

Abstract
With the Breakthrough Therapy Designation program adding to the tools that the U.S. Food and Drug Administration (FDA) has for expediting drug development, the FDA reassessed the endpoints needed for approval of transformative therapies. Although the demonstration of an improvement in overall survival remains the gold standard for drug approval, innovation in cancer research has led to use of other endpoints in regulatory decision‐making. These endpoints include substantially delaying tumor progression or extending progression‐free survival, substantially reducing tumor size for a prolonged time, improving objective response rate and duration of response, or improving cancer‐related symptoms and patient function.
In the past few decades, with improved understanding of the genomic and immunologic underpinnings of cancer, better molecular characterization of tumors, and more precisely targeted agents, new and innovative therapeutics have altered the natural histories of certain cancer types such as chronic myeloid leukemia (CML), multiple myeloma, and melanoma. Recognizing a need to further expedite development of drugs that show promising early clinical evidence of benefit over available therapy, the U.S. Congress, in 2012, established the Breakthrough Therapy Designation program [1]. The U.S. Food and Drug Administration (FDA) uses this program frequently for transformative therapies that show great promise in early clinical trials [2]. Table 1 provides examples of new FDA‐approved therapies that were developed based on the improved understanding of tumor biology and that markedly changed the therapeutic landscape of certain cancer types. Figure 1 highlights certain cancer types where 5‐year survival has improved in recent decades, which is at least partially attributed to better therapeutic options.
Table 1. Examples of cancers where the therapeutic landscape has substantially changed.
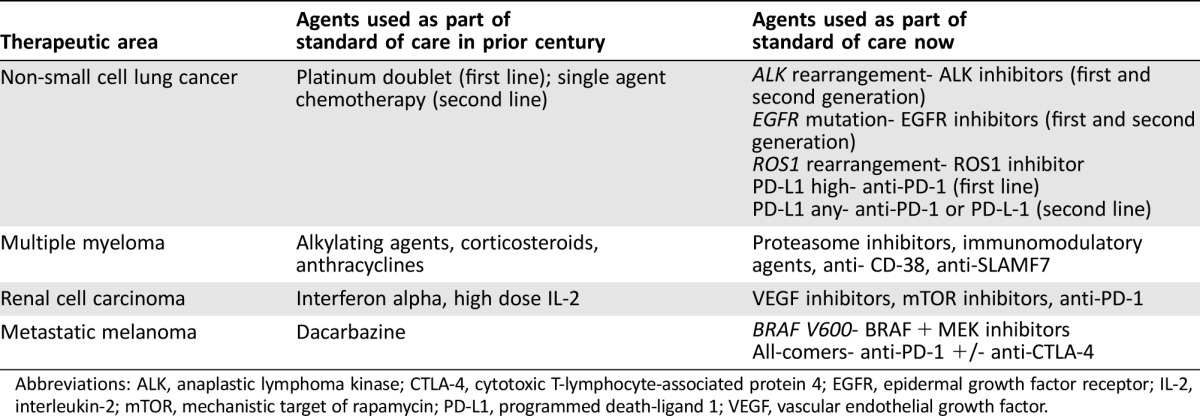
Abbreviations: ALK, anaplastic lymphoma kinase; CTLA‐4, cytotoxic T‐lymphocyte‐associated protein 4; EGFR, epidermal growth factor receptor; IL‐2, interleukin‐2; mTOR, mechanistic target of rapamycin; PD‐L1, programmed death‐ligand 1; VEGF, vascular endothelial growth factor.
Figure 1.

Five‐year relative survival by year of diagnosis in selected malignancies [33]. Includes all ages, all races, both sexes, 1975–2012. Cancer sites include invasive cases only unless otherwise noted. The 5‐year survival estimates are calculated using monthly intervals.
With the Breakthrough Therapy Designation program adding to the tools that the FDA has for expediting drug development, the FDA reassessed the endpoints needed for approval of transformative therapies. Although the demonstration of an improvement in overall survival remains the gold standard for drug approval, innovation in cancer research has led to use of other endpoints in regulatory decision‐making. These endpoints include substantially delaying tumor progression or extending progression‐free survival, substantially reducing tumor size for a prolonged time, improving objective response rate and duration of response, or improving cancer‐related symptoms and patient function.
Oncology Endpoints: A Historical Perspective
In the 1970s, the FDA usually approved drugs on the basis of objective response rate [3]. Combinations of cytotoxic chemotherapy produced durable and deep responses and cures for patients with malignancies such as metastatic testicular cancer, Hodgkin lymphoma, and acute lymphoblastic leukemia [4], [5], [6]. In other tumor types, the benefits of cytotoxic chemotherapy were incremental, and in certain settings the risks of these agents outweighed the benefits. In the 1980s, after discussions with the FDA Oncologic Drugs Advisory Committee (ODAC), the FDA determined that drug approval should be primarily based on improvements in overall survival. This was, in part, due to the benefit‐risk evaluation of many cytotoxic agents with modest efficacy and marked toxicity.
With the introduction into clinical practice of targeted therapies and, more recently, immunotherapies, the benefit‐risk evaluation of novel agents required a reassessment. Imatinib was approved for CML based on cytogenetic response rate, with subsequent trials showing a clear advantage over interferon and cytarabine, not only with respect to efficacy, but also safety [7], [8]. Due to high rates of cross‐over of patients initially allocated to interferon and cytarabine to imatinib, and the long survival of patients on both arms treated with the abl inhibitor, more than 10 years of follow‐up was required to provide a nominally (but not statistically) significant improvement in overall survival [9]. In non‐small cell lung cancer (NSCLC), crizotinib was approved for patients whose tumors harbor ALK rearrangements based on unprecedented response rates demonstrated in single‐arm trials, with a more favorable tolerability profile compared with platinum‐based chemotherapy in subsequent randomized controlled trials [10], [11].
Over the past decade, through numerous discussions about endpoints in several tumor types with external stakeholders, including oncology professional societies, patient advocacy groups, and ODAC, the FDA received advice to re‐evaluate approval endpoints. Endpoints including objective response rate of sufficient duration and progression‐free survival can be clinically relevant and meaningful to patients and treating oncologists, and may be acceptable for either accelerated or regular approval, depending on the circumstance [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25].
The FDA has acknowledged that substantially delaying tumor progression or significantly reducing tumor size can be beneficial to patients if the overall benefit‐risk profile is favorable [26]. Through numerous interactions with drug sponsors throughout the drug development process, the FDA has worked with sponsors to tailor drug development programs to the unique context of the patient population and disease under study. For breakthrough therapies, the interactions between the FDA and sponsors are even more extensive. This has led to approvals of drugs that have provided meaningful treatments to patients who previously had few therapeutic options, including patients with renal cell carcinoma, Merkel cell carcinoma, medullary thyroid cancer (MTC), gastrointestinal stromal tumor (GIST), metastatic basal cell carcinoma, pancreatic neuroendocrine tumor, multiple myeloma, CML, chronic lymphocytic leukemia (CLL), and EGFR‐ and ALK‐driven NSCLC, to name a few. Table 2 lists examples of therapies that were approved on endpoints other than overall survival in their registration trials, but provided substantial improvements to patients and are widely thought to be transformative. Table 3 highlights examples of recent approvals for disease areas that previously did not have any approved therapies available.
Table 2. Examples of novel drugs approved on non‐survival endpoints that have likely impacted the natural history of selected cancers.

Abbreviations: ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; mNSCLC, metastatic non‐small cell lung cancer; FDA, U.S. Food and Drug Administration.
Table 3. Examples of recent approvals where there were previously no satisfactory approved drugs.

Abbreviations: FDA, U.S. Food and Drug Administration.
Overall Survival Endpoint: Limitations
An improvement in overall survival may be impractical or unreasonable to demonstrate in randomized controlled trials in selected disease areas. Unlike other therapeutic areas where placebo‐controlled trials can provide a comparator for the demonstration of overall survival, the use of placebo‐controlled trials is limited in life‐threatening diseases, especially for novel drugs demonstrating improved biologic activity in early drug development. For drugs demonstrating unprecedented activity in early clinical development in cancers with few effective options, the ability to randomly allocate patients to either an agent with markedly improved durable response rates or to a toxic and marginally effective comparator may not be feasible because equipoise may not exist [27], [28]. If a randomized trial is conducted, many investigators and patients request a cross‐over to the investigational arm. Cross‐over may confound the demonstration of improvement in overall survival and require that the trial have an alternate endpoint. As was observed in trials randomizing patients with EGFR‐mutant or ALK rearranged advanced NSCLC to either targeted therapy or chemotherapy, high cross‐over to the experimental arm confounded the interpretation of overall survival [29], [30], [31], [32]. The use of alternative endpoints, such as progression‐free survival, provides a clinically relevant endpoint and allows expeditious access of important drugs to patients.
A demonstration of improvement in overall survival may not be practical in cancers with long natural histories. In part due to improved systemic treatments, patients with advanced cancers such as CLL, MTC, multiple myeloma, and CML now have 5‐year survivals upwards of 50% [33]. In these diseases, the time required and large patient numbers needed to power a trial to detect a survival improvement would not be practical and could deny effective treatment to patients, which would also negatively impact innovation.
As cancers are re‐classified by their underlying genomic properties, subsets of even common cancers such as NSCLC are classified as orphan diseases. For example, ROS1 rearrangements occur in approximately 1% of patients with advanced NSCLC, approximately 2,000 U.S. patients yearly. Even if ROS1 screening occurred in all patients, a randomized clinical trial powered to detect a survival difference may be challenging to conduct and complete. These three features—high activity in early clinical development compromising the equipoise of a randomized controlled trial, the long natural history of certain cancers, and the rarity of a cancer due to enhanced re‐classification based on genomic factors—may limit the use of overall survival as a registration endpoint and requires a re‐evaluation of other endpoints.
Health Related Quality of Life Metrics: Limitations
In addition to overall survival, many trials assess patient‐reported outcome (PRO) measurements of a patient's health‐related quality of life (HRQL), physical functioning, or tumor‐related symptoms. Health‐related quality of life is important to patients, but is challenging to assess, and has components (e.g., social and emotional well‐being) that can be affected by life circumstances outside of a drug's effect. These concerns, coupled with methodological challenges, open‐label and single arm trial designs, and a lack of standard analytic methods, have limited the use of HRQL as a statistically tested efficacy endpoint in cancer clinical trials.
Because HRQL is a broad and multi‐dimensional endpoint, it may be less responsive to a drug's effect. Concentrating PRO analyses on key disease symptoms, treatment side effects, and physical function may provide more sensitive measurement of how a patient experiences a cancer therapy. While rarely used as a primary efficacy endpoint, measurement of patient‐reported symptoms and function can complement radiographic, molecular, and survival endpoints. For example, the approval of ruxolitinib for myelofibrosis was based on reduction in spleen size and improvement of myelofibrosis symptoms using a PRO instrument, the modified myelofibrosis symptom assessment form version 2.0 [34]. Exploration of the tolerability of a cancer therapy through longitudinal PRO assessment of symptomatic side effects and their impact on the patient is also of interest. New technologies such as wearable devices may also provide important data on a patient's function in daily life. The FDA continues to actively investigate novel and existing PRO tools, methodologies, and other clinical outcome assessments to rigorously assess the patient experience, and consider these outcomes in benefit‐risk assessments [35].
Sequencing Therapies to Transform the Natural History of a Malignancy
Drugs approved on endpoints other than overall survival have provided clinically meaningful treatments to many patients. In CML, approval of the abl tyrosine kinase inhibitors based on biomarker changes, such as major molecular response, has transformed this once‐fatal disease to a chronic disease, with patients now having a life expectancy approaching that of the general population [36]. In multiple myeloma, approval of drugs such as proteasome inhibitors, immunomodulatory drugs, anti‐CD38 antibodies, and anti‐SLAMF7 antibodies based on endpoints of progression‐free survival and objective response rate has altered the disease's natural history and markedly improved 10‐year survival [37], [38]. In advanced colorectal cancer, sequential therapeutic gains with fluoropyrimidines, irinotecan‐ and oxaliplatin‐based regimens, along with monoclonal antibodies targeting vascular endothelial growth factor (VEGF) and EGFR, have contributed to a doubling of expected survival exceeding 2 years [39], [40].
Approval of RET inhibitors for MTC, kit inhibitors for GIST, hedgehog inhibitors for advanced basal cell carcinoma, anti‐interleukin‐6 for multicentric Castleman's disease, and anti‐programmed death‐ligand 1 for Merkel cell carcinoma have provided therapeutic options in diseases where there were no approved drugs. These drugs were approved on either progression‐free survival or objective response rate because of the limitations of using overall survival as described above.
Evolution of the Benefit‐Risk Assessment with Improved Scientific Understanding of Cancer
Although the vast majority of agents that are approved under expedited programs such as accelerated approval subsequently confirm benefit and receive regular approval, there are infrequent examples of drugs granted accelerated approval on the basis of non‐survival endpoints such as objective response rate or progression‐free survival that required withdrawal (either voluntary or involuntary) from the market [41], [42], [43]. These include bevacizumab for metastatic breast cancer, gefitinib for “all‐comer” (not selected by EGFR mutation status) refractory NSCLC, and gemtuzumab ozogamicin for acute myeloid leukemia. There are also infrequent cases of drugs granted regular approval based on an overall survival endpoint that require modification of the indicated population as understanding of science evolves.
The development of EGFR tyrosine kinase inhibitors gefitinib and erlotinib for refractory NSCLC is illustrative. In 2003, gefitinib was granted accelerated approval in an “all‐comer” patient population based on objective response rate, but was withdrawn from the market after several post‐marketing studies failed to confirm clinical benefit. In 2004, erlotinib was granted regular approval based on an incremental improvement in overall survival over placebo in an “all‐comer” refractory patient population. Improved knowledge of the underlying biology of cancer led to an understanding that the patients who benefit from EGFR tyrosine kinase inhibitors were those whose tumors harbored sensitizing kinase domain mutations in the EGFR gene. This led to the approval of erlotinib in 2013 and gefitinib in 2015 for patients whose NSCLC tumors harbor EGFR exon 19 deletion or exon 21 L858R substitution mutations based on endpoints of objective response rate and progression‐free survival [44], [45]. In 2016, the erlotinib indication was modified to remove the indication for patients with EGFR "wild type" NSCLC based on a study that did not demonstrate erlotinib's benefit over placebo in patients whose tumors lacked sensitizing EGFR mutations [46].
Real World Evidence: A Continuum of Drug Development
Given that more agents will likely be approved earlier in the cycle of evidence generation (such as in expansion cohorts of phase I trials) “real world evidence” may provide a better understanding of chronic safety and long‐term efficacy of oncology drugs used in the clinic [47], [48]. This evidence can be generated from multiple sources, including electronic health records, patient registries, mobile health applications, and social media [49]. Real world evidence may enhance the generalizability of clinical trial results to patient populations not studied in registration trials, and should improve the understanding of dosing and safety issues encountered in clinical practice. Real world evidence could be generated through pragmatic randomized clinical trials conducted at the point of care in community settings where traditional clinical trials are infrequently conducted. As the methodologies develop, continued discussion will be needed regarding optimal endpoints to assess efficacy in these pragmatic trials, such as assessment of mortality, real world response and progression, time to treatment failure, or time to switch in therapy. Ultimately, the success of new oncology therapies in the 21st century will not be judged solely on their performance in pre‐marketing studies, but, more importantly, on how they change outcomes for patients actually treated in clinical practice—the definitive metric of their contribution to reducing the burden of cancer for patients and for society.
Footnotes
Editor's Note: See the related editorial by Bruce A. Chabner on pages 757–758 of this issue.
Disclosures
The authors indicated no financial relationships.
References
- 1. Sherman RE, Li J, Shapley S et al. Expediting drug development—The FDA's new “breakthrough therapy” designation. N Engl J Med 2013;369:1877–1880. [DOI] [PubMed] [Google Scholar]
- 2. Shea M, Ostermann L, Hohman R et al. Regulatory Watch: Impact of breakthrough therapy designation on cancer drug development. Nat Rev Drug Discov 2016;15:152. [DOI] [PubMed] [Google Scholar]
- 3. Pazdur R. Endpoints for assessing drug activity in clinical trials. The Oncologist 2008;13:19–21. [DOI] [PubMed] [Google Scholar]
- 4. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med 2015;373:1541–1552. [DOI] [PubMed] [Google Scholar]
- 5. Einhorn LH, Donohue J. Cis‐diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann Intern Med 1977;87:293–298. [DOI] [PubMed] [Google Scholar]
- 6. Hanna NH, Einhorn LH. Testicular cancer—Discoveries and updates. N Engl J Med 2014;371:2005–2016. [DOI] [PubMed] [Google Scholar]
- 7. Cohen MH, Moses ML, Pazdur R. Gleevac for the treatment of chronic myelogenous leukemia: U.S. Food and Drug Administration regulatory mechanisms, accelerated approval, and orphan drug status. The Oncologist 2002;7:390–392. [DOI] [PubMed] [Google Scholar]
- 8. O'Brien SG, Guilhot F, Larson RA et al. Imatinib compared with interferon and low‐dose cytarabine for newly diagnosed chronic‐phase chronic myeloid leukemia. N Engl J Med 2003;348:994–1004. [DOI] [PubMed] [Google Scholar]
- 9. Hochhaus A, Larson RA, Guilhot F et al. Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med 2017;376:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malik SM, Maher VE, Bijwaard KE et al. U.S. Food and Drug Administration approval: Crizotinib for treatment of advanced or metastatic non‐small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res 2014;20:2029–2034. [DOI] [PubMed] [Google Scholar]
- 11. Kazandjian D, Blumenthal GM, Chen HY et al. FDA approval summary: Crizotinib for the treatment of metastatic non‐small cell lung cancer with anaplastic lymphoma kinase rearrangements. The Oncologist 2014;19:e5–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.U.S. Food and Drug Administration. FDA Briefing Document, Oncologic Drugs Advisory Committee Meeting April 12, 2011, NDA 22334 Afinitor® (everolimus), Applicant: Novartis Pharmaceuticals. Available at https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/oncologicdrugsadvisorycommittee/ucm250378.pdf. Accessed March 30, 2017.
- 13.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee Meeting, Morning Session, March 20, 2012. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM304478.pdf. Accessed March 30, 2017.
- 14.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee Meeting, Morning Session, December 7, 2011. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM288716.pdf. Accessed March 30, 2017.
- 15.U.S. Food and Drug Administration. Meeting of the Oncologic Drugs Advisory Committee, Morning Session, July 14, 2011. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM268656.pdf. Accessed March 30, 2017.
- 16.U.S. Food and Drug Administration. Meeting of the Oncologic Drugs Advisory Committee, Afternoon Session, July 14, 2011. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM268657.pdf. Accessed March 30, 2017.
- 17.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee, April 12, 2011, Morning Session. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM256370.pdf. Accessed March 30, 2017.
- 18.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee, April 12, 2011, Afternoon Session. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM256372.pdf. Accessed March 30, 2017.
- 19.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee, December 2, 2010. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM237499.pdf. Accessed March 30, 2017.
- 20.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee, Morning Session, October 5, 2009. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM191020.pdf. Accessed March 30, 2017.
- 21.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee, Afternoon Session, October 5, 2009. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM191021.pdf. Accessed March 30, 2017.
- 22.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee Discussion of New Drug Application (NDA) 022–393, Proposed Trade Name Istodax (romidepsin) Injection for treatment of cutaneous T‐Cell lymphoma (CTCL), Gloucester Pharmaceuticals, Inc. September 2, 2009. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM183276.pdf. Accessed March 30, 2017.
- 23.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee Discussion of New Drug Application (NDA) 022‐468, Proposed Trade Name Folotyn (pralatrexate) Injection for treatment of patients with relapsed or refractory (recurring and/or not responsive to other treatments) peripheral T‐cell lymphoma (PTCL), Allos Therapeutics, Inc. September 2, 2009. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM183277.pdf. Accessed March 30, 2017.
- 24.U.S. Food and Drug Administration. Summary Minutes of the Oncologic Drugs Advisory Committee May 29, 2009. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM170152.pdf. Accessed March 30, 2017.
- 25.U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee, Avastin (bevacizumab) for the Treatment of Previously Treated Glioblastoma Multiforme. March 31, 2009. Available at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM148933.pdf. Accessed March 30, 2017.
- 26. Blumenthal GM, Pazdur R. Response rate as an approval end point in oncology: Back to the future. JAMA Oncol 2016;2:780–781. [DOI] [PubMed] [Google Scholar]
- 27. Kurzrock R, Stewart DJ. Equipoise abandoned? Randomization and clinical trials. Ann Oncol 2013;24:2471–2474. [DOI] [PubMed] [Google Scholar]
- 28. Simon R, Blumenthal GM, Rothenberg ML et al. The role of nonrandomized trials in the evaluation of oncology drugs. Clin Pharmacol Ther 2015;97:502–507. [DOI] [PubMed] [Google Scholar]
- 29. Blumenthal GM, Karuri SW, Zhang H et al. Overall response rate, progression‐free survival, and overall survival with targeted and standard therapies in advanced non‐small‐cell lung cancer: US Food and Drug Administration trial‐level and patient‐level analyses. J Clin Oncol 2015;33:1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shaw AT, Kim DW, Nakagawa K et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2013;368:2385–2394. [DOI] [PubMed] [Google Scholar]
- 31. Sequist LV, Yang JC, Yamamoto N et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–3334. [DOI] [PubMed] [Google Scholar]
- 32. Rosell R, Carcereny E, Gervais R et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): A multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012;13:239–246. [DOI] [PubMed] [Google Scholar]
- 33.Fast Stats: An interactive tool for access to SEER cancer statistics. Surveillance Research Program, National Cancer Institute. Available at https://seer.cancer.gov/faststats. Accessed April 5, 2017.
- 34. Deisseroth A, Kaminskas E, Grillo J et al. U.S. Food and Drug Administration approval: Ruxolitinib for the treatment of patients with intermediate and high‐risk myelofibrosis. Clin Cancer Res 2012;18:3212–3217. [DOI] [PubMed] [Google Scholar]
- 35. Kluetz PG, Slagle A, Papadopoulos EJ et al. Focusing on patient‐reported outcomes in cancer clinical trials: Symptomatic adverse events, physical function, and disease‐related symptoms. Clin Cancer Res 2016;22:1553–1558. [DOI] [PubMed] [Google Scholar]
- 36. Bower H, Bjorkholm M, Dickman PW et al. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol 2016;34:2851–2857. [DOI] [PubMed] [Google Scholar]
- 37. Kumar SK, Rajkumar SV, Dispenzieri A et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Drawid A, Kaura S, Kiely D et al. Impact of novel therapies on multiple myeloma survival in the US: Current and future outcomes. J Clin Oncol 2015. [Epub ahead of print]. [Google Scholar]
- 39. Jawad I, Wilkerson J, Prasad V et al. Colorectal survival gains and novel treatment regimens: A systemic review and analysis. JAMA Oncol 2015;1:787–795. [DOI] [PubMed] [Google Scholar]
- 40. Kopetz S, Chang GJ, Overman MJ et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 2009;27:3677–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lowes, R. Avastin no longer approved for breast cancer, FDA says. Medscape, November 18, 2011. Available at http://www.medscape.com/viewarticle/753862. Accessed May 12, 2017.
- 42.AstraZeneca Pharmaceuticals LP; Withdrawal of Approval of a New Drug Application for Iressa. Federal Register, April 25, 2012. Available at https://www.federalregister.gov/documents/2012/04/25/2012-9944/astrazeneca-pharmaceuticals-lp-withdrawal-of-approval-of-a-new-drug-application-for-iressa. Accessed May 12, 2017.
- 43. Nelson, R. Gemtuzumab voluntarily withdrawn from US Market. Medscape, June 21, 2010. Available at http://www.medscape.com/viewarticle/723957. Accessed May 12, 2017.
- 44. Khozin S, Blumenthal GM, Jiang X et al. U.S. Food and Drug Administration approval summary: Erlotinib for the first‐line treatment of metastatic non‐small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. The Oncologist 2014;19:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kazandjian D, Blumenthal GM, Yuan W et al. FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation‐positive non‐small cell lung cancer. Clin Cancer Res 2016;22:1307–1312. [DOI] [PubMed] [Google Scholar]
- 46. Stenger, M. Modified Indication for Erlotinib in Advanced Non‐Small Cell Lung Cancer. ASCO Post, November 10, 2016. Available at http://www.ascopost.com/issues/november-10-2016/modified-indication-for-erlotinib-in-advanced-non-small-cell-lung-cancer/. Accessed May 12, 2017.
- 47. Chabner BA. Approval after phase 1: Ceritinib runs the three‐minute mile. The Oncologist 2014;19:577–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prowell TM, Theoret MR, Pazdur R. Seamless oncology‐drug development. N Engl J Med 2016;374:2001–2003. [DOI] [PubMed] [Google Scholar]
- 49. Sherman, RE, Anderson SA, Dal Pan GJ et al. Real‐world evidence—What is it and what can it tell us? N Engl J Med 2016;375:2293–2297. [DOI] [PubMed] [Google Scholar]