjugated ant-Nlrp3 and Alexa555-conjugated anti-ASC or anti-caspase-1 antibodies. The co-localization is shown by yellow and Nlrp3 signal is green with ASC signal in red. or Nlrp3 in green vs.) caspase-1 in red. B and C) Summarized data showing the co-localization coefficiency of Nlrp3 with ASC or with caspase-1 (N=4). D-E) Western blot analysis of cleaved caspase-1 and pro-caspase-1 in MVECs (N=3). F) IL-1beta production in MVECs by ELISA (N=5). *P < 0.0.5 vs. vehicle control; #P < 0.0.5 vs. vehicle + LCWE.
4.6. FasL enhanced LCWE-induced inflammasome formation and activation in MVECs
We next determined whether FasL-stimulated formation of MR platforms enhanced Nlrp3 inflammasome activation by LCWE in cultured MVECs. As shown in Figure 6A-C, FasL enhanced LCWE-induced Nlrp3 inflammasome formation in MVECs, which was prevented by MR disruptor MCD. Consistently, Western blot analysis also confirmed that FasL enhanced LCWE-induced caspase-1 activation in MVECs, as shown by increased production of cleaved caspase-1 and IL-1beta level. In the presence of MCD, however, FasL-enhanced inflammasome activation was prevented (Figure 6D-F). These results demonstrate that FasL-induced MR clustering induces deficiency of cell membrane repair and thereby enhances LCWE-induced Nlrp3 inflammasome formation and activation.
Figure 6.
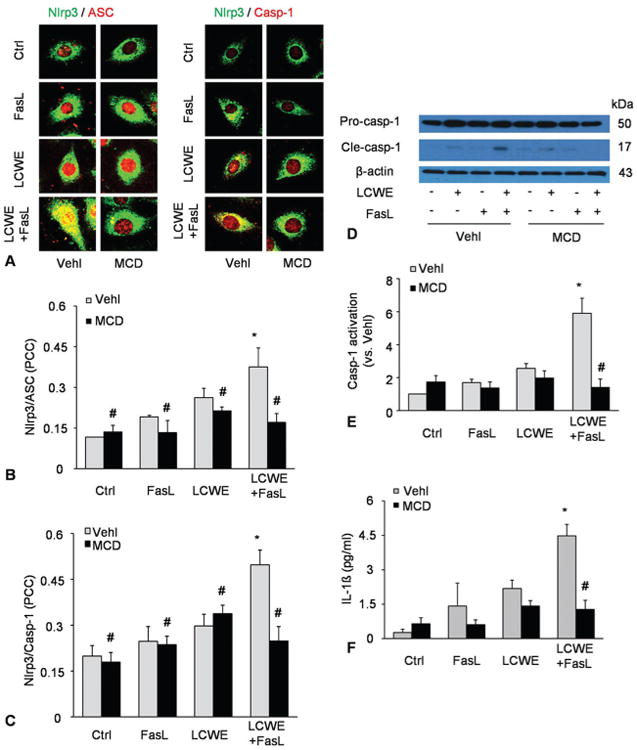
FasL enhanced LCWE-induced inflammasome formation and activation in MVECs. MVECs were stimulated without (control: ctrl) or with LCWE (15 μg/ml, 4 h) in the absence and presence of MR clustering inducer (FasL) and MR clustering inhibitor (MCD). A-C) Confocal microscopic images of MVECs stained with Alexa488-conjugated ant-Nlrp3 and Alexa555-conjugated anti-ASC or anti-caspase-1 antibodies. B and C) The co-localization coefficiency of Nlrp3 with ASC or Nlrp3 with caspase-1 (N=5). D-E) Western blot analysis of cleaved caspase-1 and pro-caspase-1 in MVECs (N=4). (F) IL-1beta production in MVECs by ELISA (N=3). *P < 0.0.5 vs. vehicle control; #P < 0.0.5 vs. vehicle + LCWE.
4.7. Blockade by ASM gene silencing of FasL-enhanced Nlrp3 inflammasome activation upon LCWE stimulation
Given that ASM product, ceramide is importantly involved in MRs clustering, we determined whether FasL enhancement of LCWE-induced Nlrp3 inflammasome activation is associated with ASM-mediated ceramide production. As shown in Figure 7A-C, confocal microscopy showed that when ASM gene was silenced, FasL failed to enhance the formation of Nlrp3 inflammasome in MVECs upon LCWE stimulation as shown by the lack of colocalization of Nlrp3 vs ASC or caspase-1. This ASM gene silencing also blocked increases in LCWE-induced cleaved caspase-1 amount and IL-1beta level in the presence of FasL (Figure 7D-E).
Figure 7.
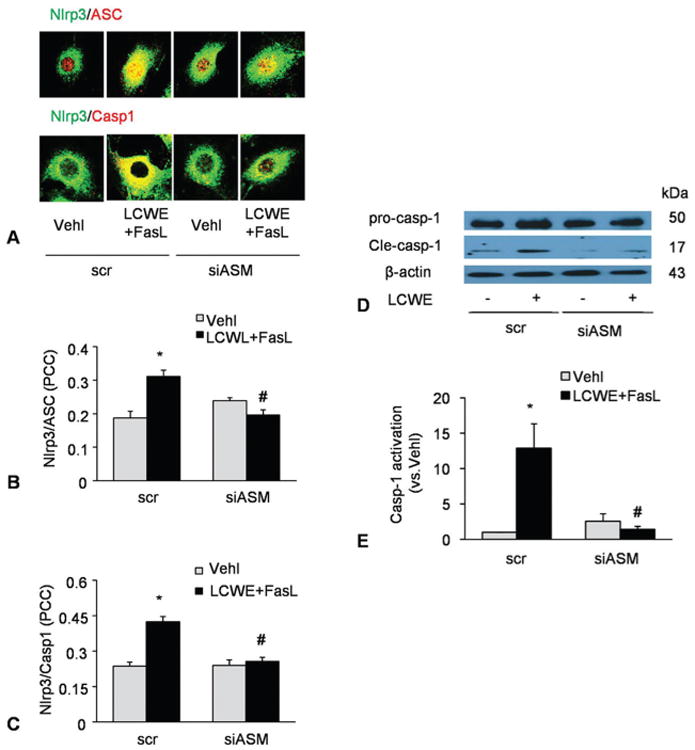
Blockade by ASM gene silencing of FasL-enhanced Nlrp3 inflammasome activation upon LCWE stimulation. MVECs were transfected with scrambled (scr) or ASM siRNA (ASM si) for 24 h and then stimulated with or without FasL (10 ng/ml, 6 h)-LCWE (15 μg/ml, 4 h). A-C) Confocal microscopic images and summarized co-localization coefficient showing the co-localization (yellow) between Nlrp3 (green) and caspase-1(red) or Nlrp3 (green) and ASC (red) (N=5). D-E) Western blot analysis of cleaved caspase-1 and pro-caspase-1 (N=4). *P < 0.0.5 vs. scramble control; #P < 0.0.5 vs. scramble + FasL + LCWE.
4.8. FasL enhancement of LCWE-induced lysosome membrane permeability and cathepsin B activation
It has been reported that LCWE-induced Nlrp3 inflammasome activation in ECs is due to damaged lysosome membrane permeability and increased cathepsin B activity (13). In the present study, we investigated whether FasL enhanced LCWE-induced inflammasome activation is also due to downstream lysosome membrane permeability increase and consequent cathepsin B activation. As shown in Figure 8, acridine orange (AO) staining in MVECs was shown in normal lysosomes by red fluorescence (2 left panels). In response to LCWE, the red fluorescence in the cells pretreated with FasL disappeared, while almost all cells had only show green fluorescence, which was due to lysosomal alkalization, suggesting increases in lysosome permeability. When ASM gene in these cells was silenced, the red fluorescence from AO staining remained unchanged no matter whether LCWE was used in the absence or presence of FasL, suggesting that ASM gene silencing blocked LCWE-induced increase in lysosome permeability.
Figure 8.
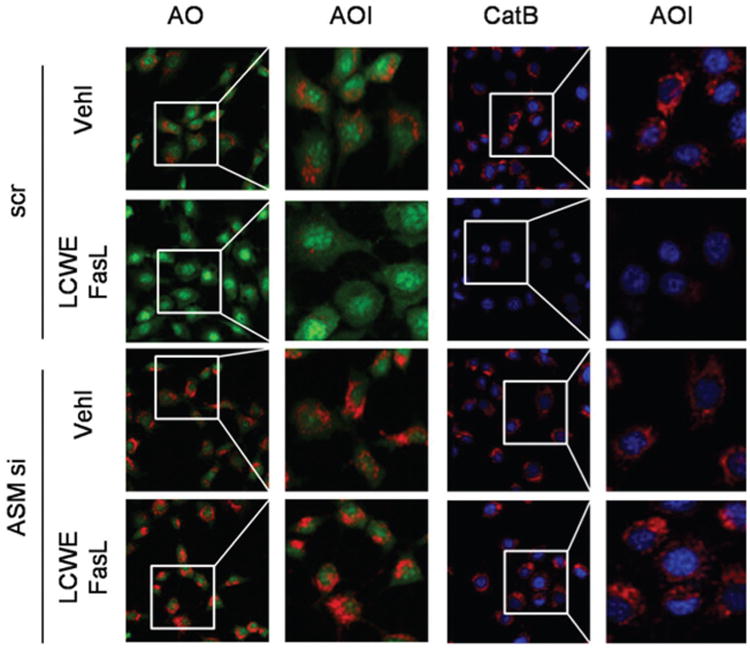
FasL enhancement of LCWE-induced lysosome membrane permeability and cathepsin B activation in MVECs. MVECs were transfected with scrambled (scr) or ASM siRNA (ASM si) for 24 h and then stimulated with or without FasL or FasL+LCWE. Lysosome membrane permeability and cathepsin B activation were stained with acridine orange (AO) and z-Arg-Arg-cresyl violet, resepectively, which was visualized by fluorescent microscopy. Enlarged images of area of interest (AOI) are indicated with a box, indicating lysosome permeability or cathepsin B activity.
By loading a fluorescent substrate, z-Arg-Arg-cresyl violet into MVECs, we detected cathespin B activity mainly in lysosomes as shown by red fluorescence when cells were treated only with vehicle. However, z-Arg-Arg-cresyl violet fluorescence disappeared from lysosomes when these cells were stimulated by LCWE with or without FasL pretreatment (2 right panels). This disappearance of z-Arg-Arg-cresyl violet fluorescence suggests the leakage of cathepsin B even out of cells. In MVECs with ASM gene silencing, LCWE caused much less cathepsin B leakage even in the presence of FasL.
5. Discussion
The major goal of the present study is to determine whether instant membrane resealing associated with MRs protects ECs from inflammatory challenges. We demonstrated that MR clustering impaired the instant plasma membrane resealing capability in ECs, which initiated or enhanced the formation and activation of Nlrp3 inflammasomes. Failure of membrane resealing led to lysosomal instability and cathepsin B activity, resulting in activation of Nlrp3 inflammasomes in response to LCWE stimulation. FasL-enhanced impairment of membrane resealing upon LCWE was almost completely blocked by a plasma membrane resealing agent, V64. It has been suggested that an normal instant membrane repairing mechanism importantly protects ECs from danger factors, which helps these cells from the canonical and uncanonical injurious effects induced by Nlrp3 inflammasome activation and may prevent cardiovascular diseases associated with endothelial dysfunction or injury.
We first observed the dynamic changes in plasma membrane injury and repair when ECs are challenged by membrane damaging factors or disruption agents such as LCWE and saponin. Using the high speed wavelength-switching fluorescent microscopy, we demonstrated that upon LCWE or saponin stimulation, a Ca2+-dependent membrane resealing mechanism mediates membrane repair when these ECs were challenged by both membrane disruptors. It is interesting that this membrane resealing occurred when intracellular Ca2+ increased as shown by Ca2+ transient peak precedes df/dt of PI fluorescence, suggesting that Ca2+ may exert an activating effect of this membrane resealing in ECs. Our results are similar to some previous reports that a Ca2+ mediated plasma membrane repairing mechanism also exists in fibroblast cells (41). It has been proposed that this instant cell membrane resealing is an active process that requires extracellular Ca2+, intracellular vesicles and a step of Ca2+-dependent exocytosis (38).
The present study also tested whether the instant membrane resealing of ECs in response to LCWE or saponin is associated with ceramide-enriched microdomains or MRs and their clustering. It has been indicated that MRs are present in a variety of mammalian cells and that this special small lipid structure is considered to be an important signaling component in the plasma membrane of a variety of mammalian cells. In this regard, MR clustering is shown activate, facilitate or amplify signal transduction including the signaling elicited by danger factors (42). In other studies, acid sphingomyelinase and its product ceramide have been reported to importantly participate in the wound healing of different cells (43, 44). Although these studies indicated that ceramide or related MRs are critical to the wound healing of cell membrane injury, the present study showed that clustering of these ceramide-based MRs by FasL, a strong inducer of MR cluster formation in ECs (6) significantly attenuated the cell membrane resealing when ECs are exposed to LCWE or saponin. In the presence of rafts clustering inhibitor, this attenuation of the cell membrane resealing in ECs was almost completely blocked. These results suggest that the clustering of MRs abolishes the capability of ceramide or its MRs to mediate cell membrane resealing or wound healing when facing danger factors.
One of the important findings from the present study is that failure of membrane resealing led to the formation and activation of Nlrp3 inflammasomes in MVECs, which was blocked by membrane-resealing agent VA64. It is suggested that an intact membrane resealing process protects ECs from the activation of Nlrp3 inflammasomes. This membrane resealing mechanism may be of importance in the development of vascular diseases associated with inflammation because the activation of Nlrp3 inflammasomes is recently considered as a major pathogenic mechanism during the development of atherosclerosis or other vascular diseases through its inflammatory and non-inflammatory actions (13, 16, 23, 24, 45-47). This activation of endothelial Nlrp3 inflammasomes due to disrupted membrane resealing was also confirmed by induction of MR clustering during FasL stimulation. In this regard, FasL as an MR clustering activator was found to impair membrane resealing and consequently result in Nlrp3 inflammation activation, which was blocked by MR disruptor, MCD or silencing of ASMase, an enzyme promoting MR clustering in ECs (48). To our knowledge, these results provide the first evidence that enhanced MR clustering leads to failure of membrane resealing in ECs when exposed to danger factors, whereby Nlrp3 inflammasomes are formed and activated. This activation of Nlrp3 may be one of the important mechanisms resulting in endothelial dysfunction and related vascular diseases such as atherosclerosis, vascular inflammation or hypertension.
To explore the mechanisms by which impairment of membrane resealing produces the Nlrp3 inflammasome formation and activation in ECs, we tested the contribution of the lysosomal instability and cathepsin B release. In a previous study, we demonstrated that LCWE activated the Nlrp3 inflammasome by increased lysosomal permeability and cathepsin B activation (13). However, it remains unknown whether this increase in lysosomal permeability and consequent cathepsin B activation are associated with impaired membrane resealing process. It was demonstrated that FasL enhanced LCWE-induced increases in lysosomal membrane permeability and activation of lysosomal cathepsin B release or activity. ASM gene silencing and MR clustering inhibition abolished FasL-enhanced lysosomal membrane permeability and activation of lysosomal cathepsin B activation. These results provide evidence that impaired membrane resealing due to FasL-induced MR clustering induces Nlrp3 inflammasome activation through lysosome-cathepsin B pathway. The present study did not attempt to further explore the mechanism by which impaired membrane resealing induces lysosomal permeabilization. Based on previous studies, however, increased lysosomal permeabilization during impaired plasma membrane resealing may be associated with release of lipid mediators, free radicals or some receptors activation or loss of function (49, 50).
In summary, the present study demonstrated that a rapid membrane resealing in ECs in response to membrane disrupting stimuli such as LCWE or saponin. This membrane resealing mechanism was found to be a Ca2+ dependent and ceramide regulated instant event in these cells. MR clustering induced by FasL impaired this endothelial membrane resealing process, which activated Nlrp3 inflammasome via a lysosomal permeabilization and cathepsin B-mediated pathway. It is concluded that MR clustering or ceramide-enrichment macrodomain formation in EC membrane damages the instant resealing or repairing process in ECs upon injury factors, thereby resulting Nlrp3 inflammasome action and triggering endothelial or vascular inflammation. Prevention of impairment of membrane resealing or repair may protect ECs or arteries from pathological changes associated with inflammasome activation in face of challenges from membrane injury factors.
Acknowledgments
This study was supported by grants from the National Institutes of Health (HL057244, HL122937, HL075316 and HL122769).
Abbreviations
- CMR
cell membrane resealing
- MVEC
mouse microvascular endothelial cell
- LCWE
Lactobacillus casei cell wall fragments
- MR
membrane raft
- ASM
acid sphingomyelinase
- Nlrp3
NOD-like receptor pyrin domain containing 3
References
- 1.McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. doi:10.1146/annurev.cellbio.19. 111301.140101. [DOI] [PubMed] [Google Scholar]
- 2.McNeil PL. Repairing a torn cell surface: make way, lysosomes to the rescue. J Cell Sci. 2002;115(Pt 5):873–9. doi: 10.1242/jcs.115.5.873. [DOI] [PubMed] [Google Scholar]
- 3.Abreu-Blanco MT, Watts JJ, Verboon JM, Parkhurst SM. Cytoskeleton responses in wound repair. Cell Mol Life Sci. 2012;69(15):2469–83. doi: 10.1007/s00018-012-0928-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. 2010;189(6):1027–38. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babiychuk EB, Monastyrskaya K, Draeger A. Fluorescent annexin A1 reveals dynamics of ceramide platforms in living cells. Traffic. 2008;9(10):1757–75. doi: 10.1111/j.1600-0854.2008.00800.x. doi:10.1111/j.1600-0854. 2008.00800.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006;47(1):74–80. doi: 10.1161/10.1161/01.HYP.0000196727.53300.62. doi:10.1161/10.1161/01. HYP.0000196727.53300.62. [DOI] [PubMed] [Google Scholar]
- 7.Boini KM, Zhang C, Xia M, Han WQ, Brimson C, Poklis JL, Li PL. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys Acta. 2010;1801(12):1294–304. doi: 10.1016/j.bbalip.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li PL, Zhang Y, Yi F. Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid Redox Signal. 2007;9(9):1457–70. doi: 10.1089/ars.2007.1667. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Xu M, Pitzer AL, Xia M, Boini KM, Li PL, Zhang Y. Control of autophagy maturation by acid sphingomyelinase in mouse coronary arterial smooth muscle cells: protective role in atherosclerosis. J Mol Med (Berl) 2014;92(5):473–85. doi: 10.1007/s00109-014-1120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL. Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One. 2014;9(1):e87552. doi: 10.1371/journal.pone.0087552. doi:10.1371/journal.pone. 0087552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi: 10.1016/j.redox.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–86. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, Li PL. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. 2015;1853(2):396–408. doi: 10.1016/j.bbamcr.2014.11.012. doi:10.1016/j.bbamcr. 2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JH, Johnson DA, Shyy JY. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128(6):632–42. doi: 10.1161/CIRCULATIONAHA.113.002714. doi:10.1161/CIRCULATIONAHA. 113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin Y, Pastrana JL, Li X, Huang X, Mallilankaraman K, Choi ET, Madesh M, Wang H, Yang XF. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front Biosci (Landmark Ed) 2013;18:638–49. doi: 10.2741/4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol. 2014;184(5):1617–28. doi: 10.1016/j.ajpath.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. doi:10.1016/S1097-2765(02) 00599-3. [DOI] [PubMed] [Google Scholar]
- 19.Lamkanfi M, Malireddi RK, Kanneganti TD. Fungal zymosan and mannan activate the cryopyrin inflammasome. J Biol Chem. 2009;284(31):20574–81. doi: 10.1.074/jbc.M109.0.23689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281(48):36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 21.Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis - a cell death modality of its kind? Eur J Immunol. 2010;40(3):627–30. doi: 10.1002/eji.200940160. [DOI] [PubMed] [Google Scholar]
- 22.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32(8):373–9. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicolson GL, Ash ME. Lipid Replacement Therapy: a natural medicine approach to replacing damaged lipids in cellular membranes and organelles and restoring function. Biochim Biophys Acta. 2014;1838(6):1657–79. doi: 10.1016/j.bbamem.2013.11.010. doi:10.1016/j.bbamem.2013.11 .010. [DOI] [PubMed] [Google Scholar]
- 24.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–60. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 25.Henry B, Ziobro R, Becker KA, Kolesnick R, Gulbins E. Acid sphingomyelinase. Handb Exp Pharmacol. 2013;(215):77–88. doi: 10.1007/978-3-7091-1368-4_4. [DOI] [PubMed] [Google Scholar]
- 26.Kornhuber J, Muller CP, Becker KA, Reichel M, Gulbins E. The ceramide system as a novel antidepressant target. Trends Pharmacol Sci. 2014;35(6):293–304. doi: 10.1016/j.tips.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Gulbins E, Li PL. Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol. 2006;290(1):R11–26. doi: 10.1152/ajpregu.00416.2005. [DOI] [PubMed] [Google Scholar]
- 28.Xiong J, Xia M, Xu M, Zhang Y, Abais JM, Li G, Riebling CR, Ritter JK, Boini KM, Li PL. Autophagy maturation associated with CD38-mediated regulation of lysosome function in mouse glomerular podocytes. J Cell Mol Med. 2013;17(12):1598–607. doi: 10.1111/jcmm.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang AY, Yi F, Teggatz EG, Zou AP, Li PL. Enhanced production and action of cyclic ADP-ribose during oxidative stress in small bovine coronary arterial smooth muscle. Microvasc Res. 2004;67(2):159–67. doi: 10.1016/j.mvr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–50. No DOI Found. [PubMed] [Google Scholar]
- 31.Xu M, Li X, Walsh SW, Zhang Y, Abais JM, Boini KM, Li PL. Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes. Am J Physiol Cell Physiol. 2013;304(5):C458–66. doi: 10.1152/ajpcell.00342.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu M, Li XX, Chen Y, Pitzer AL, Zhang Y, Li PL. Enhancement of dynein-mediated autophagosome trafficking and autophagy maturation by ROS in mouse coronary arterial myocytes. J Cell Mol Med. 2014;18(11):2165–75. doi: 10.1111/jcmm.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mbye LH, Keles E, Tao L, Zhang J, Chung J, Larvie M, Koppula R, Lo EH, Whalen MJ. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2012;32(3):515–24. doi: 10.1038/jcbfm.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller BF, Keles E, Tien L, Zhang J, Kaplan D, Lo EH, Whalen MJ. The pharmacokinetics and pharmacodynamics of Kollidon VA64 dissociate its protective effects from membrane resealing after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2014;34(8):1347–53. doi: 10.1038/jcbfm.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang AY, Yi F, Jin S, Xia M, Chen QZ, Gulbins E, Li PL. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal. 2007;9(7):817–28. doi: 10.1089/ars.2007.1509. [DOI] [PubMed] [Google Scholar]
- 36.Jacob MC, Favre M, Bensa JC. Membrane cell permeabilization with saponin and multiparametric analysis by flow cytometry. Cytometry. 1991;12(6):550–8. doi: 10.1002/cyto.990120612. [DOI] [PubMed] [Google Scholar]
- 37.Yi FX, Zhang AY, Campbell WB, Zou AP, Van Breemen C, Li PL. Simultaneous in situ monitoring of intracellular Ca2+ and NO in endothelium of coronary arteries. Am J Physiol Heart Circ Physiol. 2002;283(6):H2725–32. doi: 10.1152/ajpheart.00428.2002. [DOI] [PubMed] [Google Scholar]
- 38.Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. 2008;180(5):905–14. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S, Liu T, Liang H, Zhang H, Yan D, Wang N, Jiang X, Feng W, Wang J, Li P, Li Z. Lipid rafts uncouple surface expression of transmembrane TNF-alpha from its cytotoxicity associated with ICAM-1 clustering in Raji cells. Mol Immunol. 2009;46(7):1551–60. doi: 10.1016/j.molimm.2009.01.001. doi:10.1016/j.molimm.2009.01. 001. [DOI] [PubMed] [Google Scholar]
- 40.Bao JX, Xia M, Poklis JL, Han WQ, Brimson C, Li PL. Triggering role of acid sphingomyelinase in endothelial lysosome-membrane fusion and dysfunction in coronary arteries. Am J Physiol Heart Circ Physiol. 2010;298(3):H992–H1002. doi: 10.1152/ajpheart.00958.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murata N, Ito S, Furuya K, Takahara N, Naruse K, Aso H, Kondo M, Sokabe M, Hasegawa Y. Ca2+ influx and ATP release mediated by mechanical stretch in human lung fibroblasts. Biochem Biophys Res Commun. 2014;453(1):101–5. doi: 10.1016/j.bbrc.2014.09.063. [DOI] [PubMed] [Google Scholar]
- 42.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 43.Defour A, Van der Meulen JH, Bhat R, Bigot A, Bashir R, Nagaraju K, Jaiswal JK. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014;5:e1306. doi: 10.1038/cddis.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaughan EM, You JS, Elsie Yu HY, Lasek A, Vitale N, Hornberger TA, Bement WM. Lipid domain-dependent regulation of single-cell wound repair. Mol Biol Cell. 2014;25(12):1867–76. doi: 10.1091/mbc.E14-03-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. 2013;122(20):3405–14. doi: 10.1182/blood-2013-05-504449. doi:10.1182/blood-2013 -05-504449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, Yang XF. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol. 2009;22(2):311–22. doi: 10.1177/039463200902200208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang H, Yang XF. Uric acid, hyperuricemia and vascular diseases. Front Biosci (Landmark Ed) 2012;17:656–69. doi: 10.2741/3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stancevic B, Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010;584(9):1728–40. doi: 10.1016/j.febslet.2010.02.026. doi:10.1016/j.febslet.2010 .02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denamur S, Tyteca D, Marchand-Brynaert J, Van Bambeke F, Tulkens PM, Courtoy PJ, Mingeot-Leclercq MP. Role of oxidative stress in lysosomal membrane permeabilization and apoptosis induced by gentamicin, an aminoglycoside antibiotic. Free Radic Biol Med. 2011;51(9):1656–65. doi: 10.1016/j.freeradbiomed.2011.07.015. doi:10.1016/j.freeradbiomed .2011.07.015. [DOI] [PubMed] [Google Scholar]
- 50.Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal. 2012;17(5):766–74. doi: 10.1089/ars.2011.4405. [DOI] [PubMed] [Google Scholar]