

This review provides a comprehensive overview of the clinical characteristics and prognostic implications for patients with BRAF‐mutant non‐small cell lung cancer and discusses therapeutic strategies that leverage experience from BRAF‐mutant metastatic melanoma. Practical considerations for oncologists are discussed, including considerations for molecular profiling and management of potential toxicities associated with targeted agents.
Keywords: B‐Raf proto‐oncogene, serine/threonine kinase; Non‐small cell lung cancer; Dabrafenib; Trametinib; Vemurafenib
Abstract
Non‐small cell lung cancer (NSCLC) remains the leading cause of cancer‐related deaths globally. However, the identification of oncogenic driver alterations involved in the initiation and maintenance of NSCLC, such as epidermal growth factor receptor mutations and anaplastic lymphoma kinase translocation, has led to the development of novel therapies that directly target mutant proteins and associated signaling pathways, resulting in improved clinical outcomes. As sequencing techniques have improved, the molecular heterogeneity of NSCLC has become apparent, leading to the identification of a number of potentially actionable oncogenic driver mutations. Of these, one of the most promising therapeutic targets is B‐Raf proto‐oncogene, serine/threonine kinase (BRAF). Mutations in BRAF, observed in 2%–4% of NSCLCs, typically lead to constitutive activation of the protein and, as a consequence, lead to activation of the mitogen‐activated protein kinase signaling pathway. Direct inhibition of mutant BRAF and/or the downstream mitogen‐activated protein kinase kinase (MEK) has led to prolonged survival in patients with BRAF‐mutant metastatic melanoma. This comprehensive review will discuss the clinical characteristics and prognostic implications of BRAF‐mutant NSCLC, the clinical development of BRAF and MEK inhibitors from melanoma to NSCLC, and practical considerations for clinicians involving BRAF mutation screening and the choice of targeted therapy.
Implications for Practice.
Personalized medicine has begun to provide substantial benefit to patients with oncogene‐driven non‐small cell lung cancer (NSCLC). However, treatment options for patients with oncogenic driver mutations lacking targeted treatment strategies remain limited. Direct inhibition of mutant B‐Raf proto‐oncogene, serine/threonine kinase (BRAF) and/or downstream mitogen‐activated protein kinase kinase (MEK) has the potential to change the course of the disease for patients with BRAF‐mutant NSCLC, as it has in BRAF‐mutant melanoma. Optimization of screening strategies for rare mutations and the choice of appropriate agents on an individual basis will be key to providing timely and successful intervention.
摘要
非小细胞肺癌(NSCLC)一直是全球癌症相关死亡的首要原因。但目前已识别出涉及NSCLC发生和维持的致癌性驱动变异(例如表皮生长因子受体基因突变和间变性淋巴瘤激酶基因易位), 由此催生了直接靶向突变蛋白和相关信号通路的新型治疗, 从而改善了NSCLC的临床预后。随着测序技术的进步, NSCLC的分子异质性日渐显现, 在此基础之上识别出多种具有潜在可操作性的致癌性驱动突变。其中, 最具前景的治疗靶点之一是B‐Raf原癌基因丝氨酸/苏氨酸蛋白激酶(BRAF)。2%‐4%的NSCLC携带BRAF突变, 其通常导致蛋白结构性活化, 进而激活丝裂原活化蛋白激酶信号通路。研究表明, 直接抑制突变BRAF和/或下游的丝裂原活化蛋白激酶激酶(MEK)可延长BRAF突变型转移性黑色素瘤患者的生存期。本综述围绕以下问题进行讨论:BRAF突变型NSCLC的临床特征和预后意义;从黑色素瘤BRAF和MEK抑制剂到NSCLC同类抑制剂的临床开发历程;参与筛查BRAF突变和选择靶向治疗的临床医生所需考虑的实际问题。
Introduction
The identification of actionable oncogenic driver mutations has changed the therapeutic landscape in non‐small cell lung cancer (NSCLC). Notably, targeted inhibitors of epidermal growth factor receptor (EGFR; e.g., erlotinib, afatinib, and gefitinib) and anaplastic lymphoma kinase (ALK; e.g., crizotinib, ceritinib, and alectinib) have enhanced the survival of patients with activating mutations in EGFR and rearrangements of ALK, respectively [1], [2]. Additionally, crizotinib has demonstrated clinical activity in patients with ROS1‐rearranged NSCLC and has recently been approved by the U.S. Food and Drug Administration (FDA) for this indication [3]. The success of these agents has led to a paradigm shift, whereby targeted therapeutics have replaced platinum‐based chemotherapy as the frontline treatment for these patients with targetable driver mutations.
As genomic sequencing techniques have improved, the complex molecular heterogeneity of NSCLC has become more apparent. Consequently, several novel, potentially actionable mutations have been elucidated through the use of whole exome sequencing, including a number of additional driver alterations (i.e., RET rearrangement, MET exon 14 skipping mutations, and HER2 mutations) that are under active clinical investigation [4]. One of the most promising novel targets in NSCLC is mutant B‐Raf proto‐oncogene, serine/threonine kinase (BRAF)—a member of the mitogen‐activated protein kinase (MAPK) pathway [5]. BRAF mutations, the majority of which result in activation of the MAPK pathway, occur in 2%–4% of patients with NSCLC, with the most common resulting in a glutamate substitution for valine at codon 600 (V600E) [1], [2], [6], [7]. Non‐V600E BRAF mutations make up the remaining BRAF mutations and may be either activating (i.e., G469A/V, K601E, L597R) or inactivating (i.e., D594G, G466V) [8], [9], [10], [11]. Typically, BRAF mutations are mutually exclusive from other known oncogenic driver mutations, and, therefore, they may provide an actionable target in a patient population with otherwise limited therapeutic options (Fig. 1) [1], [2]. Pharmacological inhibition of mutant BRAF alone or in combination with downstream inhibition of mitogen‐activated protein kinase kinase (MEK) has demonstrated marked efficacy in patients with BRAF V600‐mutant metastatic melanoma (MM), providing strong rationale for the application of this strategy to BRAF V600‐mutant NSCLC [12], [13], [14], [15]. On the other hand, the utility of BRAF inhibitors (BRAFi) in patients with non‐V600 mutations is not well established.
Figure 1.

BRAF mutations in the context of mitogen‐activated protein kinase (MAPK) molecular alterations. The approximate observed frequencies of common driver mutations in the MAPK pathway in lung cancer are shown on the left of the figure. BRAF valine at codon 600 (V600E) mutations leading to constitutive activation of BRAF are relatively rare, occurring in 1%–2% of lung cancers. For patients with activating BRAF mutations, direct inhibition of BRAF alone or in combination with downstream MEK inhibition is currently under clinical evaluation. Notable BRAF and MEK inhibitors under development are depicted on the right.
Abbreviations: BRAF, B‐Raf proto‐oncogene, serine/threonine kinase; EGFR, epidermal growth factor receptor; ERK, extracellular signal‐regulated kinase; HER2, human epidermal growth factor receptor 2; KRAS, KRAS proto‐oncogene, GTPase; MEK, mitogen‐activated protein kinase kinase; MET, MET proto‐oncogene, receptor tyrosine kinase; NRAS, NRAS proto‐oncogene, GTPase; ROS, ROS proto‐oncogene 1, receptor tyrosine kinase.
This review provides a comprehensive overview of the clinical characteristics and prognostic implications for patients with BRAF‐mutant NSCLC and discusses therapeutic strategies that leverage experience from BRAF‐mutant MM. Finally, practical considerations for oncologists will be discussed, including considerations for molecular profiling and management of potential toxicities associated with targeted agents.
Materials and Methods
PubMed was searched for English‐language articles published before June 30, 2016, using the search terms “oncogene,” “BRAF,” “epidermal growth factor receptor,” “anaplastic lymphoma kinase,” “non‐small cell lung cancer,” “dabrafenib,” “trametinib,” “vemurafenib,” and “melanoma.” Abstracts were searched from recent congresses, including the American Society of Clinical Oncology and the European Society for Medical Oncology. Agents in clinical development were compiled through a search of active trials on ClinicalTrials.gov.
Clinical Characteristics of Patients with BRAF‐Mutant NSCLC
Defining the clinical characteristics of patients with specific oncogenic driver mutations can help to determine patient selection for screening and treatment. For instance, EGFR mutations are more frequently observed in patients with no or light smoking history, female patients, and those with lung adenocarcinoma, whereas ALK rearrangement is commonly observed in younger patients and those with no history of smoking [16], [17], [18], [19], [20]. However, the clinical characteristics of patients with BRAF‐mutant NSCLC are less well defined, perhaps due to the lower frequency of BRAF mutations.
One characteristic associated strongly with BRAF‐mutation frequency is adenocarcinoma histology. More than 85% of BRAF mutations are observed in adenocarcinomas, although they have also been reported in other histological subtypes, including squamous cell carcinoma (SCC) and large‐cell carcinoma [1], [8], [11], [21], [22]. The association between BRAF mutation status and patient age or sex appears to be less clear. The median age of patients presenting with BRAF‐mutant NSCLC (≈65 years) is similar to the reported median age of patients presenting with no known actionable mutation or mutations in EGFR or KRAS proto‐oncogene, GTPase (KRAS) [1], [2], [6], [8], [9], [10], [11], [21], [22], [23]. With respect to sex, one study [9] observed a significant (p < .001) association between female sex and BRAF mutation frequency; however, this result has not been confirmed in other studies: two separate studies reported no significant sex differences between patients with BRAF‐mutant NSCLC and patients with wild‐type tumors or unknown mutational status [1], [10].
Although EGFR mutations and ALK rearrangements are primarily associated with no or a light history of smoking [18], [20], several studies have shown that the majority of BRAF‐mutant patients are former or current smokers [1], [8], [9], [10], [11], [21], [23]. However, limited evidence exists suggesting that patients with BRAF V600E mutations may be less likely to have a smoking history compared with those with non‐V600E mutations [11]. Overall, approximately 20%–30% of patients with BRAF‐positive tumors report no history of smoking [1], [21], [22].
Differences in the frequency of BRAF mutations based on ethnicity have been observed in other tumor types, including a higher incidence of BRAF‐mutant colorectal cancer in patients of Anglo‐Saxon descent versus Southern European descent. In prostate cancer, BRAF mutations have been identified in Asian patients but not in white patients [24], [25], [26], [27]. Data are limited in NSCLC, but studies suggest that BRAF mutations may occur at a lower frequency in Asian patients (0.8%–2.0%) [23], [28], [29] compared with white patients primarily from France and the U.S. (2%–4%) [1], [2]. Additionally, the proportion of BRAF‐mutant NSCLC harboring a V600E mutation appears to be lower in Asian patients (30%–40%) compared with white patients (≈50%–70%) [2], [8], [11], [23], [28]. Data regarding the frequency of BRAF mutations among other ethnic cohorts are lacking and should be a focus of future research.
Therefore, aside from adenocarcinoma histology, the clinical characteristics that define patients likely to harbor BRAF mutations are not readily apparent. As opposed to other oncogenic mutations, mutations in BRAF occur in a more heterogeneous population; thus, screening for BRAF should not be limited by factors such as age, sex, or smoking status. Additionally, differences in the clinical characteristics associated with BRAF V600E and non‐V600E mutations add further complexity to the characterization of BRAF mutations as a whole.
Prognostic Significance of BRAF Mutations
Based on current literature, the prognostic significance of BRAF mutation positivity is not entirely clear; studies of patients with BRAF‐mutant NSCLC report largely conflicting results (Table 1). The inconsistencies are likely the result of small patient numbers and patient heterogeneity across various studies.
Table 1. Summary of outcomes for patients with BRAF‐mutant NSCLC [1, 8–11].
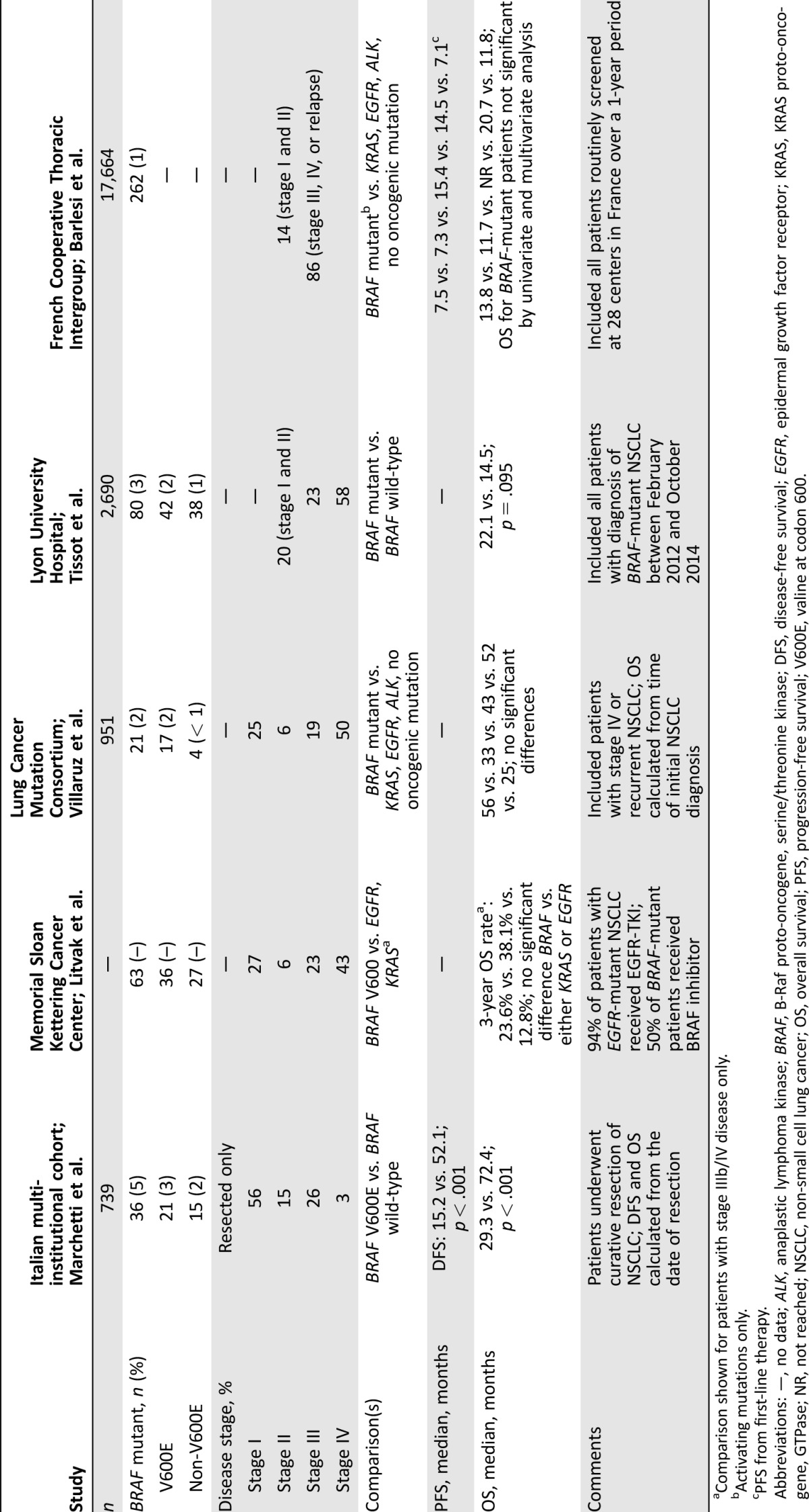
Comparison shown for patients with stage IIIb/IV disease only.
Activating mutations only.
PFS from first‐line therapy.
Abbreviations: —, no data; ALK, anaplastic lymphoma kinase; BRAF, B‐Raf proto‐oncogene, serine/threonine kinase; DFS, disease‐free survival; EGFR, epidermal growth factor receptor; KRAS, KRAS proto‐oncogene, GTPase; NR, not reached; NSCLC, non‐small cell lung cancer; OS, overall survival; PFS, progression‐free survival; V600E, valine at codon 600.
In an Italian cohort, patients with resected BRAF V600E‐mutant lung adenocarcinoma (n = 21) had a significantly shorter median disease‐free survival (15.2 vs. 52.1 months; p < .001) and overall survival (OS; 29.3 vs. 72.4 months; p < .001) compared with patients with tumors that were BRAF wild‐type (n = 310). No significant difference in disease‐free survival or OS was observed between patients with and without BRAF non‐V600E mutations [9].
Conversely, other studies have demonstrated a trend toward superior OS in patients with BRAF‐mutant NSCLC compared with other oncogene‐driven NSCLC or wild‐type tumors. In a French cohort, the median OS for patients with stage I–IV BRAF‐mutant NSCLC was 22.1 months versus 14.5 months in patients with BRAF wild‐type tumors (p = .095) [11]. Among a cohort of patients from the U.S. with advanced lung adenocarcinoma, those with BRAF‐mutant tumors (n = 15) had the longest OS (56 months) compared with patients with other known oncogenic drivers or no known oncogenic mutations, although this finding was not statistically significant [10].
Limited data are available with regard to the prognostic significance of BRAF positivity compared with that of other oncogene‐driven NSCLCs. A study examining patients with unresectable stage IIIB/IV BRAF V600‐mutant lung adenocarcinoma (n = 20) demonstrated a 3‐year OS of 23.6% compared with 38.1% in patients with EGFR mutations (n = 130; p = .25) and 12.8% in patients with KRAS‐mutant tumors (n = 142; p = .12) [8]. In addition, findings from a French National Cancer Institute study showed that patients with NSCLC with activating BRAF mutations (n = 132) had a median OS of 13.8 months compared with 11.7 months for patients with KRAS‐mutant NSCLC. The median OS values for patients with EGFR mutations (not reached) or those with ALK rearrangement (20.7 months) were much greater [1]. The above studies included heterogeneous groups of patients, and further studies are needed to better elucidate the prognostic significance of BRAF‐mutant NSCLC.
While the prognostic implications of BRAF mutation remain unclear, case reports have clearly demonstrated that durable disease control in patients with metastatic BRAF V600E‐mutant NSCLC is possible [30], [31]. However, data from an observational cohort in a real‐world setting showed that in the second‐line setting, 57% of patients with activating BRAF mutations received best supportive care only [1]. This observation indicates that there is an unmet need in patients with BRAF‐mutant NSCLC in whom rationally designed targeted therapy could provide benefit.
Clinical Development of BRAF and MEK Inhibitors
Melanoma.
Due to the high frequency of BRAF mutations in MM (≈50%), therapeutic development of specific BRAFi (dabrafenib or vemurafenib) and MEK inhibitors (MEKi; trametinib or cobimetinib) was initiated for this molecular subtype. In preclinical models, both BRAFi and MEKi reduced extracellular signal‐regulated kinase (ERK) signaling specifically in BRAF V600E‐mutant melanoma cell lines, leading to cell cycle arrest and apoptosis. In mouse xenograft models, these inhibitors also led to tumor growth delay and regression [32], [33]. Additionally, combinations of BRAFi and MEKi demonstrated synergistic antitumor activity and could prevent or delay the acquired resistance observed with BRAFi treatment [34], [35].
This work provided the foundation for clinical evaluation of BRAFi, MEKi, and combination therapy in patients with BRAF V600‐mutant MM. In similar randomized phase III trials, vemurafenib and dabrafenib each significantly reduced the risk of disease progression or death compared with dacarbazine in patients with previously untreated BRAF V600E‐mutant MM (hazard ratio [HR]), 0.26 and 0.30, respectively; p < .001), clearly demonstrating the efficacy of BRAFi in this setting [12], [13].
Despite the success of BRAFi monotherapy, 50% of the patients treated with these inhibitors experienced disease progression within 6–7 months following treatment initiation [13], [36], [37]. In order to achieve complete blockade of the MAPK pathway and reduce or delay resistance, combination BRAFi and MEKi was evaluated. In two randomized phase III trials, the combination of dabrafenib plus trametinib significantly prolonged progression‐free survival (PFS) and OS compared with either vemurafenib monotherapy (PFS: HR, 0.61; p < .001; and OS: HR, 0.66; p < .001) or dabrafenib plus placebo (PFS: HR, 0.67; p = .0004; and OS: HR, 0.71; p = .0107) in patients with previously untreated, unresectable BRAF V600‐mutant melanoma [14], [38]. The combination of vemurafenib plus cobimetinib has also demonstrated enhanced OS compared with vemurafenib monotherapy in a phase III trial of patients with BRAF V600‐mutant MM (22.3 vs. 17.4 months; HR, 0.70; p = .005) [39]. Across all aforementioned trials, combination therapy was associated with a significantly higher proportion of patients achieving a response compared with BRAFi monotherapy [14], [38], [39]. Based on the success of these trials, combinations of BRAFi and MEKi have become the standard of care for patients with BRAF V600‐mutant MM. The experience from clinical trials in melanoma provided the proof of concept that BRAF mutations are actionable targets for therapeutic intervention.
Metastatic NSCLC.
Although only 2%–4% of patients with NSCLC harbor a BRAF mutation, the vast number of patients each year with a diagnosis of NSCLC suggests that the BRAF‐mutant subgroup could represent a substantial number of patients for whom effective treatment options may be lacking. Evidence from preclinical evaluation suggests that, as in BRAF‐mutant melanoma, inhibition of BRAF or MEK alone is sufficient to induce cell cycle arrest and apoptosis in BRAF V600E‐mutant NSCLC cell lines. Furthermore, the combination of BRAFi and MEKi has demonstrated synergistic enhancement of apoptosis compared with either agent alone [40].
In an analysis of primarily (86%) previously treated patients with advanced‐stage, BRAF‐mutant (83% V600E) NSCLC who received BRAFi therapy (sorafenib [n = 1], vemurafenib [n = 29], or dabrafenib [n = 9]) outside of a clinical trial setting, the overall response rate (ORR; complete response [CR] + partial response [PR]) was 53%, and the disease control rate (DCR; CR + PR + stable disease [SD]) was 85% [41]. Patients had a median PFS of 5.0 months and a median OS of 10.8 months.
The initial report of BRAFi therapy in patients with BRAF‐mutant NSCLC from any prospective clinical trial came from a cohort of patients treated with vemurafenib as part of a basket trial [42]. Twenty patients with BRAF‐mutant NSCLC (90% V600E) were enrolled. The median age was 61 years, the majority of patients were male (70%), and nearly all had received one or more prior systemic therapies (95%). Among the 19 patients with one or more post‐baseline assessments, the ORR was 42% (all PRs), and an additional eight patients had SD for a DCR of 84%. Notably, responses in this trial did not require a second post‐baseline scan for confirmation. The median PFS was 7.3 months (95% confidence interval [CI], 3.5–10.8 months), and the 12‐month OS was 66% (95% CI, 36%–85%). Nearly all patients (95%) experienced at least one adverse event (AE), including rash (65%), photosensitivity (25%), and cutaneous SCC (35%).
Evaluation of dabrafenib in patients with BRAF‐mutant NSCLC is also under way in an open‐label, multicohort phase II trial (BRF113928; NCT01336634). Cohort A of this trial was designated for evaluation of the clinical activity of dabrafenib monotherapy, primarily in patients with previously treated BRAF V600E‐mutant NSCLC [22]. In cohort B, the combination of dabrafenib plus trametinib was investigated in patients with BRAF V600E‐mutant NSCLC who had received one or more prior platinum‐based chemotherapies [43]. Cohort C is ongoing and is designated for evaluation of the clinical activity of dabrafenib plus trametinib in patients with no prior systemic therapy.
Patients in cohort A (previously treated; n = 78) had a median age of 66 years, 50% were male, and the majority were white (76%); 79% had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≥1, 96% had adenocarcinoma histology, and 63% were current or former smokers. The investigator‐assessed confirmed ORR was 33% (all PRs), with a DCR of 58%. Responses were durable, with a median duration of response (DOR) of 9.6 months (95% CI, 5.4–15.2 months) and a median PFS of 5.5 months (95% CI, 3.4–7.3 months). The median OS was 12.7 months (95% CI, 7.3–16.9 months).
Notably, the safety profile of dabrafenib monotherapy in this cohort was similar to that of clinical experience in melanoma. Nearly all patients (99%) experienced one or more AEs. The most common AEs (≥30%) were pyrexia (36%), asthenia (30%), and hyperkeratosis (30%). The most common serious AEs were pyrexia (6%), decreased ejection fraction (2%), and pneumonia (2%). The development of cutaneous SCC was observed in 12% of patients. AEs led to dabrafenib discontinuation in 6% of patients and dose interruption in 43%; however, the majority of patients received the intended daily dose of the drug. One patient receiving a factor Xa inhibitor died from an intracranial hemorrhage that was deemed by the investigator to be related to dabrafenib treatment. Despite a high proportion of patients experiencing AEs, dabrafenib appears to have a manageable safety profile, with low rates of serious AEs.
While BRAFi demonstrated clinical activity in patients with BRAF‐mutant NSCLC, the response rates were modest compared with the >50% response typically observed with targeted therapies in other oncogene‐driven NSCLCs [44], [45], [46]. Furthermore, nearly 30% of patients treated with dabrafenib monotherapy in BRF113928 cohort A had disease progression as their best response [22]. These data suggest that more potent inhibition of the MAPK pathway could provide additional benefit in this treatment setting.
Cohort B of the BRF113928 trial evaluated the clinical activity and safety of combination dabrafenib plus trametinib in patients with previously treated BRAF V600E‐mutant metastatic NSCLC [43]. Of 57 previously treated patients, 51% were male, and the majority were white (86%); 70% had an ECOG PS ≥1, 98% had adenocarcinoma histology, and 72% were former or current smokers. Thirty‐eight (67%) of the 57 patients had received one prior treatment, and 19 (33%) had received 2–3 prior treatments. By investigator assessment, two patients had a CR, and 34 patients had a PR, for an ORR of 63% (95% CI, 49%–76%). The DCR was 79% (95% CI, 66%–89%), with 9 patients having prolonged SD (defined as SD for ≥12 weeks). The median DOR was 9.0 months (95% CI, 6.9–18.3 months), although this result could be an underestimation, as 50% of responses were ongoing at data cutoff. Patients receiving dabrafenib plus trametinib had a median PFS of 9.7 months (95% CI, 6.9–19.6 months). OS data were immature, however; 82% of patients were alive 6 months after the start of therapy. This trial was not designed for active comparison across cohorts, and such comparisons should be interpreted with caution. However, the data with combination therapy in cohort B appear to compare favorably with the monotherapy data for vemurafenib or dabrafenib, as outlined in Table 2.
Table 2. Clinical activity of targeted therapy in BRAF‐mutant metastatic non‐small cell lung cancer.
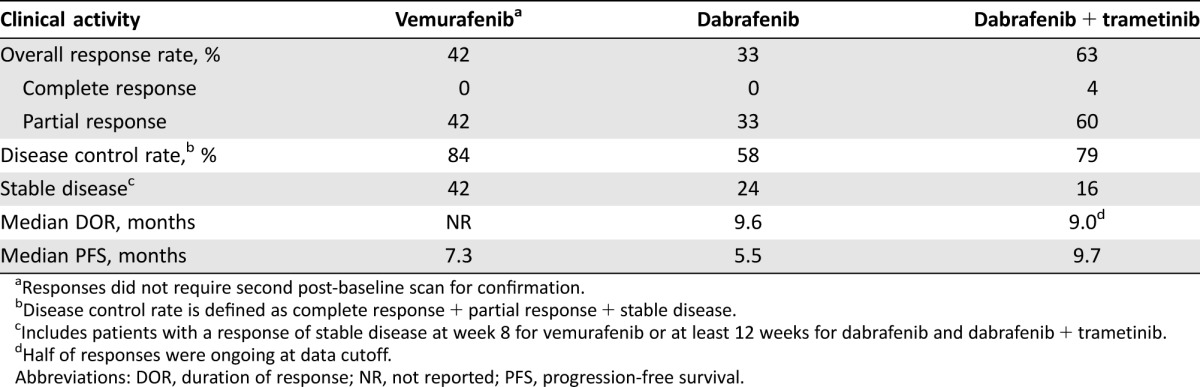
Responses did not require second post‐baseline scan for confirmation.
Disease control rate is defined as complete response + partial response + stable disease.
Includes patients with a response of stable disease at week 8 for vemurafenib or at least 12 weeks for dabrafenib and dabrafenib + trametinib.
Half of responses were ongoing at data cutoff.
Abbreviations: DOR, duration of response; NR, not reported; PFS, progression‐free survival.
The combination was tolerable, with a safety profile similar to that observed in patients with MM treated with dabrafenib plus trametinib [14]. AEs led to permanent discontinuation, dose interruption, and dose reduction in 12%, 61%, and 35% of patients, respectively. When compared indirectly, the incidence of pyrexia observed in this cohort (46%) [43] was higher than that observed with dabrafenib monotherapy (36%) [22]. However, the incidence of cutaneous SCC was 4% with dabrafenib plus trametinib versus 12% with dabrafenib monotherapy [22], [43]. A comparison of grade 3/4 AEs for BRAFi monotherapy versus dabrafenib plus trametinib is presented in Table 3. Four patients died due to serious AEs in this cohort, but no deaths were determined to be related to the study drugs.
Table 3. Common grade 3/4 adverse events with vemurafenib, dabrafenib, and dabrafenib plus trametinib.
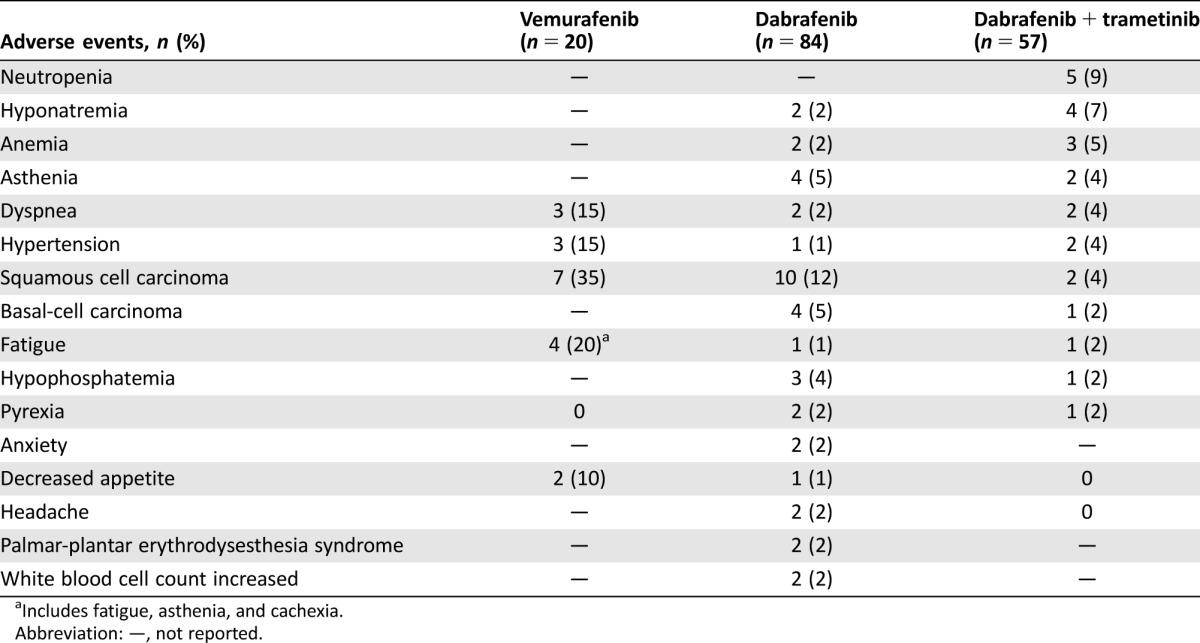
Includes fatigue, asthenia, and cachexia.
Abbreviation: —, not reported.
Cohort C is evaluating the combination of dabrafenib plus trametinib in patients with previously untreated BRAF V600E‐mutant metastatic NSCLC. In addition, studies evaluating a number of BRAFi and MEKi in patients with BRAF‐mutant NSCLC or in patients with a range of BRAF‐mutant solid tumors are ongoing (Table 4).
Table 4. Trials evaluating targeted therapies in BRAF‐mutant non‐small cell lung cancer.
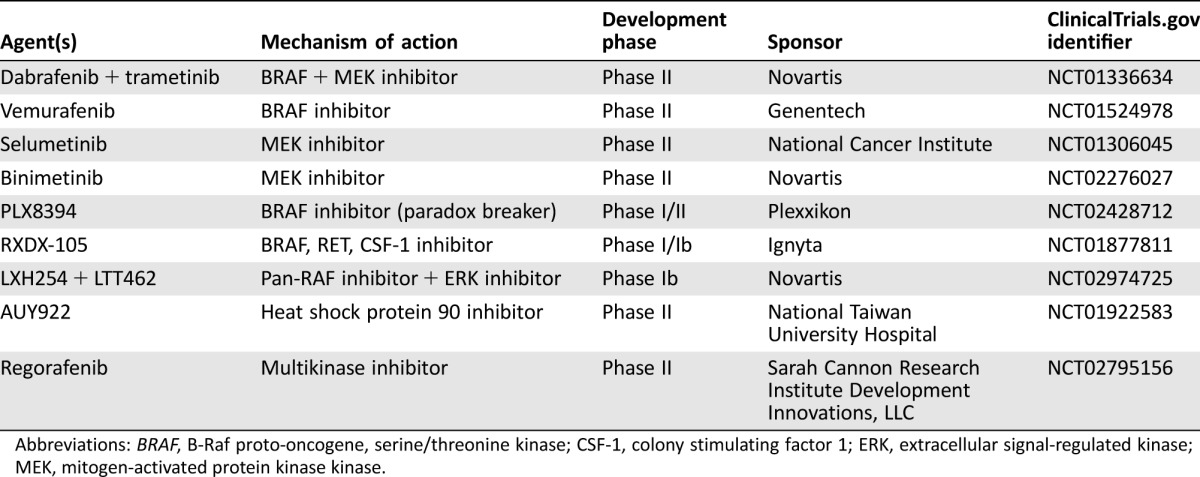
Abbreviations: BRAF, B‐Raf proto‐oncogene, serine/threonine kinase; CSF‐1, colony stimulating factor 1; ERK, extracellular signal‐regulated kinase; MEK, mitogen‐activated protein kinase kinase.
Acquired Resistance to BRAF/MEK‐Targeted Therapy.
Although a high rate of response and low rate of primary resistance has been observed in patients with BRAF V600E‐mutant NSCLC treated with BRAFi alone or in combination with MEKi, acquired resistance eventually ensues. Recently, mechanisms mediating acquired resistance to BRAFi via reactivation of MAPK pathway signaling have begun to emerge. Continuous treatment of a human BRAF V600E‐mutant NSCLC cell line with vemurafenib resulted in development of acquired resistance mediated at least partially through two discrete mechanisms: (a) loss of full‐length BRAF V600E in concert with expression of a truncated form of the mutant protein or (b) enhanced EGFR signaling through autocrine activation induced through BRAF‐independent c‐Jun signaling [47]. Notably, resistance mediated through expression of a BRAF V600E splice variant has been shown to be prevented through combined BRAF and MEK inhibition or use of second‐generation BRAFi such as PLX8394 [47, 48]. Determining mechanisms of resistance and targeted strategies should be an area of focused research over the coming years and has the potential to vastly improve the outcomes in patients in this setting.
Notably, resistance mediated through expression of a BRAF V600E splice variant has been shown to be prevented through combined BRAF and MEK inhibition or use of second‐generation BRAFi such as PLX8394. Determining mechanisms of resistance and targeted strategies should be an area of focused research over the coming years and has the potential to vastly improve the outcomes in patients in this setting.
Practical Considerations
Identification of BRAF‐Positive Patients.
The discovery of oncogenic driver mutations in NSCLC and the development of targeted therapeutics have revolutionized the treatment of these patients. However, the complex molecular heterogeneity of the disease has also created a challenge for oncologists in selecting the optimal method for mutational testing so that clinically relevant information can be obtained in a timely manner, especially with limited biopsy samples. Currently, patients with newly diagnosed adenocarcinoma routinely undergo molecular testing for the presence of activating EGFR mutations and ALK and ROS1 translocations, as there are effective targeted therapies approved by the FDA for patients with these genetic alterations. However, no consensus currently exists as to how testing should be conducted, with some clinicians opting for sequential testing and others testing for all three alterations simultaneously [49]. Sequential testing, which often starts with EGFR testing and continues to ALK and ROS1 assessment if a patient is EGFR‐mutation‐negative, may be more cost effective, but it is more likely to lead to problems due to insufficient tissue and delays in identifying the other, less common genetic alterations. Furthermore, as the number of actionable genetic alterations increases, including BRAF mutations, sequential testing will further increase these challenges. Due to these limitations, a recent survey of National Cancer Institute centers found that the majority perform concurrent upfront testing of multiple mutations despite higher associated costs [50].
Multiplex testing will likely be the most efficient method to simultaneously test for actionable targets, particularly as more clinically relevant targets (e.g., MET, RET, HER2) are identified. Multiplex testing, such as next‐generation sequencing recommended by the National Comprehensive Cancer Network guidelines in NSCLC [50], has the ability to detect multiple markers using a single sample [51]. Furthermore, it allows for the best systematic identification of rare mutations that might otherwise go undetected. Current barriers to broad integration of multiplex testing into clinical application include challenges related to heterogeneity of available assays, variable levels of clinical validation, and reimbursement for the assays. Further consensus on the clinical value and cost effectiveness of multiplex testing will help to ensure broader coverage and facilitate more widespread clinical integration [52].
Liquid biopsy methods could also change the landscape of molecular profiling in NSCLC by allowing for examination of circulating tumor cells, circulating tumor DNA (ctDNA), circulating RNA, or microRNAs [53]. Liquid biopsies have the potential to address the limitations on the number of tests that could be performed due to scarce biopsy samples. Because acquired resistance is often observed with most targeted therapies, liquid biopsy methods could also be used to evaluate resistance mutations and potentially avoid repeat biopsies in some patients. Additionally, on‐treatment monitoring of BRAF‐mutant allele fraction has the potential to measure depth of response (i.e., identification of residual disease) and provide early indication of progression as has been observed in other malignancies, including BRAF‐mutant melanoma [54], [55]. Currently, several ctDNA assays that are likely to expand in usage are undergoing clinical validation [56], [57]. However, it is important to recognize the limited sensitivity and specificity of these assays and limited clinical data in resistance settings [57], [58]. The use of ctDNA is unlikely to replace tumor biopsies, but it may be an important adjunct to current modalities for molecular testing.
Because acquired resistance is often observed with most targeted therapies, liquid biopsy methods could also be used to evaluate resistance mutations and potentially avoid repeat biopsies in some patients.
Potential Choice of Targeted Therapy in BRAF‐Mutant NSCLC.
With limited data and no head‐to‐head comparisons, a conclusion cannot be definitively reached regarding the optimal therapy in BRAF‐mutant patients, and, as of the time of writing, no targeted therapies are approved by the FDA for use in this indication. Consistent with results from randomized trials in melanoma [14], [15], the ORR and PFS were numerically higher in patients treated with dabrafenib plus trametinib [43] compared with either dabrafenib [22] or vemurafenib [42] monotherapy. However, in the absence of a randomized comparison in NSCLC, the potential choice among clinically active agents (vemurafenib, dabrafenib, or dabrafenib plus trametinib) could reasonably be guided by patient comorbidities and tolerability of these agents.
Although the toxicities associated with vemurafenib and dabrafenib are largely overlapping, there are notable exceptions associated with each agent. For instance, vemurafenib is associated with phototoxicity that is thought to be related to its chemical structure [59]. On the other hand, dabrafenib has been associated with an increased incidence of pyrexia potentially related to off‐target effects of a dabrafenib metabolite [60]. Notably, the combination of dabrafenib plus trametinib was not associated with any new safety signals, but it may increase or decrease the incidence and severity of a number of notable BRAFi‐ or MEKi‐related AEs. The rate and severity of pyrexia and some gastrointestinal toxicities were higher with dabrafenib plus trametinib versus dabrafenib monotherapy. Conversely, the incidence of a number of cutaneous AEs, particularly development of SCC, was reduced with combination therapy due to blockade of paradoxical MAPK activation in BRAF wild‐type cells through MEK inhibition (Table 3) [14].
The extensive experience with BRAFi and MEKi in melanoma has provided a wealth of information on how to properly manage common toxicities associated with these regimens. Typically, AEs can be managed through dose interruption or reduction without the need for permanent discontinuation of therapy. Dose reduction steps for BRAFi and MEKi derived from experience in melanoma are outlined in Figure 2. Comprehensive guidelines for management of BRAFi‐ and MEKi‐associated toxicities for patients with melanoma have been previously published and should help to inform thoracic oncologists who may not be familiar with these AEs [61], [62].
Figure 2.

Recommended dose reduction steps due to adverse events (AEs). aDose interruptions are generally due to grade 3 toxicity, and dose reductions occur upon reinitiation of treatment when AEs resolve to grade 1 or baseline levels. bDoses for combination dabrafenib plus trametinib are the same as for monotherapy. If dose reduction is necessary, the drug most likely to be contributing to the AE should be reduced in the same manner as monotherapy.
Abbreviations: BID, twice daily; QD, once daily.
The use of targeted inhibition in the treatment of BRAF‐mutant NSCLC has garnered deserved enthusiasm. However, ongoing research will likely focus on optimization of targeted strategies and enhancement of the risk‐benefit profile of agents in this setting. These efforts may be served by evaluating strategies with a clear rationale in other BRAF‐mutant tumors. For instance, in some BRAF‐mutant melanomas, resistant cells can become reliant on continued BRAFi treatment for their sustained proliferation [63]. This has led to the evaluation of intermittent dosing strategies in patients with BRAF‐mutant melanoma, with the intended goal of providing sustained tumor regression and avoiding undue toxicity from continuous drug exposure. There are case reports of prolonged disease control using intermittent dosing [64], and the clinical utility of this strategy in patients with melanoma is being evaluated in a randomized phase II trial comparing the efficacy of continuous versus intermittent dabrafenib and trametinib (NCT02196181) that should provide evidence pertaining to clinical feasibility.
Potential lessons could be learned from the treatment of BRAF‐mutant colorectal cancer as well. BRAF‐mutant colorectal cancers have demonstrated marked resistance to BRAFi that is driven, at least in part, through EGFR‐mediated reactivation of MAPK pathway signaling [65]. In an effort to overcome this resistance, triple combination of BRAFi plus MEKi plus EGFR tyrosine kinase inhibitors is currently under evaluation in patients with BRAF‐mutant colorectal cancer (NCT01750918). Preclinical data suggest that this strategy may have utility in patients with NSCLC, as acquired resistance to PLX8394 could be prevented by upfront combination with EGFR or mTOR inhibitors [48].
Treatment Approaches in Patients with Non‐V600E Mutations.
Approximately half of all patients with BRAF‐mutant NSCLC present with non‐V600E mutations [8], [9], [10], [11]. However, efforts targeting BRAF‐mutant NSCLC to date have almost exclusively focused on patients with V600E‐mutant disease. In the study by Gautschi et al., a total of six patients with non‐V600E mutations were enrolled (G466V, G469A, G469L, G596V, V600K, K601E) [41]. One patient with a G596V mutation experienced a PR with vemurafenib therapy.
Although clinical data are limited among patients with non‐V600E mutations in NSCLC, combination of dabrafenib plus trametinib has demonstrated antiproliferative and proapoptotic effects in cell lines harboring both activating and inactivating BRAF non‐V600 mutations [66]. Additionally, PLX8394 has demonstrated preclinical activity in both V600E and non‐V600E lung adenocarcinoma models [48].
Similarly, clinical activity of targeted therapy in patients with non‐V600E mutations has been observed in patients with MM. For example, trials in melanoma evaluating combination targeted therapy have routinely included patients with BRAF V600E or V600K mutations. These trials have demonstrated similar response to combination therapy between genotypes [67], [68] and a similar survival benefit compared with BRAFi monotherapy in patients with V600E and V600K mutations [14], [39]. Mutations other than V600E/K are rare in patients with melanoma; however, clinical activity (five of six patients with a clinical response with BRAFi monotherapy) has been observed in a limited sample of patients with BRAF V600R mutations as well [69].
Due to the large proportion of non‐V600E mutations in patients with NSCLC and the as yet unknown clinical activity of targeted BRAF and MEK inhibition in these patients, additional clinical investigation of BRAFi alone or in combination with MEKi and other therapeutic strategies in this setting is warranted. Several trials are currently ongoing to evaluate novel strategies in patients with BRAF‐mutant NSCLC, including patients with non‐V600E mutations. These trials include evaluation of combined pan‐RAF inhibitor (LXH254) + ERK inhibitor (LTT462; NCT02974725), heat shock protein 90 inhibitor (AUY922; NCT01922583), MEKi (AZD6244; NCT01306045), and multikinase inhibitor (regorafenib; NCT02795156).
Conclusion
Novel agents directly targeting oncogene‐driven NSCLC have changed the natural history of the disease in patients with EGFR mutations and ALK rearrangement [1], [2]. Vemurafenib and dabrafenib have demonstrated clinical activity in patients with previously treated BRAF V600‐mutant NSCLC, with responses observed in 33%–42% of patients and a median PFS of 5.5–7.3 months [22], [42]. Similar to experience in melanoma, a numerically higher ORR (63%) and longer PFS (9.7 months) have been observed in patients with NSCLC treated with combination BRAFi and MEKi compared indirectly with BRAFi monotherapy [22], [43]. To date, difficulties in the identification of BRAF‐positive patients have limited the power of studies examining clinical characteristics and prognostic implications of BRAF‐mutant NSCLC and have precluded conduct of large‐scale randomized trials in this indication. The relatively rare nature of BRAF mutations and lack of obvious association with clinical characteristics aside from adenocarcinoma histology suggest that identification of BRAF‐positive patients will improve with broad application of multiplex screening techniques. Although extended follow‐up and additional studies will be needed to understand whether BRAFi and MEKi can change the natural history of BRAF‐mutant NSCLC, data thus far have demonstrated that targeted therapy has clear clinical activity in this patient population with an unmet need.
Acknowledgments
The authors would like to thank Michael Demars at ArticulateScience, LLC, for medical writing support funded by Novartis Pharmaceuticals Corporation. Neither Novartis Pharmaceuticals Corporation nor ArticulateScience, LLC, influenced the content of this manuscript, nor did the author receive financial compensation for authorship.
Footnotes
For Further Reading: Pascale Tomasini, Preet Walia, Catherine Labbe et al. Targeting the KRAS Pathway in Non‐Small Cell Lung Cancer. The Oncologist 2016;21:1450–1460.
Implications for Practice: The identification of oncogene‐addicted cancers and specific inhibitors has revolutionized non‐small cell lung cancer (NSCLC) treatment and outcomes. One of the most commonly mutated genes in adenocarcinoma is KRAS, found in approximately 30% of lung adenocarcinomas, and thus it is an appealing target for new therapies. This review provides an overview of the KRAS pathway and related targeted therapies under investigation in NSCLC. Some of these agents may play a key role in KRAS‐mutant NSCLC treatment in the future.
Author Contributions
Conception/Design: Christina S. Baik
Collection and/or assembly of data: Christina S. Baik, Nathaniel J. Myall, Heather Wakelee
Data analysis and interpretation: Christina S. Baik, Nathaniel J. Myall, Heather Wakelee
Manuscript writing: Christina S. Baik, Nathaniel J. Myall, Heather Wakelee
Final approval of manuscript: Christina S. Baik, Nathaniel J. Myall, Heather Wakelee
Disclosures
Christina S. Baik: Clovis (C/A), Clovis, Genentech, GlaxoSmithKline, Incyte, Loxo Oncology, Millennium, Novartis, SWOG (RF); Heather Wakelee: ACEA Biosciences, Genentech, Helsinn, Peregrine, Pfizer (C/A), AstraZeneca, Bristol‐Myers Squibb, Celgene, Clovis, Exelixis, Genentech/Roche, Gilead, Lilly, MedImmune, Novartis, Pfizer, Pharmacyclics, Xcovery (RF). The other author indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Barlesi F, Mazieres J, Merlio JP et al. Routine molecular profiling of patients with advanced non‐small‐cell lung cancer: Results of a 1‐year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–1426. [DOI] [PubMed] [Google Scholar]
- 2. Kris MG, Johnson BE, Berry LD et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shaw AT, Ou SH, Bang YJ et al. Crizotinib in ROS1‐rearranged non‐small‐cell lung cancer. N Engl J Med 2014;371:1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dhillon AS, Hagan S, Rath O et al. MAP kinase signalling pathways in cancer. Oncogene 2007;26:3279–3290. [DOI] [PubMed] [Google Scholar]
- 6. Paik PK, Arcila ME, Fara M et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–954. [DOI] [PubMed] [Google Scholar]
- 8. Litvak AM, Paik PK, Woo KM et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marchetti A, Felicioni L, Malatesta S et al. Clinical features and outcome of patients with non‐small‐cell lung cancer harboring BRAF mutations. J Clin Oncol 2011;29:3574–3579. [DOI] [PubMed] [Google Scholar]
- 10. Villaruz LC, Socinski MA, Abberbock S et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tissot C, Couraud S, Tanguy R et al. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer 2016;91:23–28. [DOI] [PubMed] [Google Scholar]
- 12. Chapman PB, Hauschild A, Robert C et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hauschild A, Grob JJ, Demidov LV et al. Dabrafenib in BRAF‐mutated metastatic melanoma: A multicentre, open‐label, phase 3 randomised controlled trial. Lancet 2012;380:358–365. [DOI] [PubMed] [Google Scholar]
- 14. Long GV, Stroyakovskiy D, Gogas H et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF‐mutant melanoma: A multicentre, double‐blind, phase 3 randomised controlled trial. Lancet 2015;386:444–451. [DOI] [PubMed] [Google Scholar]
- 15. Larkin J, Ascierto PA, Dréno B et al. Combined vemurafenib and cobimetinib in BRAF‐mutated melanoma. N Engl J Med 2014;371:1867–1876. [DOI] [PubMed] [Google Scholar]
- 16. Inamura K, Takeuchi K, Togashi Y et al. EML4‐ALK lung cancers are characterized by rare other mutations, a TTF‐1 cell lineage, an acinar histology, and young onset. Mod Pathol 2009;22:508–515. [DOI] [PubMed] [Google Scholar]
- 17. Kwak EL, Bang YJ, Camidge DR et al. Anaplastic lymphoma kinase inhibition in non‐small‐cell lung cancer. N Engl J Med 2010;363:1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pao W, Miller V, Zakowski M et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004;101:13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosell R, Moran T, Queralt C et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–967. [DOI] [PubMed] [Google Scholar]
- 20. Wong DW, Leung EL, So KK et al. The EML4‐ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild‐type EGFR and KRAS. Cancer 2009;115:1723–1733. [DOI] [PubMed] [Google Scholar]
- 21. Cardarella S, Ogino A, Nishino M et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non‐small cell lung cancer. Clin Cancer Res 2013;19:4532–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Planchard D, Kim TM, Mazieres J et al. Dabrafenib in patients with BRAF(V600E)‐positive advanced non‐small‐cell lung cancer: A single‐arm, multicentre, open‐label, phase 2 trial. Lancet Oncol 2016;17:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kinno T, Tsuta K, Shiraishi K et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–142. [DOI] [PubMed] [Google Scholar]
- 24. English DR, Young JP, Simpson JA et al. Ethnicity and risk for colorectal cancers showing somatic BRAF V600E mutation or CpG island methylator phenotype. Cancer Epidemiol Biomarkers Prev 2008;17:1774–1780. [DOI] [PubMed] [Google Scholar]
- 25. Cho NY, Choi M, Kim BH et al. BRAF and KRAS mutations in prostatic adenocarcinoma. Int J Cancer 2006;119:1858–1862. [DOI] [PubMed] [Google Scholar]
- 26. Köllermann J, Albrecht H, Schlomm T et al. Activating BRAF gene mutations are uncommon in hormone refractory prostate cancer in Caucasian patients. Oncol Lett 2010;1:729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu T, Willmore‐Payne C, Layfield LJ et al. Lack of BRAF activating mutations in prostate adenocarcinoma: A study of 93 cases. Appl Immunohistochem Mol Morphol 2009;17:121–125. [DOI] [PubMed] [Google Scholar]
- 28. Kobayashi M, Sonobe M, Takahashi T et al. Clinical significance of BRAF gene mutations in patients with non‐small cell lung cancer. Anticancer Res 2011;31:4619–4623. [PubMed] [Google Scholar]
- 29. Sasaki H, Kawano O, Endo K et al. Uncommon V599E BRAF mutations in Japanese patients with lung cancer. J Surg Res 2006;133:203–206. [DOI] [PubMed] [Google Scholar]
- 30. Goldman JM, Gray JE. BRAF V600E mutations: A series of case reports in patients with non‐small cell lung cancer. Cancer Genet 2015;208:351–354. [DOI] [PubMed] [Google Scholar]
- 31. Myall NJ, Neal JW, Cho‐Phan CD et al. Long‐term survival of a patient with non‐small‐cell lung cancer harboring a V600E mutation in the BRAF oncogene. Clin Lung Cancer 2016;17:e17–e21. [DOI] [PubMed] [Google Scholar]
- 32. Solit DB, Garraway LA, Pratilas CA et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006;439:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsai J, Lee JT, Wang W et al. Discovery of a selective inhibitor of oncogenic B‐Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 2008;105:3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paraiso KH, Fedorenko IV, Cantini LP et al. Recovery of phospho‐ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 2010;102:1724–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. King AJ, Arnone MR, Bleam MR et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS One 2013;8:e67583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Flaherty KT, Robert C, Hersey P et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med 2012;367:107–114. [DOI] [PubMed] [Google Scholar]
- 37. Sosman JA, Kim KB, Schuchter L et al. Survival in BRAF V600‐mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012;366:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robert C, Karaszewska B, Schachter J et al. Two year estimate of overall survival in COMBI‐v, a randomized, open‐label, phase III study comparing the combination of dabrafenib (D) and trametinib (T) with vemurafenib (Vem) as first‐line therapy in patients (pts) with unresectable or metastatic BRAF V600E/K mutation‐positive cutaneous melanoma. Eur J Cancer 2015;51(suppl 3):3301a. [Google Scholar]
- 39. Ascierto PA, McArthur GA, Dréno B et al. Cobimetinib combined with vemurafenib in advanced BRAFV600‐mutant melanoma (coBRIM): Updated efficacy results from a randomised, double‐blind, phase 3 trial. Lancet Oncol 2016;17:1248–1260. [DOI] [PubMed] [Google Scholar]
- 40. Joshi M, Rice SJ, Liu X et al. Trametinib with or without vemurafenib in BRAF mutated non‐small cell lung cancer. PLoS One 2015;10:e0118210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gautschi O, Milia J, Cabarrou B et al. Targeted therapy for patients with BRAF‐mutant lung cancer: Results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–1457. [DOI] [PubMed] [Google Scholar]
- 42. Hyman DM, Puzanov I, Subbiah V et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Planchard D, Besse B, Groen HJ et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)‐mutant metastatic non‐small cell lung cancer: An open‐label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosell R, Carcereny E, Gervais R et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): A multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012;13:239–246. [DOI] [PubMed] [Google Scholar]
- 45. Maemondo M, Inoue A, Kobayashi K et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380–2388. [DOI] [PubMed] [Google Scholar]
- 46. Shaw AT, Kim DW, Nakagawa K et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2013;368:2385–2394. [DOI] [PubMed] [Google Scholar]
- 47. Lin L, Asthana S, Chan E et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci USA 2014;111:E748–E757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Okimoto RA, Lin L, Olivas V et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF‐mutant lung cancer. Proc Natl Acad Sci USA 2016;113:13456–13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Levy BP, Chioda MD, Herndon D et al. Molecular testing for treatment of metastatic non‐small cell lung cancer: How to implement evidence‐based recommendations. The Oncologist 2015;20:1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.NCCN clinical practice guidelines in oncology (NCCN Guidelines) . Non‐small cell lung cancer, version 5. 2017. Available at https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed April 13, 2017. [DOI] [PubMed] [Google Scholar]
- 51. Dietel M, Jöhrens K, Laffert MV et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: A review focussing on clinical relevance. Cancer Gene Ther 2015;22:417–430. [DOI] [PubMed] [Google Scholar]
- 52. Kurzrock R, Colevas AD, Olszanski A et al. NCCN Oncology Research Program's Investigator Steering Committee and NCCN Best Practices Committee molecular profiling surveys. J Natl Compr Canc Netw 2015;13:1337–1346. [DOI] [PubMed] [Google Scholar]
- 53. Ilie M, Hofman V, Long E et al. Current challenges for detection of circulating tumor cells and cell‐free circulating nucleic acids, and their characterization in non‐small cell lung carcinoma patients. What is the best blood substrate for personalized medicine? Ann Transl Med 2014;2:107. [DOI] [PMC free article] [PubMed]
- 54. Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene‐driven solid cancers. Nat Med 2016;22:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schreuer M, Meersseman G, Van Den Herrewegen S et al. Quantitative assessment of BRAF V600 mutant circulating cell‐free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J Transl Med 2016;14:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weber B, Meldgaard P, Hager H et al. Detection of EGFR mutations in plasma and biopsies from non‐small cell lung cancer patients by allele‐specific PCR assays. BMC Cancer 2014;14:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee J, Kim ST, Kim KM et al. Cell‐free DNA sequencing‐guided therapy in a prospective clinical trial: NEXT‐2 trial—A feasibility analysis. J Clin Oncol 2016;34:11534a. [Google Scholar]
- 58. Thompson JC, Yee SS, Troxel AB et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next‐generation sequencing of cell‐free circulating tumor DNA. Clin Cancer Res 2016;22:5772–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemurafenib therapy. N Engl J Med 2012;366:480–481. [DOI] [PubMed] [Google Scholar]
- 60. Lee CI, Menzies AM, Haydu LE et al. Features and management of pyrexia with combined dabrafenib and trametinib in metastatic melanoma. Melanoma Res 2014;24:468–474. [DOI] [PubMed] [Google Scholar]
- 61. Welsh SJ, Corrie PG. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther Adv Med Oncol 2015;7:122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Livingstone E, Zimmer L, Vaubel J et al. BRAF, MEK and KIT inhibitors for melanoma: Adverse events and their management. Chin Clin Oncol 2014;3:29. [DOI] [PubMed] [Google Scholar]
- 63. Das Thakur M, Salangsang F, Landman AS et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jain T, Bryce A. Intermittent BRAF inhibition can achieve prolonged disease control in BRAF mutant melanoma. Cureus 2015;7:e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Corcoran RB, Ebi H, Turke AB et al. EGFR‐mediated re‐activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Noeparast A, Teugels E, Giron P et al. Non‐V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of trametinib and dabrafenib. Oncotarget 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 67. Long GV, Stroyakovskiy D, Gogas H et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–1888. [DOI] [PubMed] [Google Scholar]
- 68. Robert C, Karaszewska B, Schachter J et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30–39. [DOI] [PubMed] [Google Scholar]
- 69. Klein O, Clements A, Menzies AM et al. BRAF inhibitor activity in V600R metastatic melanoma–response. Eur J Cancer 2013;49:1797–1798. [DOI] [PubMed] [Google Scholar]