

It is unknown if and to what extent the clinical course and therapeutic response of BRCA‐associated cholangiocarcinomas are distinct from non‐BRCA carriers. To gain insight, this multicenter retrospective study on BRCA‐associated cases was initiated and is reported here.
Keywords: Cholangiocarcinoma, BRCA-associated, Germline, somatic, PARPi
Abstract
Background.
Biliary tract malignancies, in particular cholangiocarcinomas (CCA), are rare tumors that carry a poor prognosis. BRCA2 mutation carriers have an increased risk of developing CCA with a reported relative risk of ∼5 according to the Breast Cancer Linkage Consortium. In addition to this risk, there are potential therapeutic implications in those harboring somatic and/or germline (GL) BRCA mutations. Therefore, it is important to define the clinical characteristics of GL/somatic BRCA1/2 variants in CCA patients.
Materials and Methods.
We performed a multicenter retrospective analysis of CCA patients diagnosed between January 2000 and December 2013 with GL or somatic variants in BRCA1/2 genes detected by GL mutations testing and/or by tumor next generation sequencing. Cases were identified from clinical databases at participating institutions. Data including demographics, clinical history, surgical procedures, and systemic chemotherapy or radiation were extracted from patients' records.
Results.
Overall, 18 cases were identified: 5 carriers of GL BRCA1/2 mutations (4 BRCA2; 1 BRCA1) and 13 harboring somatic variations (7 BRCA1; 6 BRCA2). Mean age at diagnosis was 60, SD ± 10 years (range 36–75 years), with male and female prevalence rates of 61.2% and 38.8%, respectively. Stage at diagnosis was I (n = 4), II (n = 3), III (n = 3), and IV (n = 8). Six patients had extrahepatic CCA and the rest intrahepatic CCA. Thirteen patients received platinum‐based therapy and four were treated with poly ADP ribose polymerase inhibitors, of whom one experienced sustained disease response with a progression‐free survival of 42.6 months. Median overall survival from diagnosis for patients with stage I/II in this study was 40.3 months (95% confidence interval [CI], 6.73–108.15) and with stages III/IV was 25 months (95% CI, 15.23–40.57).
Conclusion.
BRCA‐associated CCA is uncommon. This multicenter retrospective study provides a thorough clinical analysis of a BRCA‐associated CCA cohort, which can serve as a benchmark for future development and design of expanded analyses and clinical trials.
Implications for Practice.
BRCA‐associated CCA is uncommon but a very important subtype of hepatic malignancies, due to its rising prevalence. Better clinical characterization of this subtype might allow application of targeted therapy for CCA patients with germline or somatic mutations in BRCA1/2 genes, especially due to previously reported success of such therapies in other BRCA‐associated malignancies. Thus this study, first of its kind, provides a basis for future multi‐centered analyses in larger cohorts, as well as clinical trials. Additionally, this study emphasizes the importance of both germline and somatic genotyping for all CCA patients.
摘要
背景. 胆道恶性肿瘤, 尤其是胆管癌(CCA)是一类预后不良的罕见肿瘤。BRCA2突变携带者罹患CCA的风险增加, 乳腺癌联合协作组报告的相对风险约为5。除上述风险以外, 这对于携带体细胞和/或胚系(GL)BRCA突变的患者而言还具有潜在治疗意义。因此, 确定CCA患者中GL/体细胞BRCA1/2变异体的临床特征至关重要。
材料和方法. 我们针对2000年1月至2013年12月期间诊断的携带GL或体细胞BRCA1/2基因变异体(通过GL突变检测方法和/或新一代肿瘤测序技术检测)的CCA患者进行了一项多中心回顾性分析。从参与机构的临床数据库中检索病例。研究数据包括人口统计学、临床病史、手术方式以及全身化疗或放疗, 以上信息摘自患者记录。
结果. 共计检索到18例病例:5例携带GL BRCA1/2突变(4例BRCA2;1例BRCA1), 13例携带体细胞变异(7例BRCA1;6例BRCA2)。诊断时的平均年龄为60岁, SD±10岁(范围:36‐75岁), 男性和女性的患病率分别为61.2%和38.8%。诊断时的疾病分期包括I期(n=4)、II期(n=3)、III期(n=3)和IV期(n=8)。6例患者为肝外CCA, 其余为肝内CCA。13例患者接受以铂类药物为基础的治疗, 4例患者接受多聚ADP核糖聚合酶抑制剂(PARPi)治疗, 其中1例患者获得了持久性疾病缓解, 无进展生存期为42.6个月。在本研究中, 自诊断时起, I/II期患者的中位总生存期为40.3个月[95%置信区间(CI):6.73‐108.15], III/IV期患者的中位总生存期为25个月(95% CI:15.23‐40.57)。
结论. BRCA相关性CCA并不常见。本项多中心回顾性研究针对一个BRCA相关性CCA队列进行了全面的临床分析, 在日后开发和设计扩展分析和临床试验时可以此作为基准。
Introduction
Cholangiocarcinomas (CCA) are adenocarcinomas that arise from the malignant transformation of bile duct epithelium anywhere along the biliary tree from small bile ducts and bile ductules (intrahepatic CCA [ICC]) to large bile ducts at the hilum of the liver or outside the liver (extrahepatic CCA [ECC]) [1]. Although CCA is a relatively uncommon tumor, with incidence rates ranging from 0.8 to 2 per 100,000 in the Western world [2], [3], it is the second most common primary hepatic malignancy after hepatocellular carcinoma and accounts for 3% of malignant tumors of the gastrointestinal system and 15% of primary hepatic malignancies [4], [5], [6]. Most patients are diagnosed with inoperable disease, and median survival is ∼6 months for ICC patients and less than a year for ECC [1]. Even when deemed operable, only 20%–40% patients who undergo surgery achieve clear (R0) margins [7], [8]. This translates to a dismal prognosis, with 5‐year overall survival (OS) rates of less than 5% [9]. Moreover, CCA's rates are increasing globally in recent years with no effective targeted molecular therapies currently approved [10], [11], [12].
Recent discoveries of somatic genomic alterations have led to exploration of new potential therapeutic targets [13]. In particular, a comprehensive analysis published by Nakamura et al. reported a high rate (93/239—38.9%) of potentially targetable somatic genetic alterations in analyzed CCA cases [14]. The potential targets included kinases (FGFR1, FGFR2, FGFR3, PIK3CA, ALK, EGFR, ERBB2, BRAF, and AKT3), oncogenes (IDH1, IDH2, CCND1, CCND3, and MDM2), and, notably, tumor‐suppressor genes BRCA1 and BRCA2.
In another study, 75 CCA cases were genotyped for targetable somatic mutations, revealing that 16% and 40% of detected alterations in ICC and ECC cases, respectively, were affecting genes associated with DNA repair pathways, including MSH6, BAP1, ATM, MLH1, MSH2, and BRCA1 and BRCA2 [15].
The contribution of germline (GL) mutations in BRCA1/2 genes to the development of bile duct malignancies has previously been reported. Data from the early 2000s by the Breast Cancer Linkage Consortium (BCLC) reported that the relative risk (RR) of developing gall bladder or bile duct cancer among BRCA2 carriers is 4.97 (95% confidence interval [CI] 1.50–16.52), whereas other established RR factors for CCA development such as infection with liver parasites, hepatitis C virus, and hepatitis B virus are 4.8, 1.8, and 2.6, respectively [16], [17].
BRCA1 and BRCA2 proteins are involved in the DNA damage response mediated via homologous recombination (HR) [18], [19]. BRCA1/2‐mutated cells are HR deficient and hence accumulate DNA double‐strand breaks, resulting in genomic instability and increased predisposition to malignant transformation [20], rendering BRCA1/2 mutation carriers with a distinct clinical phenotype of increased sensitivity to DNA damaging therapies [21], [22], [23]. Additionally, somatic biallelic inactivation of the BRCA1 or BRCA2 genes confers sensitivity to poly ADP ribose polymerase (PARP) inhibition [24].
It is unknown if and to what extent the clinical course and therapeutic response of BRCA‐associated CCA are distinct from non‐BRCA carriers. To gain insight, this multicenter retrospective study on BRCA‐associated cases was initiated and is reported herein.
Materials and Methods
Study Population
A multicenter retrospective analysis was performed. Patients with GL or somatic BRCA1/2‐associated CCA diagnosed between January 2000 and December 2013 were identified from clinical databases at five participating institutions: Sheba Medical Center, MD Anderson Cancer Center, Mount Sinai Hospital Toronto, The Ohio State University Medical Center, and University of California, San Francisco Helen Diller Family Comprehensive Cancer Center.
Data Collection
Data on participants’ demographics, clinical history, personal and family history of cancer, past surgical procedures specifically pertaining to CCA, systemic chemotherapy, and response to treatment were extracted from patients' records or from existing institutional review board (IRB)‐approved institutional databases. Clinical stage was classified according to the seventh edition of the American Joint Committee on Cancer staging criteria [25]. The IRB of each participating institute approved this study and/or the collection of data within an institutional database for future nonhuman subject research.
DNA Analysis
At Sheba Medical Center, GL BRCA1/2 mutational status analysis was performed at the Oncogenetics Unit, and each patient was genotyped for at least 3 to a maximum of 14 predominant BRCA1 and BRCA2 mutations using previously described assays [26], [27].
Somatic mutational analysis from extracted cancerous tissue was performed commercially using next‐generation sequencing technique according to each institution's laboratory practice. Samples collected at Mount Sinai Hospital were analyzed in a clinical Advanced Molecular Diagnostics lab [28]; samples collected at MD Anderson, The Ohio State University Medical Center, and University of California, San Francisco Medical Center were analyzed at Foundation Medicine [29].
Statistical Analysis
OS was defined as the time from the date of diagnosis to the date of death from any cause using GraphPad Prism software. If a patient is not known to have died, the OS was censored until the date of last follow‐up. Progression‐free survival (PFS) was censored as well and defined as the time elapsed until recurrence or appearance of a new metastatic lesion.
Results
Demographic Features and Clinical Characteristics
Overall, we identified 18 cases of CCA harboring either GL (n = 5) or somatic (n = 13) BRCA1/2 variations. Mean age at the time of diagnosis was 60 (SD ± 10), range 36–75 years; 61.2% were males and the majority (15/18) were white, of whom four were of Jewish Ashkenazi origin. Distribution of stage at diagnosis was as follows: stage I (n = 4), stage II (n = 3), stage III (n = 3), and stage IV (n = 8). Six patients had ECC and twelve patients had ICC (Table 1).
Table 1. Study population demographic and clinical characteristics.
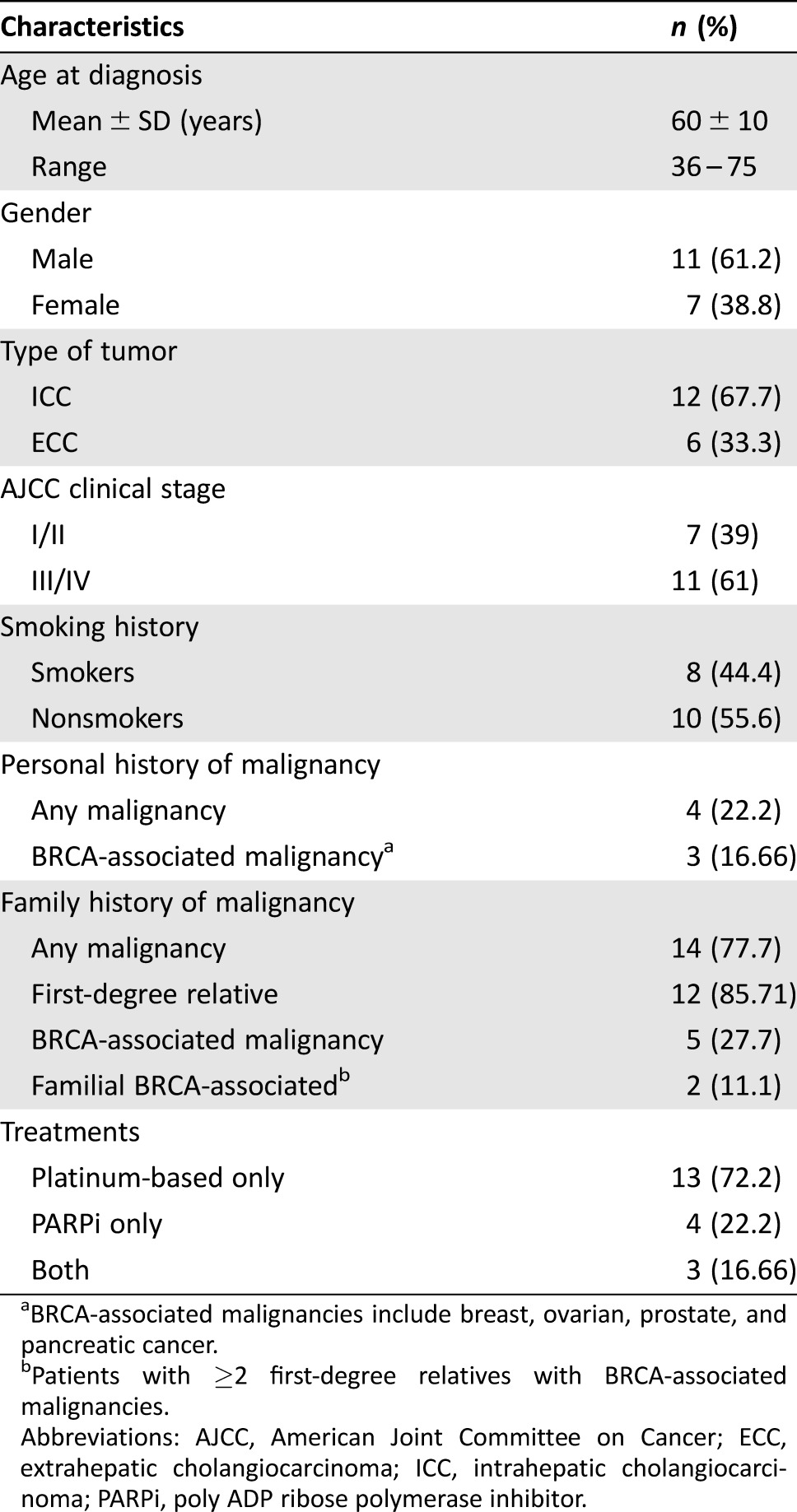
BRCA‐associated malignancies include breast, ovarian, prostate, and pancreatic cancer.
Patients with ≥2 first‐degree relatives with BRCA‐associated malignancies.
Abbreviations: AJCC, American Joint Committee on Cancer; ECC, extrahepatic cholangiocarcinoma; ICC, intrahepatic cholangiocarcinoma; PARPi, poly ADP ribose polymerase inhibitor.
Family and Personal History of Malignancies
Four patients were diagnosed with cancer prior to their current CCA diagnosis, and three of them had BRCA‐associated tumors: two breast and one pancreatic cancer. Most cases (14/18–77.8%) had a first‐ (85.71%) or second‐ (14.3%) degree relative diagnosed with cancer, and in 5/14 cases, BRCA‐associated malignancies (breast, ovarian, prostate, and pancreatic) were noted in these affected family members (Table 1).
Characterization of Identified Mutations
GL Mutations in BRCA1/2.
All identified GL mutations are known pathogenic mutations and are predominant in either the Jewish Ashkenazi or Irish Scottish ancestry (Fig. 1A) [23], [24], [30].
Figure 1.

Distribution of all analyzed mutations according to their origin and type (A) and characterization of all analyzed mutation based on known pathogenicity and origin (B).
Abbreviations: GL, germline; UNK, unknown; VUS, variants of unknown significance.
Somatic Variations in BRCA1/2.
Thirteen additional mutations were somatically identified; these included previously reported and known deleterious mutations, variants with suspected pathogenicity, and variants of unknown significance (VUS). All identified somatic variations are summarized in Figure 1A and 1B.
OS and PFS of Analyzed Patients
Median OS for patients with stage I/II was 40.27 months (95% CI, 6.73–108.15) and with stages III/IV was 25 months (95% CI, 15.23–40.57, Fig. 2A). Median OS in ICC and ECC was 24.67 (95% CI, 16.42–37.15) and 47.65 (95%CI, 3.96–125.21) months, respectively (Fig. 2B).
Figure 2.

Overall survival of study population. (A): Patients diagnosed with cholangiocarcinoma (CCA) at stages I/II versus III/IV. (B): Extra versus Intra hepatic CCA.
Treatment with Platinum/PARP Inhibitors
Overall, 13 patients received platinum‐based treatment, and four patients received PARP inhibitor (PARPi) therapy. All these latter cases were diagnosed with advanced stage disease. Treatment with PARPi resulted in a favorable response, with one patient's OS censored at 64.76 months and a PFS of 42.6 months. Data are summarized in Table 2.
Table 2. PFS and OS of patients receiving PARPi.
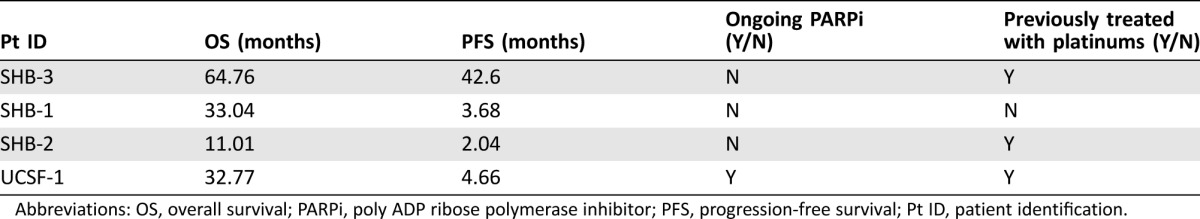
Abbreviations: OS, overall survival; PARPi, poly ADP ribose polymerase inhibitor; PFS, progression‐free survival; Pt ID, patient identification.
Known and/or Predicted Deleterious Mutations in BRCA1/2 Versus Unknown Variants
Of the 18 mutations detected, nine were previously reported either with a known founder effect or predicted/suspected to be pathogenic. An additional six were unknown variants with nonverified knowledge of pathogenic potential. Patients bearing either known pathogenic mutations or unknown variants and treated with either platinum‐based therapy or PARPi demonstrated more favorable OS, ranging from 11.01 to 64.78 months for all patients censored (Table 3).
Table 3. OS in patients treated with platinum compounds and/or PARPi.
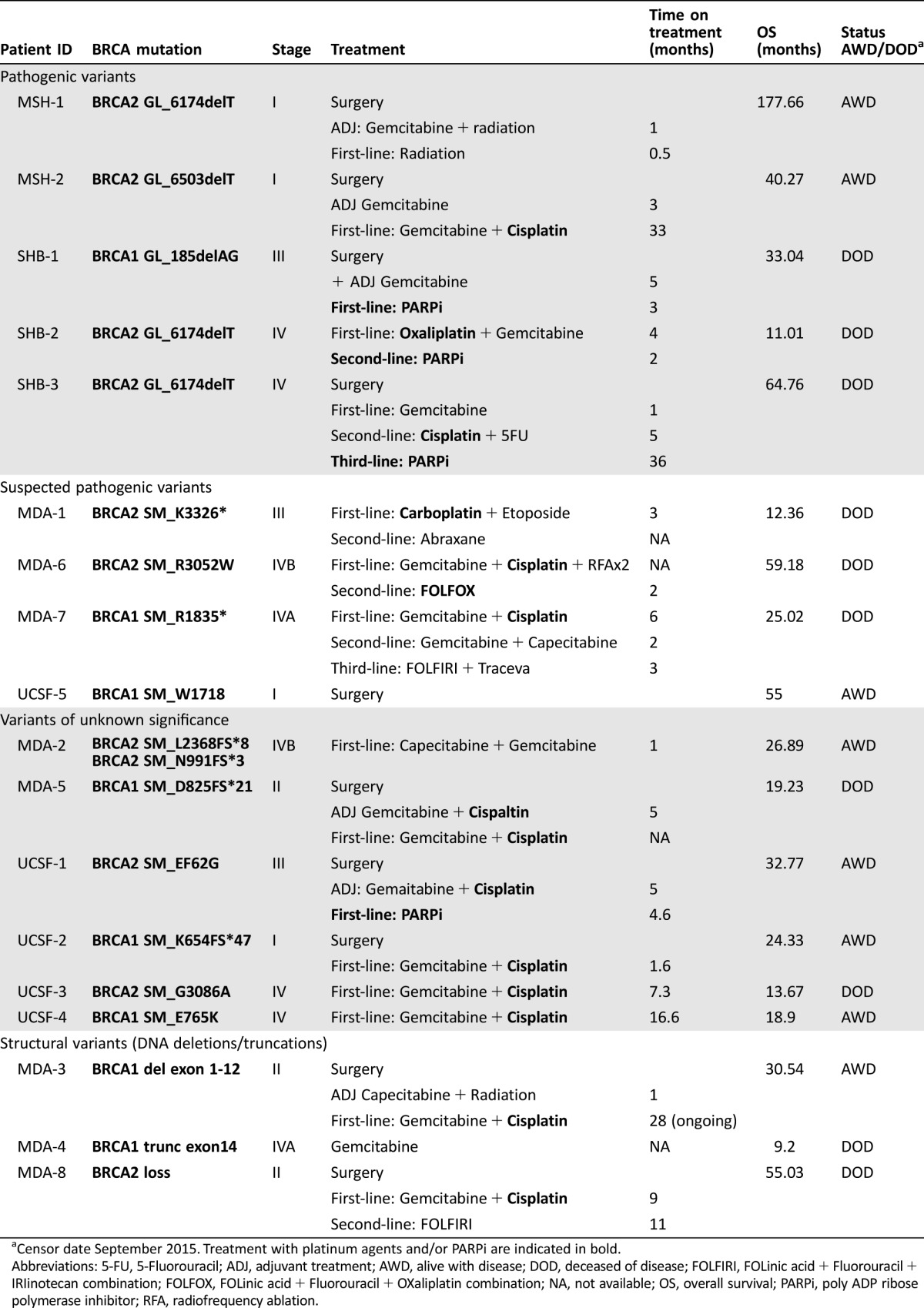
Censor date September 2015. Treatment with platinum agents and/or PARPi are indicated in bold.
Abbreviations: 5‐FU, 5‐Fluorouracil; ADJ, adjuvant treatment; AWD, alive with disease; DOD, deceased of disease; FOLFIRI, FOLinic acid + Fluorouracil + IRIinotecan combination; FOLFOX, FOLinic acid + Fluorouracil + OXaliplatin combination; NA, not available; OS, overall survival; PARPi, poly ADP ribose polymerase inhibitor; RFA, radiofrequency ablation.
Discussion
In the presented retrospective study, we report 18 cases of BRCA‐associated CCA from five participating institutions. All patients evaluated harbored either GL or somatic variations in BRCA1/2 genes.
The founder mutations of Ashkenazi Jewish origin, such as 185delAG in the BRCA1 gene and 6174delT in the BRCA2 gene, were identified in the GL of four of the enrolled patients. An additional founder mutation, 6503delTT in BRCA2, the frequency of which is enriched in Central and Northern European populations, was identified in the GL DNA of one patient.
We also identified a variety of somatic variants derived from tumor tissue of participating patients, some of which were previously described as pathogenic or suspected to be pathogenic and also VUS. The potentially pathogenic variants identified here include a known BRCA2 polymorphism variant K3326X (10204 A > T substitution), which occurs with a higher frequency in individuals with familiar pancreatic adenocarcinoma [31]. The K3326X variant was also demonstrated to be associated with the risk of developing breast and ovarian cancers independent of other pathogenic variants in BRCA2 [32]. The reported substitution leads to the appearance of a premature stop codon, resulting in the loss of the final 93 amino acids in the BRCA2 protein. Importantly, the C‐terminus of BRCA2 is thought to be functional [33]. Another variant identified in BRCA1‐R1835X and associated with breast/ovarian cancer in populations of Ashkenazi, Philippines, and Western European origin [34] eliminates the last 29 amino acid residues of BRCA1, which might lead to impaired interactions with various BRCA1 binding proteins [35].
Several cases of substitutions of R3052 in BRCA2 to glutamine (R3052Q) or tryptophan (R3052W) have been reported. The R3052W variant specifically has been identified in multiple breast cancer families by Myriad Genetic Laboratories [36]. Farrugia et al. describe a large family with seven cases of breast cancer, all harboring the R3052W mutation [37]. Arginine 3052 is located in the interface between oligonucleotide/oligosaccharide‐binding folds and makes hydrogen bonds with neighboring amino acid residues, thus linking them together [38]. Hence, substitution at this position might have a deleterious effect given that the newly created amino acid has different chemical characteristics. Indeed, the BRCA2 R3052W variant's pathogenic potential was demonstrated in various designated in vitro assays. For instance, mouse embryonic stem (ES) cell functional analysis of both the R3052Q and R3052W variants demonstrated that ES cells expressing the R3052W variant didn't survive, whereas R3052Q expression had no effect. Additionally, R3052W variant's influence of HR repair was analyzed using an in vitro green fluorescent protein‐dependent homology‐directed repair reporter assay, in which R3052W displayed reduced homologous recombination‐dependent (HRD) activity compared with wild‐type BRCA2, which supports the notion of its deleterious effect in DNA repair activity [37].
VUS identified in this study need further verification and analysis to confirm their pathogenic potential or lack thereof. Additionally, whether patients and their families should be screened for VUS in the GL is controversial.
In this multicenter retrospective cohort, the median age of diagnosis, stage, and gender prevalence were similar to the Surveillance, Epidemiology, and End Results program (SEER) [39]. For CCA patients harboring pathogenic variations in BRCA1/2 genes who were treated with platinum‐based and/or PARPi therapy, survival outcomes appear longer than SEER historical controls. Superior response to treatment with platinum‐based agents and/or PARPi was demonstrated in patients with breast, ovarian, and pancreatic cancer harboring mutations in BRCA1/2 genes in clinical trials and retrospective analyses [40]. A number of PARPi agents are being pursued and hold promise for personalized treatment for patients with GL or somatic BRCA mutations. Therefore, genetic testing for known founder mutations is currently recommended for high‐risk populations [41], [42], [43], [44]. Subsequently, family members of patients with identified GL mutations are offered to undergo genetic screening.
The results of this study support the rationale for somatic and/or GL BRCA genotyping in all patients diagnosed with CCA, the latter initially in populations in which founder mutations occur, such as Jewish Ashkenazi and Irish Scottish, and/or when family history or other clinical features indicate BRCA testing.
The limitations of this study, including the small sample size, its retrospective nature, the combination of somatic and GL variants, and the nondifferentiation between clearly pathogenic and VUS, make any conclusions tentative at best. Most importantly, we do not have sufficient information on non‐BRCA‐associated CCA cases identified in each participating institution with which we can compare our findings.
Our findings in this BRCA carriers CCA cohort are further reinforced by recently reported genomic similarity between CCA tumors and tumors of pancreatic, diffuse glioma, and small cell (SC) lung origins, which demonstrates that tumor genotype can impact treatment response and must be incorporated along with anatomic site of origin into treatment decisions [45]. CCA and pancreatic tumors bearing variants in the same DNA repair pathway may share molecular burden with each other, and thus approaching BRCA‐associated CCA similarly to BRCA‐associated pancreatic ductal adenocarcinoma may be beneficial to a population of selected patients.
The data presented here are plausible to suggest conducting a prospective multicenter basket trial for BRCA‐associated CCA, with either GL or somatic identified variations, and applying PARPi as a potential study arm, allowing for a more profound analysis of larger patients’ cohorts [46]. The clinical relevance of somatic BRCA mutations can thus be analyzed in the setting of a prospective clinical trial. A similar approach was previously demonstrated in the Biomarker‐Integrated Approaches of Targeted Therapy for lung cancer elimination trial for personalizing therapy for lung cancer, which represents the first completed, prospective, biomarker‐based study in which non‐small cell lung cancer patients were adaptively randomized to different therapeutic options based on relevant molecular biomarkers analyzed in fresh core needle biopsy specimens [47].
Conclusion
The data presented herein provide the first clinical characterization of a multicenter cohort of CCA patients with GL or somatic BRCA variants. With the rising prevalence of CCA and the success of targeted therapy in other BRCA‐associated tumors, this study provides a framework for future multicenter cohort analyses in BRCA‐associated CCA and supports the development of a genotype‐directed clinical trial in this important population. Additionally, this study demonstrates a rationale for both GL/tumor genotyping for CCA patients and genetic screening for individuals with a known family history of BRCA‐associated malignancies, especially those with known genetic predispositions and ethnic backgrounds.
Contributed equally.
Author Contributions
Conception/Design: Talia Golan, Maria Raitses‐Gurevich, Milind Javle
Provision of study material or patients: Talia Golan, Robin K. Kelley, Andrea G. Bocobo, Ayelet Borgida, Rachna T. Shroff, Spring Holter, Steven Gallinger, Daniel H. Ahn, Dan Aderka, Jain Apurva, Tanois Bekaii‐Saab, Milind Javle
Collection and/or assembly of data: Talia Golan, Maria Raitses‐Gurevich, Robin K. Kelley, Andrea G. Bocobo, Ayelet Borgida, Rachna T. Shroff, Spring Holter, Steven Gallinger, Daniel H. Ahn, Dan Aderka, Jain Apurva, Tanois Bekaii‐Saab, Milind Javle
Data analysis and interpretation: Talia Golan, Maria Raitses‐Gurevich, Milind Javle
Manuscript writing: Talia Golan, Maria Raitses‐Gurevich, Eitan Friedman, Milind Javle
Final approval of manuscript: Talia Golan, Maria Raitses‐Gurevich, Robin K. Kelley, Andrea G. Bocobo, Ayelet Borgida, Rachna T. Shroff, Spring Holter, Steven Gallinger, Daniel H. Ahn, Dan Aderka, Jain Apurva, Tanois Bekaii‐Saab, Eitan Friedman, Milind Javle
Disclosures
Robin K. Kelley: Agios, Arqule (C/A), Agios, Eli Lilly and Co., Merck, Novartis, (RF); Rachna T. Shroff: Amgen, Codiak Biosciences, Celgene (C/A), Agios, Celgene, Eli Lilly (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Sempoux C, Jibara G, Ward SC et al. Intrahepatic cholangiocarcinoma: New insights in pathology. Semin Liver Dis 2011;31:49–60. [DOI] [PubMed] [Google Scholar]
- 2. Rajagopalan V, Daines WP, Grossbard ML et al. Gallbladder and biliary tract carcinoma: A comprehensive update, Part 1. Oncology (Williston Park) 2004;18:889–896. [PubMed] [Google Scholar]
- 3. Augustine MM, Fong Y. Epidemiology and risk factors of biliary tract and primary liver tumors. Surg Oncol Clin N Am 2014;23:171–188. [DOI] [PubMed] [Google Scholar]
- 4. Poultsides GA, Zhu AX, Choti MA et al. Intrahepatic cholangiocarcinoma. Surg Clin North Am 2010;90:817–837. [DOI] [PubMed] [Google Scholar]
- 5. Vasilieva LE, Papadhimitriou SI, Dourakis SP. Modern diagnostic approaches to cholangiocarcinoma. Hepatobiliary Pancreat Dis Int 2012;11:349–359. [DOI] [PubMed] [Google Scholar]
- 6. Bartlett DL. Intrahepatic cholangiocarcinoma: A worthy challenge. Cancer J 2009;15:255–256. [DOI] [PubMed] [Google Scholar]
- 7. Geynisman DM, Catenacci DV. Toward personalized treatment of advanced biliary tract cancers. Discov Med 2012;14:41–57. [PubMed] [Google Scholar]
- 8. Yamamoto M, Ariizumi S. Surgical outcomes of intrahepatic cholangiocarcinoma. Surg Today 2011;41:896–902. [DOI] [PubMed] [Google Scholar]
- 9. Mosconi S, Beretta GD, Labianca R et al. Cholangiocarcinoma. Crit Rev Oncol Hematol 2009;69:259–270. [DOI] [PubMed] [Google Scholar]
- 10. Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001;33:1353–1357. [DOI] [PubMed] [Google Scholar]
- 11. Patel T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer 2002;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taylor‐Robinson SD, Toledano MB, Arora S et al. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968–1998. Gut 2001;48:816–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ross JS, Wang K, Gay L et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next‐generation sequencing. The Oncologist 2014;19:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura H, Arai Y, Totoki Y et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–1010. [DOI] [PubMed] [Google Scholar]
- 15. Churi CR, Shroff R, Wang Y et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS One 2014;9:e115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breast Cancer Linkage Consortium , Easton D, Thompson D et al. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 1999;91:1310–1316. [DOI] [PubMed] [Google Scholar]
- 17. Shin HR, Oh JK, Masuyer E et al. Epidemiology of cholangiocarcinoma: An update focusing on risk factors. Cancer Sci 2010;101:579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002;108:171–182. [DOI] [PubMed] [Google Scholar]
- 19. Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci 2001;114:3591–3598. [DOI] [PubMed] [Google Scholar]
- 20. Tutt A, Ashworth A. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med 2002;8:571–576. [DOI] [PubMed] [Google Scholar]
- 21. Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–921. [DOI] [PubMed] [Google Scholar]
- 22. Tutt AN, Lord CJ, McCabe N et al. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol 2005;70:139–148. [DOI] [PubMed] [Google Scholar]
- 23. Cass I, Baldwin RL, Varkey T et al. Improved survival in women with BRCA‐associated ovarian carcinoma. Cancer 2003;97:2187–2195. [DOI] [PubMed] [Google Scholar]
- 24. Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005;434:913–917. [DOI] [PubMed] [Google Scholar]
- 25. Edge SB, Compton CC. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010;17:1471–1474. [DOI] [PubMed] [Google Scholar]
- 26. Abeliovich D, Kaduri L, Lerer I et al. The founder mutations 185delAG and 5382insC in BRCA1 and 6174delT in BRCA2 appear in 60% of ovarian cancer and 30% of early‐onset breast cancer patients among Ashkenazi women. Am J Hum Genet 1997;60:505–514. [PMC free article] [PubMed] [Google Scholar]
- 27. Schayek H, De Marco L, Starinsky‐Elbaz S et al. The rate of recurrent BRCA1, BRCA2, and TP53 mutations in the general population, and unselected ovarian cancer cases, in Belo Horizonte, Brazil. Cancer Genet 2016;209:50–52. [DOI] [PubMed] [Google Scholar]
- 28. Bedard PL, Oza AM, Tsao MS et al. Princess Margaret Cancer Centre (PMCC) Integrated Molecular Profiling in Advanced Cancers Trial (IMPACT) using genotyping and targeted next‐generation sequencing (NGS). J Clin Oncol 2013;31:11002a. [Google Scholar]
- 29. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scottish/Northern Irish BRCA1/BRCA2 Consortium. BRCA1 and BRCA2 mutations in Scotland and Northern Ireland. Br J Cancer 2003;88:1256–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin ST, Matsubayashi H, Rogers CD et al. Increased prevalence of the BRCA2 polymorphic stop codon K3326X among individuals with familial pancreatic cancer. Oncogene 2005;24:3652–3656. [DOI] [PubMed] [Google Scholar]
- 32. Meeks HD, Song H, Michailidou K et al. BRCA2 polymorphic stop codon K3326X and the risk of breast, prostate, and ovarian cancers. J Natl Cancer Inst 2015;108:pii djv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Biswas K, Das R, Eggington JM et al. Functional evaluation of BRCA2 variants mapping to the PALB2‐binding and C‐terminal DNA‐binding domains using a mouse ES cell‐based assay. Hum Mol Genet 2012;21:3993–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rashid MU, Zaidi A, Torres D et al. Prevalence of BRCA1 and BRCA2 mutations in Pakistani breast and ovarian cancer patients. Int J Cancer 2006;119:2832–2839. [DOI] [PubMed] [Google Scholar]
- 35. Magnard C, Bachelier R, Vincent A et al. BRCA1 interacts with acetyl‐CoA carboxylase through its tandem of BRCT domains. Oncogene 2002;21:6729–6739. [DOI] [PubMed] [Google Scholar]
- 36. Easton DF, Deffenbaugh AM, Pruss D et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer‐predisposition genes. Am J Hum Genet 2007;81:873–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farrugia DJ, Agarwal MK, Pankratz VS et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res 2008;68:3523–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang H, Jeffrey PD, Miller J et al. BRCA2 function in DNA binding and recombination from a BRCA2‐DSS1‐ssDNA structure. Science 2002;297:1837–1848. [DOI] [PubMed] [Google Scholar]
- 39. Howlader N, Noone AM, Krapcho M et al. SEER Cancer Statistics Review, 1975–2013, National Cancer Institute. Bethesda, MD. SEER data. 2016. Available at http://seer.cancer.gov/csr/1975_2013/. Accessed March 17, 2017. [Google Scholar]
- 40. Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther 2015;8:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gabai‐Kapara E, Lahad A, Kaufman B et al. Population‐based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci USA 2014;111:14205–14210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bancroft EK, Page EC, Castro E et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: Results from the initial screening round of the IMPACT study. Eur Urol 2014;66:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Margel D, Benjaminov O, Ozalvo R et al. Personalized prostate cancer screening among men with high risk genetic predisposition‐ Study protocol for a prospective cohort study. BMC Cancer 2014;14:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peters ML, Tseng JF, Miksad RA. Genetic testing in pancreatic ductal adenocarcinoma: Implications for prevention and treatment. Clin Ther 2016;38:1622–1635. [DOI] [PubMed] [Google Scholar]
- 45. Thomas MB. Tumor‐specific differences of biliary tract cancers. Presented at: American Society of Clinical Oncology (ASCO) 2015 Annual Meeting,; May 2015; Chicago, IL. [Google Scholar]
- 46. Redig AJ, Jänne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol 2015;33:975–977. [DOI] [PubMed] [Google Scholar]
- 47. Kim ES, Herbst RS, Wistuba II et al. The BATTLE trial: Personalizing therapy for lung cancer. Cancer Discov 2011;1:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]