

Comprehensive genomic profiling (CGP) using next‐generation sequencing was performed on a series of 41 clinical esthesioneuroblastoma samples. This study provides a view into the genomic landscape of esthesioneuroblastoma and insight into the biological mechanisms underlying this tumor type.
Keywords: Esthesioneuroblastoma, Comprehensive genomic profiling, Sunitinib, Everolimus, Pazopanib, Next‐generation sequencing
Abstract
Background.
Esthesioneuroblastoma (ENB), also known as olfactory neuroblastoma, is a rare malignant neoplasm of the olfactory mucosa. Despite surgical resection combined with radiotherapy and adjuvant chemotherapy, ENB often relapses with rapid progression. Current multimodality, nontargeted therapy for relapsed ENB is of limited clinical benefit.
Materials and Methods.
We queried whether comprehensive genomic profiling (CGP) of relapsed or refractory ENB can uncover genomic alterations (GA) that could identify potential targeted therapies for these patients. CGP was performed on formalin‐fixed, paraffin‐embedded sections from 41 consecutive clinical cases of ENBs using a hybrid‐capture, adaptor ligation based next‐generation sequencing assay to a mean coverage depth of 593X. The results were analyzed for base substitutions, insertions and deletions, select rearrangements, and copy number changes (amplifications and homozygous deletions).
Results.
Clinically relevant GA (CRGA) were defined as GA linked to drugs on the market or under evaluation in clinical trials. A total of 28 ENBs harbored GA, with a mean of 1.5 GA per sample. Approximately half of the ENBs (21, 51%) featured at least one CRGA, with an average of 1 CRGA per sample. The most commonly altered gene was TP53 (17%), with GA in PIK3CA, NF1, CDKN2A, and CDKN2C occurring in 7% of samples.
Conclusion.
We report comprehensive genomic profiles for 41 ENB tumors. CGP revealed potential new therapeutic targets, including targetable GA in the mTOR, CDK and growth factor signaling pathways, highlighting the clinical value of genomic profiling in ENB.
Implications for Practice.
Comprehensive genomic profiling of 41 relapsed or refractory ENBs reveals recurrent alterations or classes of mutation, including amplification of tyrosine kinases encoded on chromosome 5q and mutations affecting genes in the mTOR/PI3K pathway. Approximately half of the ENBs (21, 51%) featured at least one clinically relevant genomic alteration (CRGA), with an average of 1 CRGA per sample. The most commonly altered gene was TP53 (17%), and alterations in PIK3CA, NF1, CDKN2A, or CDKN2C were identified in 7% of samples. Responses to treatment with the kinase inhibitors sunitinib, everolimus, and pazopanib are presented in conjunction with tumor genomics.
Introduction
Esthesioneuroblastoma (ENB), also known as olfactory neuroblastoma, is a rare sinonasal neoplasm thought to arise from the olfactory neuroepithelium [1], [2], [3]. Although it shares common features with other small round blue cell tumors, it is a distinct entity from other tumors arising in the nasal cavity and skull base [1], [2], [3]. The rarity of ENB has limited the scope of clinical research studies, particularly those investigating the genetic and biochemical processes driving tumorigenesis. Several studies utilizing comparative genomic hybridization (CGH) have identified complex patterns of copy number abnormalities in ENB tumors, but few observations were consistent across studies [4], [5], [6], [7], [8], limiting generalizable conclusions and indicating that heterogeneous sets of alterations can drive ENB development. Even fewer studies have used DNA sequencing techniques to profile ENB [9], with three tumors in the literature evaluated by comprehensive sequencing methods [9], [10]. There are no mutations in ENB tumors reported in commonly used databases collating genomic cancer data, such as COSMIC, cBioPortal, and the ICGC portal.
This lack of genomic data, in turn, has in some ways limited the discovery of new therapeutic strategies. Current treatment strategies are based on surgical resection, in combination with radiotherapy and/or chemotherapy, as needed [2]. However, despite the successful combination of surgical resection combined with radiotherapy and adjuvant chemotherapy, ENBs can often relapse and pursue an aggressive clinical course [2], [3], [11], [12], [13]. Current multimodality nontargeted therapies for relapsed ENBs are of limited clinical benefit. The use of targeted systemic therapies is rare, but durable responses, predominantly stable disease, have been reported for sunitinib [14], sunitinib plus cetuximab [15], everolimus [16], imatinib [17], and bevacizumab [18].
In the present study, we performed comprehensive genomic profiling (CGP) using next‐generation sequencing on a series of 41 clinical ENB samples. This view into the genomic landscape of ENB provides additional insight into the biological mechanisms underlying this tumor type and may identify potential treatment targets. Our dataset comprises the largest collection of genomic profiles available to date and illustrates the heterogeneous nature of this malignancy. These findings warrant further evaluation of refractory or recurrent ENB by comprehensive profiling to identify common alterations and potentially define molecular subtypes of the disease.
Materials and Methods
A series of 41 clinical cases of ENBs were analyzed using CGP in a Clinical Laboratory Improvement Amendments‐certified, College of American Pathologists‐accredited laboratory (Foundation Medicine, Cambridge, MA). Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin stained slides, and all samples forwarded for DNA and/or RNA extraction contained a minimum of 20% tumor nuclear nuclei. Grade or stage was not available for the majority of samples.
Extensive technical descriptions and validation of the genomic profiling assays used to analyze these samples in the course of clinical care have been published previously (supplemental online Appendix 1) [19], [20]. In brief, ≥ 50 ng DNAs was extracted from 40 microns of tumor samples in formalin‐fixed, paraffin‐embedded tissue blocks. The samples were assayed by adaptor ligation hybrid capture, performed for all coding exons of 236 (v1), 315 (v2), or 405 (v3) cancer‐related genes plus select introns from 19 (v1), 28 (v2), or 31 (v3) genes frequently rearranged in cancer (supplemental online Tables 1–3) [19], [20]. For those samples for which RNA was available, targeted RNA‐seq was performed for rearrangement analysis in 265 genes [20]. RNA sequences were analyzed for the presence of rearrangements only. Sequencing of captured libraries was performed using an Illumina technology to a mean exon coverage depth of 593X, and resultant sequences were analyzed for base substitutions, insertions, deletions, copy number alterations (focal amplifications and homozygous deletions), and select gene fusions, as previously described [19], [20]. Clinically relevant genomic alterations (CRGA) were defined as alterations that are targetable by anticancer drugs currently available on the market or in registered clinical trials. Germline variants documented in the dbSNP database (dbSNP142; http://www.ncbi.nlm.nih.gov/SNP/), with two or more counts in the ExAC database (http://exac.broadinstitute.org/), or recurrent variants of unknown significance that were predicted by an internally developed algorithm to be germline were removed, with the exception of known driver germline events (e.g., documented hereditary BRCA1/2 and deleterious TP53 mutations). Known confirmed somatic alterations deposited in the Catalog of Somatic Mutations in Cancer were highlighted as biologically significant [21]. All inactivating events (i.e., truncations and deletions) in known tumor suppressor genes were also called as significant. To maximize mutation‐detection accuracy (sensitivity and specificity) in impure clinical specimens, the test was previously optimized and validated to detect base substitutions at a ≥5% mutant allele frequency (MAF), indels with a ≥10% MAF with ≥99% accuracy, and fusions occurring within baited introns/exons with >99% sensitivity [19].
Results
The 28 male (68%) and 13 female (32%) patients with ENB had an average age of 50.9 years (range 15–83 years). All tumors were stages III and IV at the time of CGP. The sequenced samples were most often obtained from the primary or recurrent ENB in the sinonasal cavity (11; 27%). Other sites included the brain (9, 22%); lymph nodes (6, 15%); bone (4, 10%); soft tissue (4, 10%); neck (3, 7%); and breast, liver, masticator space, and parotid gland (1 case each, 2.4%) (Table 1). A total of 28 ENBs harbored GA, with a mean of 1.5 GA per sample (supplemental online Table 4). Slightly more than half of the ENBs (21, 51%) featured at least one CRGA, with an average of 1 CRGA per sample. The most commonly altered gene was TP53 (17%), with GA in PIK3CA, NF1, CDKN2A, or CDKN2C each occurring in 7% of samples (Fig. 1). Potentially targetable GA were identified in several samples, including genes comprising the PI3K/mTOR pathway (PIK3CA, NF1, PTEN, PIK3R2, RICTOR, and TSC1) (11 samples; 27%) and the CDK cell‐cycle regulatory pathway (CDKN2A, CDKN2B, CDKN1B, CDKN2C, CDK6) (6 samples; 15%) (Table 2, Table 3). Targeted therapies associated with mutations in these and other genes are shown in Table 4.
Table 1. Characteristics of genomically profiled esthesioneuroblastoma.

Abbreviations: GA, genomic alterations.
Figure 1.

Alteration frequencies in genes and pathways mutated in esthesioneuroblastoma. (A): Mutation frequency in 41 esthesioneuroblastoma by gene. (B): Mutation frequency in 41 esthesioneuroblastoma by pathway.
Table 2. Genomics and clinical characteristics for profiled esthesioneuroblastomas (n = 41).

Abbreviations: F, female; M, male; MAF, minor allele frequency.
Table 3. Treatment and clinical outcomes for 6 genomically profiled esthesioneuroblastoma.
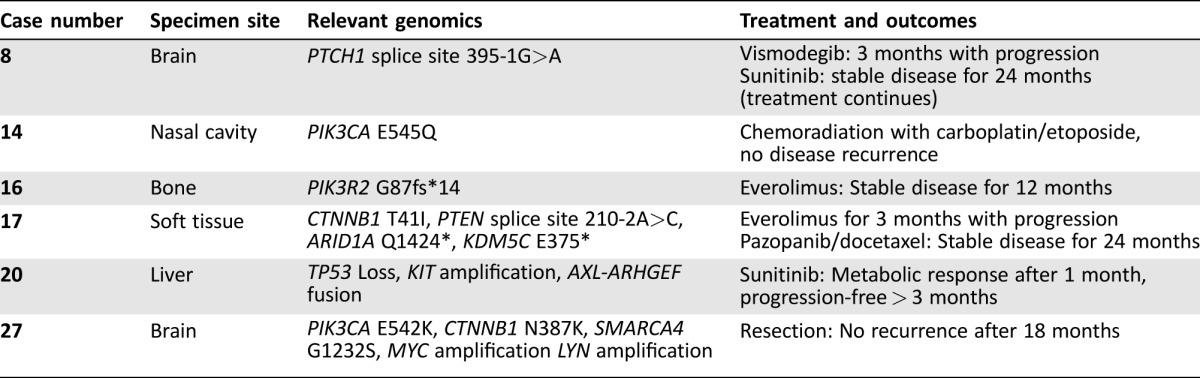
Table 4. Therapies associated with genes altered in esthesioneuroblastoma.

In addition to substitutions, small indels, focal CNAs and rearrangements, nonfocal changes in copy number could be observed in several cases. In particular, gains in chromosome 5 were observed in 3 cases (cases 5, 11, and 35), showing amplification of RICTOR (5p13.1) and FGF10 (5p13‐p12) in all 3 samples, and amplification of FLT4 (5q35.3), PDGFRB (5q33.1), and FGFR4 (5q35.2) in samples 5 and 35 (Table 2). An additional case harbored large scale amplification of chromosome 20, with increased copy numbers for ZNF217 (20q13), AURKA (20q13), ARFRP1 (20q13.33), SRC (20q12‐q13), and TOP1 (20q12–20q13.1).
Clinical history and treatment details were available for six patients whose tumors were sequenced in this series (Table 3). Case 8 is a 49‐year‐old man presenting with recurring esthesioneuroblastoma. Comprehensive genomic profiling showed an acceptor splice site mutation adjacent to exon 3 (c.395‐1G>A) that is computationally predicted to disrupt expression of PTCH1, an upstream regulator of SHH signaling. Although this mutation has not been experimentally characterized, it is annotated in the ClinVar database as predicted deleterious (RCV000149897.1). In the context of basal cell carcinoma, PTCH1 mutations predict response to treatment with vismodegib, which targets the protein smoothened that signals downstream of PTCH1. The patient received vismodegib for 3 months and experienced disease progression. Therapy was then switched to sunitinib based on a published case study [14] of successful disease control, and the patient currently continues on treatment with stable disease for 24 months.
Case 14 is a 70‐year‐old woman with a cranially invasive ENB that responded well to treatment with radiation combined with carboplatin and etoposide. There was no disease recurrence after 17 months. Comprehensive genomic profiling revealed a PIK3CA E545Q alteration that could predict sensitivity to mTOR inhibitors or PI3K inhibitors currently under investigation in clinical trials, if further treatment becomes warranted.
Case 16 is a 40‐year‐old man with recurrent ENB previously treated with radiation therapy and chemotherapy. Comprehensive genomic profiling revealed a PIK3R2 frameshift mutation (G87fs*14) predicted to disrupt the tumor suppressive function of the encoded protein, p85‐beta. The patient has received treatment with everolimus and has experienced ongoing stable disease for 12 months.
Case 17 is a 49‐year‐old woman with a history of ENB of paranasal sinus. She was initially diagnosed in 2006, then underwent complete surgical resection followed by radiation. She was disease free from 2006 to 2012. In 2012, she recurred in lungs and bones (biopsy proven ENB). Comprehensive genomic profiling of the metastasis revealed alterations affecting CTNNB1 (T41I), PTEN (splice site 210‐2A>C), ARID1A (Q1424*), and KDM5C (E375*). Since 2012, she has undergone several lines of treatment, including everolimus for 3 months with disease progression. She maintained a stable disease on pazopanib and docetaxel from April 2014 to April 2016. She progressed in April 2016 and is currently undergoing evaluations for clinical trials.
Case 20 is a 72‐year‐old man who initially presented with sinus symptoms for several months. A left neck mass developed and he underwent an magnetic resonance imaging (MRI) of the brain and head that revealed a large mass involving the nasopharyngeal mucosa space greater on the left than the right and left neck adenopathy at the level 2B. Biopsy revealed a grade 4 ENB. He underwent an anterior craniofacial resection with left neck dissections level 1B through 5. There were lymph nodes involved at level 2B, with the largest focus being 3.5 cm in maximum dimension with extension to perineural adipose tissues. A total of 3 of 9 nodes were involved. The final pathologic stage was a Kadish stage D. He was treated with adjuvant external beam radiation therapy to the nasopharynx/neck with a total of 5580 cGy. Adjuvant chemotherapy with carboplatin AUC 5 on day 1 and etoposide 100 mg/m2 on days 1–3 of a 28‐day cycle for a total of 4 cycles were administered. Unfortunately, the patient recurred in the liver 8 months later. Initial treatment consisted of radiofrequency ablation (RFA). A biopsy was obtained and sent for CGP. Testing revealed a KIT amplification, an AXL‐ARHGEF fusion, and TP53 loss (exons 1–8). The patient continued with RFA treatment intermittently over the course of a year. When local therapy was no longer an option, he commenced sunitinib at a dose of 37.5 mg continuous daily dosing aimed at the cKIT amplification and AXL‐ARHGEF fusion. The patient had positron emission tomography (PET) imaging performed after 1 month of therapy and had a metabolic response with >50% reduction in metabolic activity in the metastatic lesions (Fig. 2). The lesions were stable based on RECIST 1.1 criteria. He continued on active therapy, 3 months after initiation, but had required a sunitinib dose‐reduction to 25 mg daily due to grade 3 stomatitis.
Figure 2.

A metabolic response to treatment with sunitinib after 1 month (case 20). (A): Maximum intensity projection from positron emission tomography (PET) imaging. (B): Cross‐sectional PET images showing decreased metabolic activity between March 2016 (left) and April 2016 (right) following treatment with sunitinib.
Case 27 is a 41‐year‐old man who presented with cognitive and personality changes. A brain MRI revealed a large T2 hypointense heterogeneously enhancing basal mass with peritumoral cysts along the superolateral aspect of the mass, involving bilateral ethmoid and right sphenoid sinuses with bifrontal involvement and significant edema, and endoscopic biopsy revealed a high‐grade malignant neoplasm with neuroendocrine differentiation most consistent with ENB. The patient underwent a gross total resection 1 month later, and pathology revealed a malignant neoplasm most consistent with poorly differentiated ENB. Comprehensive genomic profiling revealed a common oncogenic mutation in PIK3CA (E542K), additional missense mutations in CTNNB1 (N387K) and SMARCA4 (G1232S), focal amplification of MYC, and a nonfocal amplification of a region containing LYN. Body and brain fluorodeoxyglucose‐PET revealed no residual or metastatic disease following resection, and the most recent MRI and PET scans remain negative for residual or recurrent disease as of 22 months from the initial diagnosis. He has not required any antineoplastic therapy.
Discussion
Esthesioneuroblastomas are a rare entity, which has limited the genomic information available to date. In this series of 41 refractory or recurrent ENBs, 68% of samples harbored at least one somatic mutation and 51% of tumors harbored potentially targetable GA, with 27% of tumors harboring alterations that may predict sensitivity to inhibitors of the PI3K/mTOR pathway (Fig. 1). The remaining 24% of samples harbored targetable mutations in a wide variety of genes associated with responses to targeted therapies.
Data from several CGH studies have shown the changes that include gains at 7q, 9p, 13q, 17q, 17p13, 20p/q, 22q, and Xp/q, and losses or deletions at 1p, 2q, 3p/q, 6q, 9p, 10p/q, 22q, and Xp/q, with high‐grade ENBs demonstrating more alterations than low‐grade tumors [4], [5], [6], [7], [9], [22]. None of these changes have been routinely observed, limiting their prognostic or diagnostic value. Although the current assay is optimized to identify short variants, focal copy number alterations, and select rearrangements, it is possible to observe larger nonfocal copy number changes in some instances. In the current study, 2 of 41 tumors showed nonfocal amplification of chromosome 5q in a region harboring the genes FLT4 and PDGFRB. Other tumors also harbored nonfocal amplifications, affecting the regions 20q12‐q13, 18q11.1‐q11.2, and 8q13; however, none of these were recurrent amplifications. Trisomy 8 has been reported previously in ENB [4], [23], but cannot be definitively detected with the current assay.
Currently, targeted therapy is not widely used to treat ENB due to a lack of available efficacy data, although case studies and small trials have reported disease stabilization in response to treatment with sunitinib [14], [15], everolimus [16], and imatinib [17], among others. In addition to the PI3K/mTOR pathway, our study identified alterations that may predict sensitivity to MEK inhibitors, such as cobimetinib and trametinib; CDK4/6 inhibitors, such as palbociclib; or DNA methyltransferase inhibitors, such as decitabine. We report here one case of stable disease in response to treatment with everolimus (case 16) for a tumor with a PIK3R2 mutation, a mutation predicted to activate the PI3K/mTOR pathway; two responses to treatment with sunitinib (cases 8 and 20); and stable disease in response to treatment with pazopanib and docetaxel (case 17). These cases, in addition to other cases in the literature [9], support further examination into the use of tyrosine kinase inhibitors such as sunitinib, imatinib, and pazopanib that target genomic alterations observed multiple times in this cohort, such as amplification of PDGFRB, FLT4, SRC, or KIT.
Conclusion
This series of 41 ENBs represents the largest collection of ENBs with comprehensive genomic profiles presented to date. Compared with the large scale chromosomal changes previously observed by CGH, this cohort of ENBs harbor relatively few focal CNAs or rearrangements. Nevertheless, nearly half of the tumors profiled harbored CRGA associated with targeted therapies, including kinases known to be targeted by small molecule inhibitors such as sunitinib. Our results suggest possible novel treatment strategies for recurrent or unresectable ENBs based on results of genomic profiling.
See http://www.TheOncologist.com for supplemental material available online.
Author Contributions
Conception/design: Laurie Gay, Sungeun Kim
Provision of study material or patients: Madappa Kundranda, Yazmin Odia, Chaitali Nangia, James Battiste, Gerardo Colon‐Otero, Steven Powell, Jeffery Russell
Collection and/or assembly of data: Laurie Gay, Kyle Fedorchak, Yazmin Odia, Chaitali Nangia, James Battiste, Gerardo Colon‐Otero, Steven Powell, Jeffery Russell, Jo‐Anne Vergilio, James H Suh, Jeffrey S Ross
Data analysis and interpretation: Laurie Gay, Kyle Fedorchak, Jeffery Russell, Julia Elvin
Manuscript writing: Laurie Gay, Jeffery Russell
Final approval of manuscript: Laurie Gay, Sungeun Kim, Madappa Kundranda, Yazmin Odia, Chaitali Nangia, James Battiste, Gerardo Colon‐Otero, Steven Powell, Jeffery Russell, Julia Elvin, Jo‐Anne Vergilio, James H Suh, Siraj M Ali, Philip J Stephens, Vincent A Miller, Jeffrey S Ross
Disclosures
Laurie M. Gay: Foundation Medicine, Inc. (E, OI); Madappa Kundranda: Bayer, Celgene (C/A, H); James Battiste: Novocure (H); Gerardo Colon-Otero: Novartis (RF); Steven Powell: Merck (RF); Jeffery Russell: EMD Serono (C/A); Julia A. Elvin: Foundation Medicine, Inc. (E, OI); Jo-Anne Vergilio: Foundation Medicine, Inc. (E, OI); James Suh: Foundation Medicine, Inc. (E, OI); Siraj M. Ali: Foundation Medicine, Inc. (E, OI); Philip J. Stephens: Foundation Medicine, Inc. (E, OI); Vincent A. Miller: Foundation Medicine, Inc. (E, OI); Jeffrey S. Ross: Foundation Medicine, Inc. (E, OI, RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supplementary Information
References
- 1. Barnes L, Weltgesundheitsorganisation, International Agency for Research on Cancer, eds. Pathology and genetics of head and neck tumours: [… reflects the views of a working group that convened for an Editorial and Consensus Conference in Lyon, France, July 16 ‐ 19, 2003], Reprinted. Lyon: IARC Press, 2007:1–430.
- 2. Schwartz JS, Palmer JN, Adappa ND. Contemporary management of esthesioneuroblastoma. Curr Opin Otolaryngol Head Neck Surg 2016;24:63–69. [DOI] [PubMed] [Google Scholar]
- 3. Faragalla H, Weinreb I. Olfactory neuroblastoma: A review and update. Adv Anat Pathol 2009;16:322–331. [DOI] [PubMed] [Google Scholar]
- 4. Holland H, Koschny R, Krupp W et al. Comprehensive cytogenetic characterization of an esthesioneuroblastoma. Cancer Genet Cytogenet 2007;173:89–96. [DOI] [PubMed] [Google Scholar]
- 5. Riazimand SH, Brieger J, Jacob R et al. Analysis of cytogenetic aberrations in esthesioneuroblastomas by comparative genomic hybridization. Cancer Genet Cytogenet 2002;136:53–57. [DOI] [PubMed] [Google Scholar]
- 6. Szymas J, Wolf G, Kowalczyk D et al. Olfactory neuroblastoma: Detection of genomic imbalances by comparative genomic hybridization. Acta Neurochir (Wien) 1997;139:839–844. [DOI] [PubMed] [Google Scholar]
- 7. Bockmuhl U, You X, Pacyna‐Gengelbach M et al. CGH pattern of esthesioneuroblastoma and their metastases. Brain Pathol 2004;14:158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guled M, Myllykangas S, Frierson HFJ et al. Array comparative genomic hybridization analysis of olfactory neuroblastoma. Mod Pathol 2008;21:770–778. [DOI] [PubMed] [Google Scholar]
- 9. Czapiewski P, Kunc M, Haybaeck J. Genetic and molecular alterations in olfactory neuroblastoma: Implications for pathogenesis, prognosis and treatment. Oncotarget 2016;7:52584–52596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiss GJ, Liang WS, Izatt T et al. Paired tumor and normal whole genome sequencing of metastatic olfactory neuroblastoma. PLoS One 2012;7:e37029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tajudeen BA, Arshi A, Suh JD et al. Esthesioneuroblastoma: An update on the UCLA experience, 2002‐2013. J Neurol Surg B Skull Base 2015;76:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bossi P, Saba NF, Vermorken JB et al. The role of systemic therapy in the management of sinonasal cancer: A critical review. Cancer Treatment Rev 2015;41:836–843. [DOI] [PubMed] [Google Scholar]
- 13. Kumar R. Esthesioneuroblastoma: Multimodal management and review of literature. World J Clin Cases 2015;3:774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Preusser M, Hutterer M, Sohm M et al. Disease stabilization of progressive olfactory neuroblastoma (esthesioneuroblastoma) under treatment with sunitinib mesylate. J Neurooncol 2010;97:305–308. [DOI] [PubMed] [Google Scholar]
- 15. Wang L, Ding Y, Wei L et al. Recurrent olfactory neuroblastoma treated with cetuximab and sunitinib: A case report. Medicine (Baltimore) 2016;95:e3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fury MG, Sherman E, Haque S et al. A phase I study of daily everolimus plus low‐dose weekly cisplatin for patients with advanced solid tumors. Cancer Chemother Pharmacol 2012;69:591–598. [DOI] [PubMed] [Google Scholar]
- 17. Kim S, Atta JR, Bergmann L et al. Imatinib mesylate as second‐line treatment in a c‐kit‐positive esthesioneuroblastoma. ASCO Meeting Abstracts 2011;29:e12513. [Google Scholar]
- 18. Dunbar EM, Pumphrey PK, Bidari S. Unexpectedly durable palliation of metastatic olfactory neuroblastoma using anti‐angiogenic therapy with Bevacizumab. Rare Tumors 2012;4:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He J, Abdel‐Wahab O, Nahas MK et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016;127:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forbes SA, Bindal N, Bamford S et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gil Z, Fliss DM. Cytogenetic analysis of skull base tumors: Where do we stand? Curr Opin Otolaryngol Head Neck Surg 2012;20:130–136. [DOI] [PubMed] [Google Scholar]
- 23. VanDevanter DR, George D, McNutt MA,et al. Trisomy 8 in primary esthesioneuroblastoma. Cancer Genet Cytogenet 1991;57:133–136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.