

Public databases have been increasingly used in a clinical setting; however, the well‐documented limitations of these databases call into question how often clinicians will encounter discordant variant classifications that may introduce uncertainty into patient management. This study evaluated discordance in BRCA1 and BRCA2 variant classifications between a single commercial testing laboratory and a public database commonly consulted in clinical practice.
Keywords: Variant classification, Public databases, BRCA1, BRCA2, Genetic testing
Abstract
Background.
There is a growing move to consult public databases following receipt of a genetic test result from a clinical laboratory; however, the well‐documented limitations of these databases call into question how often clinicians will encounter discordant variant classifications that may introduce uncertainty into patient management. Here, we evaluate discordance in BRCA1 and BRCA2 variant classifications between a single commercial testing laboratory and a public database commonly consulted in clinical practice.
Materials and Methods.
BRCA1 and BRCA2 variant classifications were obtained from ClinVar and compared with the classifications from a reference laboratory. Full concordance and discordance were determined for variants whose ClinVar entries were of the same pathogenicity (pathogenic, benign, or uncertain). Variants with conflicting ClinVar classifications were considered partially concordant if ≥1 of the listed classifications agreed with the reference laboratory classification.
Results.
Four thousand two hundred and fifty unique BRCA1 and BRCA2 variants were available for analysis. Overall, 73.2% of classifications were fully concordant and 12.3% were partially concordant. The remaining 14.5% of variants had discordant classifications, most of which had a definitive classification (pathogenic or benign) from the reference laboratory compared with an uncertain classification in ClinVar (14.0%).
Conclusion.
Here, we show that discrepant classifications between a public database and single reference laboratory potentially account for 26.7% of variants in BRCA1 and BRCA2. The time and expertise required of clinicians to research these discordant classifications call into question the practicality of checking all test results against a database and suggest that discordant classifications should be interpreted with these limitations in mind.
Implications for Practice.
With the increasing use of clinical genetic testing for hereditary cancer risk, accurate variant classification is vital to ensuring appropriate medical management. There is a growing move to consult public databases following receipt of a genetic test result from a clinical laboratory; however, we show that up to 26.7% of variants in BRCA1 and BRCA2 have discordant classifications between ClinVar and a reference laboratory. The findings presented in this paper serve as a note of caution regarding the utility of database consultation.
Introduction
Early in the development of genetic testing, public databases were established as a mechanism for sharing user‐submitted research data to facilitate the identification of causal relationships between genetic variants and patient health status [1], [2], [3], [4]. Although this data sharing has been valuable as a research tool, clinical utilization of information in these databases has been discouraged due to multiple reports of false assignments of pathogenicity [3], [5], [6], [7], [8], [9]. With the increased application of genetic testing in medicine and the use of test results to guide patient management [3], [10], [11], the accuracy of clinical variant classification is receiving increased attention. For example, individuals found to carry a pathogenic variant in BRCA1 or BRCA2 are recommended for increased breast cancer screening and may be candidates for prophylactic mastectomy or oophorectomy [10]. Accurate variant classification is a critical step in ensuring that these interventions are targeted to appropriate patients.
The American College of Medical Genetics and Genomics (ACMG) suggests that the clinical pathogenicity of a variant be evaluated using multiple lines of evidence from available literature, structural/functional data, population frequencies, and statistical analyses of clinical data [12]. Ideally, clinical variant classification should be a dynamic process based on expert review of all available evidence. Although most commercial and academic genetic testing centers use the ACMG guidelines as the foundation for variant classification, some of the elements are subjective and open to interpretation. Therefore, it is possible for testing laboratories to arrive at different clinical classifications.
Many believe that public databases can serve as valuable tools for sharing variant classifications and facilitating the large‐scale collection and curation of the evidence used for establishing clinical significance. However, it is also widely agreed that significant improvements and modifications are necessary before most existing databases can be used clinically [1], [3], [6], [9], [13], [14]. The suggested requirements to retrofit existing or develop new databases for clinical use include (a) regular maintenance of database entries by a team with varied and complimentary expertise [1], [3], [6], (b) establishment of standard procedures to evaluate variant pathogenicity [1], [6], [9], (c) systematic phenotype collection [1], [6], [13], (d) dynamic review of data [6], [14], and (e) a mechanism to inform users of changes in variant classification [9], [15].
Despite the known limitations of data quality in public databases, some health care providers routinely consult these resources following receipt of a genetic test result from a clinical laboratory with the intent of identifying discrepant classifications [16]. However, it is unclear whether database consultation by health care providers improves patient care. Although confirmation of a test result may add confidence to medical management decisions, previous studies show that there is a high frequency of discrepancies within and between databases [9]. Because databases rely on user‐submitted information, there is often inconsistent documentation of the evidence supporting classification. This may make it difficult for clinicians to critically evaluate the significance of discrepancies with a laboratory classification. In addition, there is some evidence that databases may have a higher frequency of uncertain classifications compared with commercial laboratories [16]. These issues may add uncertainty to management decisions, even when a definitive classification is provided by the testing laboratory.
Despite these known issues, there is little information available about how often a clinician will encounter discordance between a laboratory classification and the information available in a publicly accessible database. This study evaluates the extent of the discordance for the genes BRCA1 and BRCA2 by comparing the classifications for variants from a single commercial testing laboratory with those in ClinVar, a public database commonly consulted in clinical practice. In order to further assess how discordant findings may impact clinical management, reasons for discordance (i.e., uncertain database classification versus definitive laboratory classification) were also evaluated.
Materials and Methods
ClinVar Database
Variant classifications for BRCA1 and BRCA2 were obtained from the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/). ClinVar is a government archive hosted by the National Center for Biotechnology Information (NCBI) that contains contributions at many levels, from basic reports to authoritative reviews or guidelines [2]. Contributors include both academic and commercial laboratories as well as professional organizations and societies. Although ClinVar sets the editorial and structural standards for submitted data, the intellectual content comes from the submitter. As such, ClinVar may list multiple entries for a single variant that disagree with each other. ClinVar is a component of ClinGen, which was established in 2013 to be a central resource for public variant data [4]. ClinGen offers additional curation measures, such as rating the level of evidence for a variant classification, with the hope of facilitating clinical use [4].
Database Classifications
The full ClinVar database was downloaded on February 25, 2015 and filtered to show entries for BRCA1 and BRCA2 (n = 6,417). Duplicate entries were identified using the Human Genome Variation Society names, a standard nomenclature format, entered in the databases. For this analysis, database classifications were divided into three groups (pathogenic, benign, and uncertain). Variants with “pathogenic” or “likely pathogenic” database classifications were considered pathogenic. Variants with “benign” or “likely benign” classifications were considered benign. Variants with database classifications of “variant of unknown significance” or “uncertain significance” were considered uncertain.
For case examples, the evidence of pathogenicity listed by ClinVar submitters as of March 10, 2017 was included. The pathogenicity of ClinVar classifications for case examples did not change between February 2015 and March 2017. Inclusion of more recent literature enables the current classification landscape to be evaluated for the listed case examples.
Laboratory Variant Classification
The dynamic classification process utilized by the reference commercial testing laboratory examined here has been previously described [17] and is represented schematically in Figure 1. Briefly, this process involves a daily review of the functional and clinical implications of new variants by a classification committee consisting of over 30 scientists with expertise in a variety of fields, including molecular genetics, human genetics, population genetics, clinical genetics, structural biology, RNA/splicing, bioinformatics, and statistics. This committee also reviews new information for existing variants that may result in an updated classification.
Figure 1.

Overview of reference laboratory classification process.
Comparison of Variant Classifications
Clinical classifications for all variants reported by both the reference laboratory and ClinVar were identified and compared. Results were considered concordant if the pathogenicity was the same (Fig. 2). Because variants in ClinVar may have more than one classification listed, concordance and discordance were evaluated in two tiers. First, variants with only one ClinVar classification were considered as described in Figure 2. This includes variants with either a single entry or multiple entries that report the same pathogenicity (i.e., pathogenic and likely pathogenic). Second, variants with conflicting ClinVar classifications were considered partially concordant if at least one of the listed classifications agreed with the classification assigned by the reference laboratory process. For example, a variant that was classified as benign by the reference laboratory with entries in ClinVar classifying it as both a variant of uncertain significance (VUS) and benign would be considered partially concordant.
Figure 2.

Determination of classification concordance between the reference laboratory and database.
Abbreviations: VUS, variant of uncertain significance.
Discordant classifications were those with different pathogenicity (Fig. 2). For example, variants classified as uncertain by the database and benign by the laboratory would be considered discordant. The type of discordance was also documented in three potential categories based on definitive (benign or pathogenic) and uncertain classifications: database uncertain classification (the variant has an uncertain classification in the database and a definitive classification from the reference laboratory process), laboratory uncertain classification (the variant has a definitive classification in the database and an uncertain classification from the reference laboratory process), and opposite classification (the variant has opposite, definitive classifications from the database and reference laboratory process; Fig. 2).
Results
There were 4,250 unique BRCA1 and BRCA2 variants with entries in ClinVar that were eligible for analysis. Overall, 73.2% of variant classifications in ClinVar were concordant with the reference laboratory process (Table 1). This includes variants for which a single pathogenicity is reported. An additional 12.3% of variants had at least one ClinVar classification that agreed with the reference laboratory classification. This results in a total potential concordance of 85.6% between the classifications assigned by the reference laboratory process and the ClinVar database.
Table 1. Concordance between variant classifications from the reference laboratory and all database entries, as well as database entries from contributing commercial laboratories.
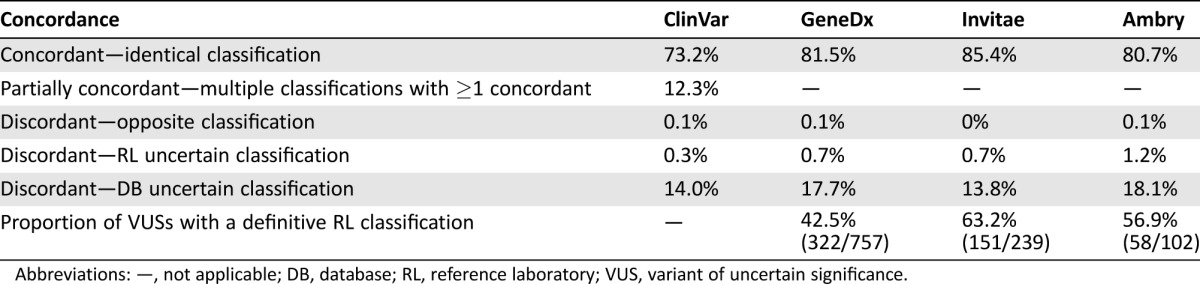
Abbreviations: —, not applicable; DB, database; RL, reference laboratory; VUS, variant of uncertain significance.
One example of a variant with partial concordance is BRCA1 c.427G>A (p.Glu143Lys), which is classified as both VUS and benign in ClinVar. Although multiple entries list different pathogenicity, Table 2 shows that both pathogenic and VUS ClinVar entries cite a study by Lindor et al. [18]. Although all listed literature [18], [19], [20], [21] was reviewed by the reference laboratory, the variant was classified as benign based on phenotypic evidence from a history‐weighting algorithm [22] and in trans observations (occurring in the same gene but different allele) with a known pathogenic variant [12].
Table 2. Case examples and evidence of pathogenicity for variants with discordant laboratory and database classification.
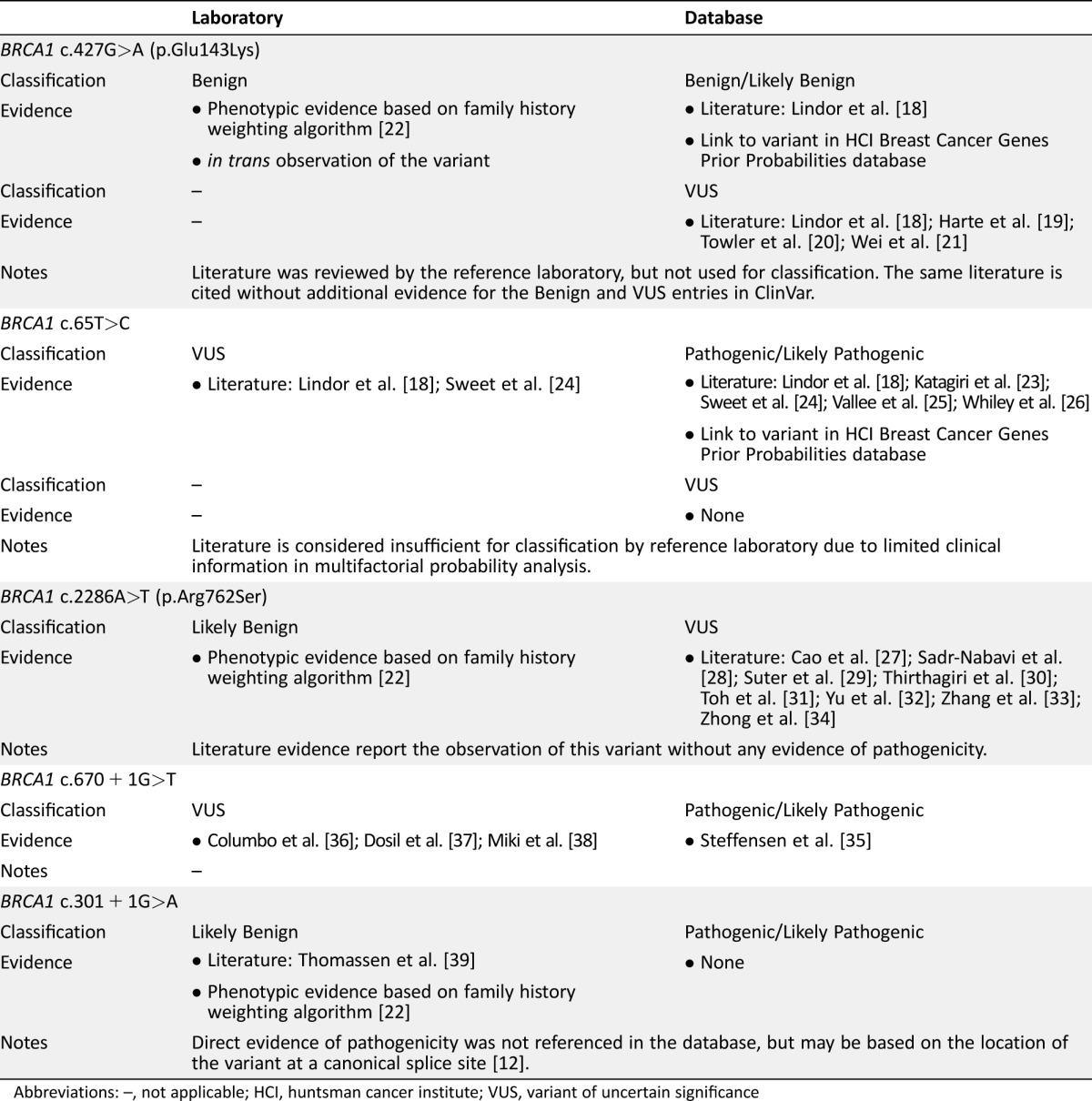
Abbreviations: –, not applicable; HCI, huntsman cancer institute; VUS, variant of uncertain significance
Another example of a partial concordance is BRCA1 c.65T>C (p.Leu22Ser), which has pathogenic/likely pathogenic and VUS entries in ClinVar. The pathogenic/likely pathogenic entries are based on multifactorial probability analyses predicting pathogenicity (Table 2) [18], [23], [24], [25], [26]. However, this evidence is not considered sufficient for a pathogenic clinical classification by the reference laboratory due to limited clinical information used in the probability analysis. Although the ClinVar submitters may have additional evidence for the pathogenicity of this variant, it is not specifically referenced in the database. In the absence of additional information, the reference laboratory continues to report BRCA1 c.65T>C as uncertain. For this variant, the same literature evidence is available for classification; however, the interpretation of the evidence differs.
The remaining 14.5% of variants had discordant classifications. The largest source of discordance was variants assigned a definitive classification (pathogenic or benign) by the reference laboratory process compared with an uncertain classification in ClinVar (14.0%; Table 1). For example, BRCA1 c.2286A>T (p.Arg762Ser) is classified as a VUS in ClinVar (Table 2). This variant has been previously reported in the literature in individuals of East Asian ancestry [27], [28], [29], [30], [31], [32], [33], [34] and has 14 observations in the Exome Aggregation Consortium database. This literature is referenced in ClinVar, and the absence of evidence of variant pathogenicity likely resulted in its uncertain classification. The reference laboratory process was able to assign a classification of likely benign based on phenotypic evidence from a history‐weighting algorithm [22].
A smaller percentage of discordant classifications were observed for variants that had a definitive database classification but were classified as VUS by the reference laboratory (0.3%). One such example is BRCA1 c.670+1G>T, which has likely pathogenic and pathogenic entries in ClinVar. This variant occurs at the +1 position, a canonical splice site at which any change is expected to disrupt gene function. As such, ACMG guidelines state that this is strong evidence of pathogenicity; however, ACMG guidelines also caution that certain caveats may impact the pathogenicity of these variants. Although a previous study has shown this variant affects splicing of exon 10 [35], there is also evidence of a naturally occurring transcript with an in‐frame deletion of exons 9‐10 in healthy individuals [36], [37], [38]. This alternate transcript may produce enough functional protein to negate the cancer risks expected for a variant at the +1 position. In the absence of clinical information to assess the actual effect BRCA1 c.670+1G>T given the alternate transcript, the reference laboratory classifies BRCA1 c.670+1G>T as uncertain.
In a small number of cases, the database and reference laboratory provide definitive but opposite classifications (0.1%). For example, BRCA1 c.301 + 1G>A is listed as likely pathogenic in ClinVar but is reported as likely benign by the reference laboratory (Table 2). Although the ClinVar entry does not include the evidence used to support a pathogenic classification, it may be based on the pathogenicity normally associated with variants that occur at the +1 position, as discussed previously. For this particular variant, published analysis of this junction reveals an alternative splice site that, if used, results in the deletion of three amino acids [39]. Additionally, phenotypic information for individuals identified as carrying this variant by the reference laboratory, as well as other variants at this junction that would also abolish the wild‐type donor, is not consistent with hereditary breast and ovarian cancer syndrome. Therefore, the use of the alternate splice site means the variant does not result in a significant risk of breast and ovarian cancer. Together, this literature and phenotype evidence result in a reference laboratory classification of likely benign.
Several commercial testing laboratories contribute to ClinVar. In order to evaluate possible differences between testing laboratories, these submissions were compared with the reference laboratory classifications. This sub‐analysis shows that only 80%–85% of the contributing laboratory classifications in ClinVar are concordant with the reference laboratory classifications (Table 1), which is similar to what is observed for the overall ClinVar database. Again, the main source of discordance arises from variants with an uncertain classification in ClinVar compared with a definitive classification from the reference laboratory process (14%–18%). Further analysis shows that 43%–63% of variants that have uncertain ClinVar entries from commercial testing laboratories have definitive classifications from the reference laboratory.
Discussion
The expansion of clinical genetic testing has substantially increased our knowledge of gene‐ and variant‐specific cancer risks. The results of clinical genetic testing now play a role in management decisions, and professional society guidelines include gene‐specific recommendations [10]. However, variability in the classification of variants has inspired some to argue that individual laboratory classifications should be routinely corroborated by comparison with public databases. Unfortunately, these databases cannot yet accommodate the standards required for clinical variant classification, including curation by experts, standardized methods, and timely notification of variant reclassification [1], [3], [6], [15]. These limitations continue to result in uncertain [9], [16] and incorrect [3], [5], [6], [7], [8], [9] variant classifications. As such, discrepancies between databases and commercial testing laboratories may add undue ambiguity and error to management decisions.
Here, we assessed how often this discordance may be encountered in clinical practice by evaluating BRCA1 and BRCA2 variant classifications from a single commercial testing laboratory and ClinVar, a commonly used database. Our findings show that 73.2% of variant classifications in ClinVar agreed with the reference laboratory (Table 1). An additional 12.3% of variants with multiple database classifications had at least one entry that agrees with the reference laboratory. Although this results in an overall potential concordance of 85.5%, the inclusion of discrepant entries within a single database may generate clinical uncertainty.
One example of a variant with partial concordance is BRCA1 c.427G>A (p.Glu143Lys), which has literature evidence listed in ClinVar supporting both VUS and benign classifications. Notably, multiple, discordant entries continue to be listed in ClinVar and cite the same literature evidence (Table 2). For variants with this partial concordance, a clinician consulting the database to evaluate laboratory findings would be faced with evaluating discrepant database entries as well. Conversely, the laboratory classification of benign is based, in part, on phenotypic data. For this data, it is vital to have a matched control group in order to accurately evaluate pathogenicity. Although this can be managed for a testing population from a single laboratory, population and ascertainment differences may convolute assessment of this data in a public database. Overall, this example highlights the potential complexity of a clinical review, as the same literature is cited for both benign and VUS classifications.
The remaining variant classifications were discordant. The majority of these variants had an uncertain database classification compared with a definitive (pathogenic or benign) laboratory classification. A separate analysis of ClinVar entries from contributing commercial testing laboratories is consistent with the overall ClinVar findings, in which 40%–60% of variants classified as uncertain by ClinVar contributing laboratories are definitively classified by the reference laboratory. This is also consistent with data published on the Prospective Registry of Multiplex Testing, which shows that 26% of variants had conflicting classifications from different testing laboratories [40].
Although professional society guidelines recommend that individuals carrying a VUS should be managed based on personal and family cancer history [10], there is variability in the management and impact of VUS test results [41], [42]. For example, some studies show increased surgical interventions [43] and anxiety [44] among patients receiving a VUS test result, whereas others show no increase in psychological distress [45]. As such, many testing laboratories expend a great deal of time and resource to reclassify VUSs and ensure appropriate patient care. Conflicting classifications between a testing laboratory and a public database likely reintroduce this uncertainty into patient care. This is of particular concern for patients with a variant classified as pathogenic by the testing laboratory and uncertain by a database, as they may not receive the appropriate screening or other recommended medical interventions.
Some of the discordance arose from differences in the evaluation and weighting of publicly available evidence. This was the case for BRCA1 c.65T>C (p.Leu22Ser), for which review of the available literature by the laboratory panel of experts determined that the literature evidence of pathogenicity was insufficient for clinical use. The same literature is cited in the ClinVar classification without any additional supporting evidence. As many ClinVar users may not have the specific expertise to independently evaluate the literature evidence, this finding will likely introduce uncertainty into medical management decisions.
A small number of discordant classifications arose from variants with opposite classifications from the reference laboratory process and databases. Although there have been no studies documenting how these discrepancies may impact patient care, it would seem that the potential for inappropriate medical interventions is much higher. For example, ACMG guidelines would appear to support a pathogenic classification of BRCA1 c.301 + 1G>A; however, expert review and additional phenotypic evidence supports a clinical laboratory classification of benign. Furthermore, the database entry does not cite any evidence supporting the pathogenic classification. Again, it is unlikely that database users would have the broad expertise required to independently evaluate the available evidence.
Overall, the classification discrepancies presented here highlight the limitations of clinical consultation of public databases. Efforts are being made to resolve discrepancies within ClinVar by voluntary review by experts; however, the time requirements for this task coupled with the volume of new genetic testing data mean that clinicians will likely continue to encounter such discrepancies. In light of these challenges, clinicians may consider how they would handle discrepant classifications before consulting a public database and the potential impact on patient management.
Conclusion
In all medical specialties, the receipt of discordant test results introduces uncertainty into patient management. Here, we show that discrepant classifications between a public database and single reference laboratory potentially account for 26.7% of variants in BRCA1 and BRCA2. The high degree of discordance observed here is likely reflective of the fact that public databases cannot currently accommodate the consistent standard of variant classification required for clinical use. Although concordant variant classifications may add confidence in management decisions, it is unclear whether any additional benefit is gleaned over receipt of the laboratory test result alone. In addition, the time and expertise needed for busy clinicians to research discordant classifications call into question the practicality of checking all test results against a database. Collectively, these findings serve as a note of caution regarding the utility of database consultation, as discordant classifications should be interpreted with these limitations in mind.
Acknowledgments
This analysis was supported by Myriad Genetic Laboratories, Inc.
Author Contributions
Conception/Design: William Gradishar, Erin Mundt, Susan Manley
Collection and/or assembly of data: KariAnne Johnson, Erin Mundt
Data analysis and interpretation: William Gradishar, KariAnne Johnson, Krystal Brown, Erin Mundt
Manuscript writing: William Gradishar, Krystal Brown, Erin Mundt, Susan Manley
Final approval of manuscript: William Gradishar, KariAnne Johnson, Krystal Brown, Erin Mundt, Susan Manley
Disclosures
Susan Manley: Myriad Genetic Laboratories, Inc. (E, OI) KariAnne Johnson: Myriad Genetic Laboratories, Inc. (E); Krystal Brown: Myriad Genetic Laboratories (E); Erin Mundt: Myriad Genetic Laboratories, Inc. (E). The other author indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Greenblatt MS, Brody LC, Foulkes WD et al. Locus‐specific databases and recommendations to strengthen their contribution to the classification of variants in cancer susceptibility genes. Hum Mutat 2008;29:1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Landrum MJ, Lee JM, Benson M et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. MacArthur DG, Manolio TA, Dimmock DP et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rehm HL, Berg JS, Brooks LD et al. ClinGen–The Clinical Genome Resource. N Engl J Med 2015;372:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bell CJ, Dinwiddie DL, Miller NA et al. Carrier testing for severe childhood recessive diseases by next‐generation sequencing. Sci Transl Med 2011;3:65ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cotton RG, Auerbach AD, Beckmann JS et al. Recommendations for locus‐specific databases and their curation. Hum Mutat 2008;29:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Norton N, Robertson PD, Rieder MJ et al. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet 2012;5:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xue Y, Chen Y, Ayub Q et al. Deleterious‐ and disease‐allele prevalence in healthy individuals: Insights from current predictions, mutation databases, and population‐scale resequencing. Am J Hum Genet 2012;91:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vail PJ, Morris B, van Kan A et al. Comparison of locus‐specific databases for BRCA1 and BRCA2 variants reveals disparity in variant classification within and among databases. J Community Genet 2015;6:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daly M, Pilarski R, Axilbund JE et al. Genetic/familial high‐risk assessment: Breast and ovarian. V2.2016. NCCN Clinical Practice Guidelines in Oncology 2016.
- 11. Provenzale D, Jasperson K, Ahnen DJ et al. Colorectal cancer screening V 1.2015. NCCN Clinical Practice Guidelines in Oncology 2015. J Natl Compr Canc Netw 2015;13:959–968. 26285241 [Google Scholar]
- 12. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitropoulou C, Webb AJ, Mitropoulos K et al. Locus‐specific database domain and data content analysis: Evolution and content maturation toward clinical use. Hum Mutat 2010;31:1109–1116. [DOI] [PubMed] [Google Scholar]
- 14. Johnston JJ, Biesecker LG. Databases of genomic variation and phenotypes: Existing resources and future needs. Hum Mol Genet 2013;22:R27–R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheon JY, Mozersky J, Cook‐Deegan R. Variants of uncertain significance in BRCA: A harbinger of ethical and policy issues to come? Genome Med 2014;6:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pepin MG, Murray ML, Bailey S et al. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet Med 2016;18:20–24. [DOI] [PubMed] [Google Scholar]
- 17. Eggington JM, Bowles KR, Moyes K, et al. A comprehensive laboratory‐based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet 2014;86:229–237. [DOI] [PubMed] [Google Scholar]
- 18. Lindor NM, Guidugli L, Wang X et al. A review of a multifactorial probability‐based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat 2012;33:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. MT Harte, GJ O'Brien, NM Ryan, et al. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res 2010;70:2538–2547. [DOI] [PubMed] [Google Scholar]
- 20. Towler WI, Zhang J, Ransburgh DJ et al. Analysis of BRCA1 variants in double‐strand break repair by homologous recombination and single‐strand annealing. Hum Mutat 2013;34:439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei L, Lan L, Hong Z et al. Rapid recruitment of BRCA1 to DNA double‐strand breaks is dependent on its association with Ku80. Mol Cell Biol 2008;28:7380–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pruss D, Morris B, Hughes E et al. Development and validation of a new algorithm for the reclassification of genetic variants identified in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat 2014;147:119–132. [DOI] [PubMed] [Google Scholar]
- 23. Katagiri T, Kasumi F, Yoshimoto M et al. High proportion of missense mutations of the BRCA1 and BRCA2 genes in Japanese breast cancer families. J Hum Genet 1998;43:42–48. [DOI] [PubMed] [Google Scholar]
- 24. Sweet K, Senter L, Pilarski R et al. Characterization of BRCA1 ring finger variants of uncertain significance. Breast Cancer Res Treat 2010;119:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vallée MP, Francy TC, Judkins MK et al. Classification of missense substitutions in the BRCA genes: A database dedicated to Ex‐UVs. Hum Mutat 2012;33:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whiley PJ, Parsons MT, Leary J et al. Multifactorial likelihood assessment of BRCA1 and BRCA2 missense variants confirms that BRCA1:c.122A>G(p.His41Arg) is a pathogenic mutation. PLoS One 2014;9:e86836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cao W, Wang X, Gao Y et al. BRCA1 germ‐line mutations and tumor characteristics in eastern Chinese women with familial breast cancer. Anat Rec (Hoboken) 2013;296:273–278. [DOI] [PubMed] [Google Scholar]
- 28. Sadr‐Nabavi A, Dastpak M, Homaei‐Shandiz F et al. Analysis of novel mutations in BRCA1 in Iranian families with breast cancer. Hereditas 2014;151:38–42. [DOI] [PubMed] [Google Scholar]
- 29. Suter NM, Ray RM, Hu YW et al. BRCA1 and BRCA2 mutations in women from Shanghai China. Cancer Epidemiol Biomarkers Prev 2004;13:181–189. [DOI] [PubMed] [Google Scholar]
- 30. Thirthagiri E, Lee SY, Kang P et al. Evaluation of BRCA1 and BRCA2 mutations and risk‐prediction models in a typical Asian country (Malaysia) with a relatively low incidence of breast cancer. Breast Cancer Res 2008;10:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toh GT, Kang P, Lee SS et al. BRCA1 and BRCA2 germline mutations in Malaysian women with early‐onset breast cancer without a family history. PLoS One 2008;3:e2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Y Yu, L Dong, D Li, et al. Targeted DNA sequencing detects mutations related to susceptibility among familial non-medullary thyroid cancer. Sci Rep 2015;5:16129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang M, Xu Y, Ouyang T et al. Somatic mutations in the BRCA1 gene in Chinese women with sporadic breast cancer. Breast Cancer Res Treat 2012;132:335–340. [DOI] [PubMed] [Google Scholar]
- 34. X Zhong, Z Dong, H Dong et al. Prevalence and prognostic role of BRCA1/2 variants in unselected chinese breast cancer patients. PloS One 2016;11:e0156789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. AY Steffensen, M Dandanell, L Jonson et al. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur J Hum Genet 2014;22:1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. M Colombo, MJ Blok, P Whiley et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: a report from the ENIGMA consortium. Hum Molec Genet 2014;23:3666–3680. [DOI] [PubMed] [Google Scholar]
- 37. V Dosil, A Tosar, C Canadas et al. Alternative splicing and molecular characterization of splice site variants: BRCA1 c.591C>T as a case study. Clin Chem 2010;56:53–61. [DOI] [PubMed] [Google Scholar]
- 38. Y Miki, J Swensen, D Shattuck-Eidens et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66–71. [DOI] [PubMed] [Google Scholar]
- 39. Thomassen M, Blanco A, Montagna M et al. Characterization of BRCA1 and BRCA2 splicing variants: A collaborative report by ENIGMA consortium members. Breast Cancer Res Treat 2012;132:1009–1023. [DOI] [PubMed] [Google Scholar]
- 40. Balmaña J, Digiovanni L, Gaddam P et al. Conflicting interpretation of genetic variants and cancer risk by commercial laboratories as assessed by the Prospective Registry of Multiplex Testing. J Clin Oncol 2016; 34:4071–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petrucelli N, Lazebnik N, Huelsman KM et al. Clinical interpretation and recommendations for patients with a variant of uncertain significance in BRCA1 or BRCA2: A survey of genetic counseling practice. Genet Test 2002;6:107–113. [DOI] [PubMed] [Google Scholar]
- 42. Richter S, Haroun I, Graham TC et al. Variants of unknown significance in BRCA testing: Impact on risk perception, worry, prevention and counseling. Ann Oncol 2013;24 (suppl 8):viii69–viii74. [DOI] [PubMed] [Google Scholar]
- 43. Culver JO, Brinkerhoff CD, Clague J et al. Variants of uncertain significance in BRCA testing: Evaluation of surgical decisions, risk perception, and cancer distress. Clin Genet 2013;84:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Neill SC, Rini C, Goldsmith RE et al. Distress among women receiving uninformative BRCA1/2 results: 12‐month outcomes. Psychooncology 2009;18:1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Dijk S, van Asperen CJ, Jacobi CE et al. Variants of uncertain clinical significance as a result of BRCA1/2 testing: Impact of an ambiguous breast cancer risk message. Genet Test 2004;8:235–239. [DOI] [PubMed] [Google Scholar]