

Abstract
Skeletal dysplasias result from disruptions in normal skeletal growth and development and are a major contributor to severe short stature. They occur in approximately 1/5,000 births, and some are lethal. Since the most recent publication of the Nosology and Classification of Genetic Skeletal Disorders, genetic causes of 56 skeletal disorders have been uncovered. This remarkable rate of discovery is largely due to the expanded use of high-throughput genomic technologies. In this review, we discuss these recent discoveries and our understanding of the molecular mechanisms behind these skeletal dysplasia phenotypes. We also cover potential therapies, unusual genetic mechanisms, and novel skeletal syndromes both with and without known genetic causes. The acceleration of skeletal dysplasia genetics is truly spectacular, and these advances hold great promise for diagnostics, risk prediction, and therapeutic design.
Keywords: Growth insufficiency, mosaicism, imprinting, primordial dwarfism, ciliopathy, epigenetics
INTRODUCTION
This review provides background information on skeletal development and a historical perspective on skeletal dysplasia genetics. Skeletal dysplasias are a vast area of investigation, and owing to space constraints, we focus mainly on selected recent genetic discoveries that illustrate the genetic heterogeneity and diversity of processes that affect skeletal development. We present disorders with unique forms of inheritance, such as mutations in imprinted genes and somatic mosaicism for deleterious mutations. We discuss mutations that affect epigenetic programming and the potential role of microRNAs (miRNAs) in severe short stature. Primordial dwarfisms and skeletal ciliopathies receive considerable attention because of the rapid rate at which their causative mutations and molecular mechanisms are being uncovered. Each of these cases highlights how these discoveries are adding to our understanding of the pathology of skeletal dysplasias and the fundamentals of normal skeletal growth and development.
OVERVIEW OF EARLY SKELETAL DEVELOPMENT AND ENDOCHONDRAL OSSIFICATION
The bones of the skeleton are formed through two processes: intramembranous ossification and endochondral ossification. Both of these developmental programs begin with the condensation of mesenchymal cells in areas of the body where a skeletal element will eventually form (67, 71, 73). The condensations that will form the cranium and parts of the clavicle and pubic bone undergo intramembranous ossification, in which mesenchymal cells differentiate directly into osteoblasts that transform the condensation into true bone tissue.
The bones of the body that form through endochondral ossification are the main determinants of final stature (71). Early on, the mesenchymal cells of the condensation differentiate into chondrocytes instead of osteoblasts. These intermediary chondrocytes proliferate and undergo terminal hypertrophic differentiation, which directs the addition of mature bone tissue (67, 71, 73). The progressive mineralization and maturation of the cartilage template occur along its length and at either end. As this new element takes on more characteristics of a true bone, undifferentiated chondrocytes become sealed within either end of the forming element. These pools of chondrocytes are known as the epiphyseal growth plates.
The growth plate is a highly organized tissue. It is composed of three distinct zones—the resting zone, the proliferative zone, and the hypertrophic zone—followed by the ossification front (67, 71, 73, 119) (Figure 1). Lesser-differentiated cells in the resting zone enter the proliferative zone, where they divide perpendicular to the plane of the growth plate, intercalate over each other, and form pillars of discoid chondrocytes (73, 84, 119, 132). Eventually, the cells receive signals that promote their differentiation into hypertrophic chondrocytes, leading to a tremendous increase in cell volume. Until recently, it was thought that the primary method for deposition of bone matrix involved chondrocyte cell cycle exit followed by cell death, concomitant with recruitment of osteoblasts to remodel the hypertrophic scaffold. New studies have shown that chondrocytes differentiate into osteoblasts during their terminal differentiation and contribute to the addition of mature bone matrix (155a, 155b, 158a). Regulation of chondrocyte proliferation, differentiation, transdifferentiation into osteoblasts, and mineralization controls the growth of each skeletal element and of the individual.
Figure 1. Overview of the growth plate.
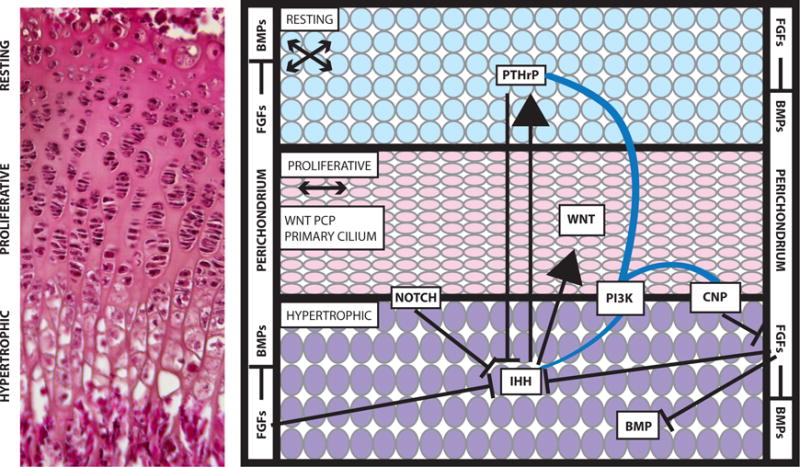
Cells within the resting, proliferative, and hypertrophic zones of the growth plate have a distinctive morphology and organization, which is visible in a section through a three-week-old mouse growth plate stained with eosin and hematoxylin (left). The Notch, WNT, FGF, Hedgehog, and BMP signaling pathways act on the cells of the growth plate (right). Arrows indicate activation of one pathway by another, and lines with a bar at the end indicate inhibition of the pathway. Blue lines indicate crosstalk between pathways. For example, PI3K is thought to affect the CNP, IHH, and PTHrP signaling pathways. Many of these pathways have been implicated in multiple skeletal dysplasias. The arrows underneath the names of the resting and proliferative zones indicate the orientation of cell division within these zones. Abbreviations: BMP, bone morphogenetic protein; CNP, C-type natriuretic peptide; FGF, fibroblast growth factor; IHH, Indian Hedgehog; PCP, planar-cell polarity; PI3K, phosphoinositide 3-kinase; PTHrP, parathyroid hormone–related peptide.
CELLULAR PROCESSES THAT CONTRIUTE TO NORMAL SKELETAL GROWTH AND SKELTAL DYSPLASIA
Several signaling pathways are known to regulate chondrocyte transition through the growth plate (Figure 1). Indian Hedgehog (IHH), parathyroid hormone–related peptide (PTHrP), fibroblast growth factor (FGF), C-type natriuretic peptide (CNP), TGF-β, bone morphogenetic protein (BMP), Notch, and WNT (canonical and noncanonical) signaling pathways all aid and guide chondrocytes through the growth plate and regulate functions in the perichondrium/periosteum (9, 67, 73) (Figure 1). The functions of these signaling pathways in growth, patterning, and mineralization are illustrated in Figure 1 and have been reviewed extensively (9, 67, 73). Here, we discuss selected recent genetic discoveries in signal transduction.
Figure 1 also highlights the direction of cell division as chondrocytes progress from the resting zone to the proliferative zone (arrows in Figure 1). In the resting zone, chondrocytes divide in roughly any orientation (84), with division occurring along the long axis of the cell (118a). Once the chondrocytes are in the proliferative zone, division occurs perpendicular to the direction of longitudinal growth, followed by chondrocyte rotation to form an ordered columnar array of cells (84). Recent work has demonstrated a requirement for noncanonical WNT signaling (84), likely through the planar-cell polarity (PCP) pathway (43a, 118a, 147a), and the appearance of a cadherin- and β-catenin-based cell adhesion surface between daughter cells (118a) for column formation. Following cell division, daughter cells remain closely associated and use the adhesion-laden surface to spread over one another (118a). The primary cilium also plays a role in regulating growth plate architecture, as defective intraflagellar transport through conditional loss of the gene encoding motor protein Kif3a leads to disordered columns in the mouse (132). The connection between the primary cilium and canonical and noncanonical WNT signaling pathways are currently mired in controversy (12a, 45a; reviewed extensively in 147). However, it is intriguing that both the primary cilium and the PCP pathway appear to be required for proper column formation. Future work on the growth plate will clarify the nature of the connection between these two regulators of growth plate architecture, if it exists.
Cell adhesion, a critical regulator of column formation (118a)], and the composition of the extracellular matrix can be directly affected by a defect in intracellular trafficking (139b). This process involves the endoplasmic reticulum, Golgi apparatus, and secretory vesicles. Without proper trafficking of extracellular matrix and cell adhesion components, as well as ligands for critical signaling pathways, the function of the growth plate can be profoundly compromised. Often, the result is profound endoplasmic reticulum stress, which is a common pathogenic mechanism in disorders of the skeleton, including osteogenesis imperfecta (87a, 105a, 116a, 136a).
THE SKELETAL DYSPLASIAS
The term skeletal dysplasia is typically used to describe bone and cartilage disorders that cause dwarfism, but this set of disorders is highly diverse and also includes overgrowth syndromes. Numerous genetic mutations have the ability to disrupt the organization and function of the growth plate (9, 59, 67, 71, 149). Studies in humans and mice have identified many genes, including those that disrupt the processes discussed in the section above (Figure 1) that can profoundly affect the growth of the individual, preventing achievement of adult height that is normal for age, ethnicity, and sex.
There are more than 400 skeletal dysplasias documented in the medical literature (149). They are predicted to occur in approximately 1/5,000 children (71) and can range from relatively mild anomalies to lethality. Most lethal forms of skeletal dysplasia result from thoracic hypoplasia, where the rib cage is not large enough to permit proper lung development (34). In such cases, the infant dies of respiratory distress soon after birth.
Many forms of skeletal dysplasia result in severe short stature but are compatible with life. In some cases, other organ systems are affected because the causative gene also has functional roles in tissues other than the skeleton. For example, many skeletal dysplasia patients have hearing loss (8, 71, 78, 138). In patients with achondroplasia, the most common nonlethal form of skeletal dysplasia (71), hearing loss could be due to chronic otitis media (138). Individuals may also have neurological involvement and vision problems (71). Patients should be carefully monitored for all of these potential complications, as they may require immediate intervention (71, 122).
There currently are no pharmacological therapies for individuals affected with skeletal dysplasia, although there are some promising candidates worthy of discussion below. Enzyme therapies available for some mucopolysaccharidoses improve lung function, but they are not effective for skeletal and joint abnormalities associated with these disorders (99). Growth hormone therapy can accelerate growth rate, but in cases of achondroplasia or idiopathic short stature, final adult height is not substantially improved (3a, 56). Controversial surgical procedures can successfully lengthen the limbs of a child, but the surgeries are numerous, expensive, and require dedicating much of the childhood years to medical care (71, 122). Molecular diagnosis of skeletal dysplasias is highly desired, and many patients and their families seek treatment options.
THE GENETICS OF SKELETAL DYSPLASIA
Historically, skeletal dysplasias have been characterized by inheritance pattern, associated symptoms, and radiological features. Mutations in the same gene can cause many different forms of skeletal dysplasia (149). One example of this is the features associated with mutations in FGFR3, the gene that encodes fibroblast growth factor receptor 3. Mutations in this gene represent an allelic series in clinical severity ranging from nonlethal achondroplasia and hypochondroplasia to thanatophoric dysplasia types I and II, which are lethal. Mutations in different genes can cause the same form of skeletal dysplasia. Osteogenesis imperfecta is a premier example of this phenomenon. Genes that encode collagens (COL1A1 and COL1A2) are a major cause of this disorder, but other cases are caused by mutations in genes that encode molecular chaperones (e.g., SERPINH1 and FKBP10) (87a).
Throughout the 1980s, a great deal of excitement arose from the discoveries that mutations in collagen genes cause conditions such as osteogenesis imperfecta (33, 115), Stickler syndrome (1a, 41), and spondyloepiphyseal dysplasia (82). The involvement of collagen genes in skeletal dysplasia was first suggested by protein analysis of cartilage (reviewed in 77). These studies made use of genetic material collected from nuclear families with multiple affected individuals and utilized such techniques as linkage analysis, positional cloning (41, 82, 137), and traditional Sanger sequencing to uncover the causative mutations. The discovery of collagen mutations in skeletal disorders heralded a new era, one where molecular genetic causes could be used for diagnostics and risk prediction in conjunction with family history, clinical examination, and radiographs (71).
These successes were quickly followed by the discovery of the genetic etiology of achondroplasia. In 1994, three groups mapped achondroplasia to a region on human chromosome 4 (42, 80, 142). All three studies included multiple unrelated nuclear families (N = 6–18) with multiple affected individuals for linkage analysis. A locus on chromosome 4p16.3 cosegregated with achondroplasia in all of the families, providing evidence for genetic homogeneity. Munnich and colleagues (80) included patients with hypochondroplasia, which was thought to be allelic to achondroplasia. Their results supported this hypothesis, because both conditions mapped to the same region on chromosome 4. This region included several candidate genes, one of which was FGFR3.
Only a few months after achondroplasia and hypochondroplasia were mapped, the achondroplasia mutation in FGFR3 was discovered (120, 129). Astonishingly, all of the patients screened had the same mutated nucleotide—the guanosine at position 1138 of the FGFR3 transcript was substituted with an adenosine or cytosine, resulting in a missense mutation that substituted an arginine for a glycine at codon 380 (p.G380R). We now know that the frequency of this mutation is higher with advanced paternal age, and the high incidence may be due to the presence of a highly mutable CpG at the site and/or to a selective advantage for sperm with this mutation (46). When the p.G380R mutation was discovered to cause achondroplasia, its detrimental effects were speculated to be due to dominant negative action. Subsequently, normal FGF signaling was shown to have an inhibitory effect on the regulation of skeletal growth (32), and the molecular mechanism underlying achondroplasia was recognized as constitutive activation of FGFR3 by the p.G380R substitution (150, 151). As predicted, mutations that activate FGFR3 by altering other functional domains cause hypochondroplasia and thanatophoric dysplasia types I and II (40, 41, 43).
The time elapsed from the initial mapping of the achondroplasia mutant locus to the discovery of mutations in FGFR3 was only approximately one year. This timeline is impressive given the technology of the time and that all of these studies took place before the release of the human genome sequence. Soon thereafter, mutations in two related genes, FGFR2 and FGFR1, were discovered to cause other types of skeletal dysplasia (9, 149). The rate of mutation discovery has accelerated with the availability of the reference genome sequence and high-throughput genomic mapping and sequencing technologies, which have revolutionized the way these studies are conducted.
HIGH-THROUGHPUT TECHNOLOGIES AND MUTATION DISCOVERY
Many genetic causes of defects in skeletal growth and/or development have been discovered since the most recent version of the Nosology and Classification of Genetic Skeletal Disorders was published in 2011 (149) (Table 1). Recent studies have made use of high-throughput technologies, including whole-genome single-nucleotide polymorphism (SNP) mapping, comparative genomic hybridization arrays, whole-exome or whole-genome next-generation sequencing, and targeted enrichment of a locus followed by next-generation sequencing. Most of the mutations listed in Table 1 were discovered by next-generation sequencing of patient genomic DNA samples. As more efficient and cost-effective sequencing technologies are developed, whole-genome sequencing will likely become the preferred strategy (10, 92). The discovery of causal variants will substantially aid in the diagnosis, understanding, and treatment of many genetic disorders, including those that affect the skeleton (10). We discuss a selection of the genetic causes listed in Table 1 in more detail below.
Table 1.
Causative mutations of skeletal disorders discovered since the most recent publication of the Nosology and Classification of Genetic Skeletal Disorders (149)
| Group | Disorder | Inheritance | MIM number | Locus | Gene | Notes/function |
|---|---|---|---|---|---|---|
| 9 | Short rib thoracic dysplasia (new form) | AR | NA | 5q23.2 | CEP120 (centrosomal protein, 120 kDa (124a) | Four separate families with same missense variant |
| 9 | Short-rib thoracic dysplasia 4 with or without polydactyly | AR | 613819 | 2q24.3 | TTC21B (Tetratricopeptide repeat domain-containing protein 21B) (29a) | Retrograde ciliary transport (IFT-A) |
| 9 15 |
Short-rib thoracic dysplasia 5 with or without polydactyly Cranioectodermal dysplasia 4 |
AR | 614376 614378 |
4p14 | WDR19 (WD-repeat containing protein 19) (22) | IFT-A complex, retrograde ciliary trafficking, ciliogenesis |
| 9 15 |
Short-rib thoracic dysplasia 7 with or without polydactyly Cranioectodermal dysplasia 2 |
AR | 614091 613610 |
2p24 | WDR35 (WD repeat-containing protein 35) (94) | Ciliogenesis and cilium maintenance |
| 9 | Short-rib thoracic dysplasia 8 with or without polydacyly | AR | 615503 | 7q36.3 | WDR60 (WD repeat-containing protein 60) (89) | Ciliogenesis, Hedgehog signaling |
| 9 | Short-rib thoracic dysplasia 9 with or without polydactyly | AR | 266920 | 16p13.3 | IFT140 (intraflagellar transport 140, Chlamydomonas, homolog of) (111) | Ciliogenesis, part of IFT-A complex (retrograde ciliary transport) |
| 9 | Short-rib thoracic dysplasia 10 with or without polydactyly | AR | 615630 | 2p23.3 | IFT172 (intraflagellar transport 172, Chlamydomonas, homolog of) (50) | Anterograde ciliary transport (IFT-B) |
| 9 | Short-rib thoracic dysplasia 11 with or without polydactyly | AR | 615633 | 9q32.11 | WDR34 (WD repeat-containing protein 34) (61, 123) | Retrograde ciliary transport (IFT-A), NF-κB signaling |
| 9 | Joubert syndrome 18 Orofaciodigital syndrome IV |
AR | 614815 258860 |
10q24.1 | TCTN3 (Tectonic family, member 3) (136) | Cilogenesis, Hedgehog signaling |
| 9 | Orofaciodigital syndrome V | AR | 174300 | 1q32.1 | DDX59 (DEAD box polypeptide 59) (127) | RNA helicase, RNA metabolism |
| 12 | Spondylometaphyseal dysplasia with cone-rod dystrophy | AR | 608940 | 3q29 | PCYT1A (phosphate cytidylyltransferase 1, choline, alpha isoform) (55, 154) | Phosphotidylcholine metabolism |
| 13 | Spondyloepimetaphyseal dysplasia with joint laxity, type 2 | AD | 603546 | 16p11.2 | KIF22 (Kinesin family member 22) (21, 95) | Unknown (speculated role in intracellular trafficking or ciliary transport) |
| 13 19 |
Spondyloepimetaphyseal dysplasia with joint laxity, type 2 Seckel Syndrome 7 |
AR | 603546 614851 |
14q22.1 | NIN (Ninein) (29, 47a) | Centrosome-mediated asymmetric cell division |
| 14 | Novel spondylodysplastic dysplasia | AR | NA | 16p13.3 | PAM16 (presequence translocase-associated motor 16) (90) | Mitochondrial transport |
| 14 | Opsismodysplasia | AD | 258480 | 11q13.4 |
INPPL1 (15, 60) Inositol polyphosphate phosphatase-like 1 |
PI3K-AKT signaling modulation |
| 15 | Acrodysostosis 1, with or without hormone resistance | AD | 101800 | 17q24.2 | PRKAR1A (protein kinase, cAMP dependent, regulatory, type 1, alpha) (86) | PKA/cAMP mediated signaling |
| 15 | Acrodysostosis 2, with or without hormone resistance | AD | 614613 | 5q12.1 | PDE4D (phosphodiesterase 4D) (83, 93) | Degradation of cAMP/inhibiting signaling pathway |
| 15 | Acromicric dysplasia Geleophysic dysplasia 2 |
AD AR |
102370 614185 |
15q21.1 | FBN1 (fibrillin 1) (79) | Integrin and TGF-β signaling |
| 18 | Bent bone dysplasia syndrome | AD | 614592 | 10q26.13 | FGFR2 (fibroblast growth factor receptor 2) (91) | Proper regulation of chondrocyte proliferation and differentiation |
| 19 | Microcephalic Primordial Dwarfism | AR | NA | 5q14.2 | XRCC4 (X-ray repair, complementing defective, in Chinese hamster, 4) (102a) | Nonhomologous end joining DNA repair |
| 19 | Microcephaly and chorioretinopathy, autosomal recessive, 2 | AR | 616171 | 4q28.2 | PLK4 (Polo-like kinase 4) (87b, 123c) | Centrosome duplication and maturation |
| 19 | New form of primordial dwarfism | AR | NA | 8q24.13 | NSMCE2 (non-SMC element 2 homolog) (110a) | E3 SUMO-ligase |
| 19 | Microcephaly 13, primary, autosomal recessive | AR | 616051 | 4q24 | CENPE (centromeric protein E) (96) | Kinetochore-associated chromosome alignment |
| 19 | Short stature, onychodysplasia, facial dysmorphism and hypotrichosis | AR | 614813 | 3p21.1–21.31 | POC1A (POC1 centriolar protein, Chlamydomonas, homolog of, A) (121, 124) | Centrosome maintenance, Golgi retrograde trafficking, proper mitotic spindle polarity, cilogenesis |
| 19 | IMAGe syndrome | AD, maternally imprinted | 614732 | 11p15 | CDKN1C (p57Kip2) (5) | Inhibition of cell cycle progression |
| 19 | Seckel syndrome 2 | AR | 606744 | 18q11.2 | RBBP8 (retinoblastoma-binding protein 8) (116) | DNA repair |
| 19 | Microcephalic osteodysplastic primordial dwarfism, type I | AR | 210710 | 2q14.2 | RNU4ATAC (RNA, U4ATAC small nuclear) (52, 37) | RNA splicing |
| 20 | Disorder of glycosylation resembling Desbuquois dysplasia | AR | NA | 6q14.1–14.2 | PGM3 (phosphoglucomutase 3) (134a) | Generates substrates for protein glycosylation |
| 20 | Desbuquois dysplasia 2 | AR | 615777 | 16p12.3 | XYLT1 (xylosyltransferase 1) (22a, 123a) | Glycosaminoglycan synthesis |
| 25 | Bone fragility with craniosynostosis, ocular proptosis, hydrocephalus, and distinctive facial features | AR | 112240 | 4q26 | SEC24D (SEC24-related gene family, member D) (43b) | Component of COPII-coated vesicles |
| 25 | Osteogenesis imperfecta, type V | AD | 610967 | 11p15.5 | IFITM5 (interferon-induced transmembrane protein 5) (26a, 123b) | Stimulates mineralization when overexpressed |
| 25 | Osteogenesis imperfecta, type XIII | AR | 614856 | 8p21.3 | BMP1 (bone morphogenetic protein 1) (7a, 87c) | BMP ligand |
| 25 | Osteogenesis imperfecta, type XIV | AR | 615066 | 9q31.2 | TMEM38B (transmembrane protein 38B) (123d) | Intracellular cation channel |
| 25 | Osteogenesis imperfecta type XV | AR | 615220 | 12q13.12 | WNT1 (wingless-type MMTV integration site family member 1) (68a) | WNT ligand |
| 25 | Osteogenesis imperfecta, type XVI | AR | 616229 | 11p11.2 | CREB3L1 (cAMP response element-binding protein 3–like 1) (134b) | Unfolded protein response |
| 30 | Proteus syndrome, somatic | AD, somatic mosaicism | 176920 | 14q32.33 | AKT1 (V-AKT murine thymoma ciral ocongene homolog 1) (85) | PI3K-AKT signaling, cell proliferation, cell growth |
| 32 | Yunis-Varon syndrome | AR | 216340 | 6q21 | FIG4 (FIG4, S. cerevisiae, homolog of) (24) | PI(3,5)P2 signaling |
| 35 | Klippel-Feil syndrome 2 | AR | 214300 | 17q21.31 | MEOX1 (mesenchyme homeobox 1) (98) | Transcription factor involved in somitogenesis |
| 36 | Genitopatellar syndrome SBBYSS syndrome |
AD | 606170 603736 |
10q22.2 | KAT6B (lysine acetyltransferase 6B) (23, 27) | Histone acetyltransferase |
| 36 | Meier-Gorlin syndrome 1 | AR | 224690 | 1p32.3 | ORC1 (origin recognition complex, subunit 1, S. cerevisiae, homolog of) (17, 16) | ORC subunit, DNA replication |
| 36 | Meier-Gorlin syndrome 2 | AR | 613800 | 2q22.3–23.1 | ORC4 (origin recognition complex, subunit 4, S. cerevisiae, homolog of) (49) (16) | ORC subunit, DNA replication |
| 36 | Meier-Gorlin syndrome 3 | AR | 613803 | 16q11.2 | ORC6 (origin recognition complex, subunit 6, S. cerevisiae, homolog of) (16) | ORC subunit, DNA replication |
| 36 | Meier-Gorlin syndrome 4 | AR | 613804 | 16q24.3 |
CDT1 (16) Chromatin licensing and DNA replication factor 1 |
Recruited to fully assembled prereplication complex |
| 36 | Meier-Gorlin syndrome 5 | AR | 613805 | 17q21.2 | CDC6 (cell division cycle 6, S. cerevisiae, homolog of) (16) | Recruited to fully assembled prereplication complex |
| 37 | Myhre syndrome | AD | 139210 | 18q21.2 | SMAD4 (mothers against decapentalplegic, Drosophila, homolog of, 4) (78) | TGF-β and BMP signaling modulation |
| 37 | Feingold syndrome 2 | AD | 614326 | 13q31.3 | MIR17HG (micro RNA 17 host gene) (31) | miRNA cluster transcriptionally regulated by N-MYC |
| 39 | Meckel syndrome 2 | AR | 603194 | 11p12–q13.3 | TMEM216 (transmembrane protein 216) (140) | Ciliogenesis, cilium maintenance, planar-cell polarity signaling |
| 39 | Meckel syndrome 10 | AR | 614175 | 19q13.2 | B9D2 (B9 domain–containing protein 2) (35) | Ciliogenesis, ciliary protein localization, Hedgehog signaling |
| 39 | Joubert syndrome 21 | AR | 615636 | 8q13.1–13.2 | CSPP1 (centrosome spindle pole associate protein 1) (2, 125, 139) | Some patient phenotypes resembled Meckel syndrome |
| ? | New syndrome with short stature and several additional clinical features, including neurodegeneration and photosensitivity | AR | NA | 20p13–p12 | PCNA (proliferating cell nuclear antigen) (10b) | Required for DNA repair |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; BMP, bone morphogenetic protein; IMAGe, intraunterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital abnormalities; MIM, Mendelian Inheritance in Man; miRNA, microRNA; NA, not applicable; ORC, origin recognition complex; PI3K, phosphoinositide 3-kinase; SBBYS, Say-Barber-Biesecker-Young-Simpson.
IMAGE SYNDROME: THE EFFECTS OF IMPRINTING
IMAGe syndrome is characterized by intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital abnormalities (145). The IMAGe phenotype encompasses many organ systems and leads to hypoplasia of affected tissues. The variable skeletal abnormalities in these patients include defects at the level of the growth plate (metaphyseal dysplasia, epiphyseal dysplasia, and short arms and legs) and the bone tissue itself (osteopenia and delayed bone age) (5, 145). This disorder is caused by mutations in CDKN1C, which encodes cyclin-dependent kinase inhibitor 1C, also known as p57Kip2. CDKN1C is imprinted, resulting in silencing of the paternal allele, which explains the apparent autosomal dominant inheritance. This phenomenon was observed in all tested patients in a large five-generation Argentinian family.
Mutations that cause IMAGe syndrome (Table 1) occur specifically in the domain of CDKN1C responsible for binding proliferating cell nuclear antigen (PCNA), which marks CDKN1C for ubiquitin-targeted protein degradation (5). The mutant form of CDKN1C does not bind PCNA, resulting in abnormal stabilization of the cell cycle inhibitor. Normally, transcriptional activation of CDKN1C by C/EPBβ is thought to regulate the transition from proliferation to hypertrophic differentiation in the growth plate (54). Thus, the mutant CDKN1C likely prematurely inhibits chondrocyte division, leading to premature differentiation and ultimately dwarfism. This mechanism underlies the pathophysiology of skeletal dysplasias associated with IHH and parathyroid hormone (PTH) mutations (146). PTHrP promotes chondrocyte proliferation and is thought to downregulate p57Kip2, making it an attractive therapeutic candidate for counteracting the effects of the stabilizing mutations (74).
PROTEUS SYNDROME: A DISORDER OF SOMATIC MOSAICISM
Proteus syndrome is a very rare, highly sporadic, and debilitating condition characterized by patches of tissue overgrowth, or asymmetrical hyperplasia, in a variety of tissues, including bone and connective tissue (18, 85). Hyperplasia of affected tissues begins approximately a year after birth (18). The bones of the limbs are particularly affected, with the hyperplasia altering normal bone patterning and sometimes leading to joint immobility and/or bowing and rotation of the long bones. There is a 20% rate of premature death, typically caused by venous thrombosis and pulmonary embolism. Vascular abnormalities and tumors also occur (18, 85).
The asymmetric nature of the hyperplasia in Proteus syndrome patients led to the hypothesis that it results from somatic mosaicism, in which a de novo mutation occurs after the fertilized egg has begun to develop, causing a subset of cells to undergo excessive cell division (51). Genetic analysis of cells from hyperplastic regions and normal regions of Proteus syndrome patients led to the discovery of the causative mutation, and additional work revealed a shared etiology between Proteus and SOLAMEN syndromes (the latter of which is characterized by segmental overgrowth, lipomatosis, arteriovenous malformation, and epidermal nevus) (25, 85). Tissues affected by Proteus syndrome harbor a missense mutation (c.49G→A, p.Glu17Lys) in AKT1 and increased phosphorylated AKT1, indicating overactivation of the phosphoinositide 3-kinase (PI3K)–AKT pathway (85). SOLAMEN syndrome patients are somatic mosaics with loss of function in the tumor suppressor PTEN, leading to AKT activation (25). The affected areas of SOLAMEN patients are nullizygous for PTEN, the result of an inherited germline mutation in PTEN and a de novo somatic mutation in the affected area. Because PTEN inhibits activation of AKT kinase, PTEN deficiency leads to overactivation of the PI3K-AKT signaling pathway in the affected areas, resulting in increased cell proliferation and reduced apoptosis (25, 85).
How does overactive AKT1 cause the skeletal phenotypes in Proteus and SOLAMEN syndromes? The role of PI3K-AKT signaling in bone is controversial (14). Most data are consistent with promotion of hypertrophic differentiation of growth plate chondrocytes (14, 40, 58, 118, 155). Overactivation of this pathway in mice through the conditional loss of PTEN activity leads to an overgrowth of skeletal elements, disorganized growth plates, and chondrocytes with altered cellular morphologies indicative of accelerated hypertrophic differentiation (14, 40, 58, 155). Known downstream effectors of the PI3K-AKT pathway in the growth plate include mTOR, SOX9 [also involved upstream (61a)], IHH, RUNX2, COL2A1, and proteoglycans (reviewed in 14), with additional targets discovered through microarray analyses (139a). Although more work is necessary to define the PI3K-AKT pathway function(s) in bone, it is clear that the primary role of AKT is to promote endochondral ossification in both humans and mice (14, 85). In Proteus syndrome patients, the mutant AKT1 protein causes accelerated disorganized bone growth in asymmetric patches, leading to the debilitating phenotype (18). Inhibitors of AKT1 or its downstream effectors may provide therapeutic targets.
Somatic mosaicism underlies some cases of McCune-Albright syndrome, a genetically heterogeneous disorder that affects the skeleton, skin, and endocrine system (152). Similarly, somatic mosaicism for mutations in isocitrate dehydrogenase (IDH1) or mitochondrial IDH2 underlies some cases of Ollier disease and Maffucci syndrome, which are characterized by periosteal cartilaginous tumors, gliomas, and acute myelogenous leukemia (110). The IDH1 tumors are associated with hypermethylation and gene silencing in cartilage tumors, suggesting an epigenetic aspect of tumorigenesis. Thus, somatic mosaicism may contribute more to genetic disorders than was previously appreciated.
EPIGENETIC CONTROL: MUTATIONS IN HISTONE ACETYLTRANSFERASE GENES CAUSE A VARIETY OF DISORDERS
The modification of histones by chromatin-modifying enzymes maintains cellular identity and regulates cell fate transitions during differentiation (15a, 148a). These processes are important to ensure proper organogenesis and growth. Chromatin-modifying enzymes include histone methyltransferases, histone demethylases, histone acetyltransferases, histone deacetylases (HDACs), and enzymes that phosphorylate and ubiquitinate histones (10a, 15a, 148a). Recently, skeletal growth disorders have been shown to result from loss of function in HDAC4 (152b) and mutations in the genes encoding other chromatin-modifying enzymes. For example, mutations in the gene encoding histone H3.3 are drivers of both chondroblastoma and giant cell tumors of bone (13), and mutations in the histone acetyltransferase gene KAT6B cause two distinct skeletal development disorders (23, 27).
KAT6B is a member of the MYST family of histone acetyltransferase genes. Mutations at the 3´ end of KAT6B have been reported in patients with Say-Barber-Biesecker-Young-Simpson (SBBYS) syndrome, a variant of Ohdo syndrome. This disorder causes intellectual disability, facial abnormalities, and distinct skeletal characteristics, including hypoplastic or absent patellae, joint laxity, and abnormally long first digits. These patients are heterozygous for de novo mutations that disrupt the RUNX2-binding domain (27). RUNX2 is a homeodomain transcription factor that regulates chondrocyte hypertrophy and osteoblast differentiation (36, 67). Thus, the skeletal phenotype in these patients could be due to defects in chondrocytes, osteoblasts, or both cell types. Specific loss-of-function mutations in KAT6B cause genitopatellar syndrome, a disorder characterized by intellectual disability, absence or hypoplasticity of the patellae, craniofacial abnormalities, and genital malformations (23). The pathogenic mutations are de novo insertions or deletions that cause frameshifts or nonsense mutations at the start of the last exon of KAT6B (23). The mutant forms of KAT6B lack the transcription-activation domain and the RUNX2-binding domain and may have dominant negative effects (23, 27). If mutant KAT6B is unable to acetylate histones, HDAC inhibitors might counteract the loss of function, provided that appropriate specificity can be achieved. Future studies will clarify the molecular mechanisms underlying the role of histone acetyltransferases and other chromatin-modifying enzymes in skeletal development and growth.
MUTATIONS IN MIRNAs AND SEVERE SHORT STATURE
Mutations in MYCN, or N-myc, and a miRNA cluster cause Feingold syndrome, a disorder that can involve multiple organ systems but consistently affects the skeleton (39). The hallmark skeletal anomalies are short stature, microcephaly, brachydactyly of the second and fifth digits of the hands, and brachysyndactyly of the toes. A nonrecombinant interval for Feingold syndrome was mapped to human chromosome 2p23–p24 (26), and subsequent analysis identified heterozygous mutations in MYCN (141). Genomic DNA from Feingold patients negative for mutations in MYCN was analyzed by comparative genomic hybridization arrays to screen for disease-causing copy-number variations. These patients are heterozygous for microdeletions on human chromosome 13q31.3 that encompass the miR-17~92 miRNA cluster, known as MIR17HG (26, 28, 31, 39). This was confirmed by the observation that mice homozygous for deletion of the MIR17HG locus develop a Feingold syndrome–like phenotype (144).
MYCN appears to directly activate transcription of this miRNA cluster (31), explaining why the hemizygous loss of function of either MYCN or the miR cluster leads to very similar skeletal phenotypes in mice and humans (31, 97, 103, 108). Defining the targets of these miRNAs will give a deeper understanding of the pathophysiology of Feingold syndrome.
THE PRIMORDIAL DWARFISMS
Many of the skeletal dysplasias are characterized by disproportionate bone growth, with long bones being most affected (69, 71). Primordial dwarfism, on the other hand, causes a proportionate reduction in growth, similar to that caused by deficiencies in insulin-like growth factor 1 (IGF-1) or growth hormone (GH) (69). Patients with IGF-1 deficiency and primordial dwarfism are smaller at birth and are sometimes ascertained in utero, whereas patients with GH deficiency are normally sized at birth and are usually identified later in childhood. Primordial dwarfisms include Seckel syndrome, Meier-Gorlin syndrome (MGS), and microcephalic osteodysplastic primordial dwarfism types I–III (MOPD I–III) and have been recently reviewed (69).
GH/IGF-1-deficient patients do not have skeletal dysplasia or microcephaly (9). They typically exhibit delayed bone age, and many patients respond to hormone supplementation (47b), unlike traditional skeletal dysplasia patients (71). Primordial dwarfisms cause reductions in the overall cell number but do not affect endocrine processes (69). It is not clear whether the reduction in cell number is due to augmentation of cell death, delay or reduction in cell proliferation, or both. Mouse models and cell culture studies should clarify the roles of mechanisms that underlie primordial dwarfisms.
The forms of primordial dwarfism vary significantly in severity (69). Seckel syndrome patients tend to have moderate to mild intellectual disability, microcephaly, and distinctive facies. MOPD I and III cause curvature of the long bones, drastic growth insufficiency, and death by three years of age, as well as skin abnormalities and sparseness of hair. MOPD II is compatible with life, and patients have normal intellect. They are predisposed to vascular abnormalities and insulin resistance, which can lead to type II diabetes. MGS causes hypoplasia or complete absence of the patellae, microtia, and microcephaly and presents with highly variable growth outcomes (20, 30). Not all forms of primordial dwarfism are included in the most recent version of the Nosology and Classification of Genetic Skeletal Disorders (149), but we have included recent genetic discoveries for all the forms in the same classification in Table 1 (group 19, which includes forms of MOPD).
Primordial dwarfism patients have genetic aberrations in genes with diverse functions that are likely to affect all of the cells in the body and are connected to the cell cycle (69) (Figure 2). Seckel syndrome is associated with mutations in the DNA-damage response genes ATR (106), RBBP8 (116), and possibly ATRIP (107), and the centriole-associated genes CPAP (CENPJ) (3) and CEP152 (65). MOPD II is caused by mutations in PCNT, which encodes a pericentriolar protein involved in the generation of the mitotic spindle and centrosome maturation. Microcephalic primordial dwarfism has been associated with mutations in NIN, which encodes the centrosomal protein ninein (29). Little is known about the function of ninein, although it is thought to regulate asymmetric cell division (70, 114, 148). Mutations in XRCC4 (encoding a component of the nonhomologous end joining DNA repair pathway), and CENPE (encoding a centromere-associated protein) are also associated with microcephalic primordial dwarfism (96, 102a). Mutations in genes that encode components of the origin recognition complex (ORC), which regulates the licensing of DNA replication origins, cause MGS (16, 17, 30, 49) (Table 1). MOPD I is caused by mutations in U4ATAC, which is involved in splicing U12 introns, present in a subset of human genes (37, 52). MOPD II is likely caused by alterations in the splicing of specific transcripts required for modulation of cellular growth and cell number (69). Thus, genes that contain U12 introns could be involved in other forms of primordial dwarfism.
Figure 2. Cellular pathways disrupted in primordial dwarfism.
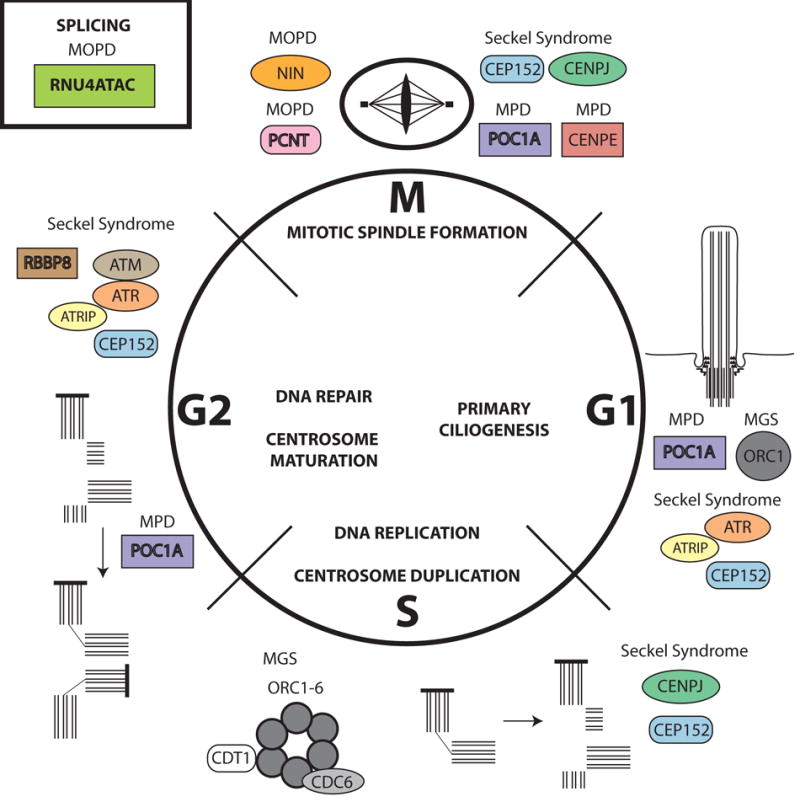
Most genes implicated in primordial dwarfism (MGS, MOPD I and II, Seckel syndrome, and SOFT syndrome) are involved in cell cycle regulation and cell division. These processes include primary ciliogenesis (G1 phase), DNA replication and centrosome duplication (S phase), DNA repair and centrosome maturation (G2 phase), and mitotic spindle formation and mitosis (M phase). Many of the genes function in more than one phase of the cell cycle. Some genes affect the rate of progression through the cell cycle, and some lead to aneuploidy, probably due to defects in DNA repair or mitotic spindle formation. In some cases, the cell cycle becomes disrupted after several rounds of cell division owing to disturbances in centriole duplication and/or maturation. All the processes shown involve replication and repair or maturation of the genome and the centrosome except RNU4ATAC. RNU4ATAC has a critical role in splicing, which could affect genes involved in cell cycle regulation or division. Abbreviations: MGS, Meier-Gorlin syndrome; MOPD, microcephalic osteodysplastic primordial dwarfism; MPD, microcephalic primordial dwarfism; SOFT, short stature, onychodysplasia, facial dysmorphism, and hypotrichosis.
Primordial dwarfisms correspond phenotypically and genetically with one of two major affected processes: DNA repair and replication or centrosome-associated proteins. MOPD I is an exception, as mutations in U4ATAC likely disrupt multiple cellular functions, causing pleiotropic effects and early lethality. Mutations of ATR and CEP152 in Seckel syndrome cause similar phenotypes: moderate learning disability and a more marked reduction in the size of the skull. Patients with mutations in CENPJ (CPAP) have clinical features similar to those of MOPD II patients with mutations in PCNT, in that both groups have a proportionate reduction in head size and body size. Intellectual disability in the Seckel CENPJ patients is mild, whereas MOPD II/PCNT patients have a normal intellect. ORC-associated proteins such as ORC1 associate with the centrosome (53, 57), and mutations in ORC1 cause more severe reductions in height and head circumference than any other cause of MGS (30, 57). All but one of these ORC1 patients had normal intellect, and the one exception had mild intellectual disability.
The effect of the centrosome-associated genes on stature may be due to the requirement of the centrosome to form the primary cilium, a structure necessary to maintain the organization of the growth plate (59, 119, 132). There are exceptions to these general correlations between clinical features and the type of gene mutated. For example, mutations in the centrosomal protein encoded by NIN cause both severe short stature and profound intellectual disability (29). Morpholino-induced knockdown of NIN in zebrafish or loss of ninein in mice reduces the number of cells in a distinct part of the developing nervous system (148). Although NIN deficiency does not appear to affect overall cell division, it could affect asymmetric divisions required for specific populations of cells, and it could have important undiscovered functions distinct from its role in centrosome biology.
Mechanistic details of MGS have emerged from studies of ORC1. ORC1 binds dimethylated lysine 20 of histone H4 (H4K20me2), a chromatin modification commonly present at replication origins, and mutation of the H4K20me2 binding domain causes MGS (75). In vitro studies suggest that slow progression through S phase is not a determinant of the MGS phenotype (133). Additional work has shown that ORC1 regulates centrosome duplication (57, 105). MGS-associated mutations of ORC1 reduce the rate of primary cilium formation and alter the collection of signaling components present within the primary cilium, but the cilia that form have a normal appearance (133). These studies demonstrate that MGS and related forms of primordial dwarfism potentially have mechanistic connections to the centrosome and the primary cilium, and these connections should be explored to determine whether these disorders represent a subset of skeletal ciliopathies.
THE SKELETAL CILIOPATHIES
Mutation of centrosome-associated genes in primordial dwarfisms could influence the formation and maintenance of the primary cilium, formation of the mitotic spindles and cell division, and/or secretory pathways (133). The primary cilium maintains the architecture of the growth plate (132), and disruptions in ciliogenesis can lead to many severe forms of skeletal dysplasia (59). These forms of skeletal dysplasia are known as the skeletal ciliopathies and may include some forms of primordial dwarfism (133). Some of these disorders spare the tissues typically damaged in ciliopathies (such as kidney, retina, brain, and liver) and are embryonic or perinatal lethal, and many of them result in short ribs and polydactyly, presumably resulting from altered IHH signaling. The short ribs may impair lung development and function, causing lethality at birth (34, 59).
The primary cilium is a microtubule-based structure present on the majority of noncycling cell types, including chondrocytes. The axoneme of this nonmotile cilium extends outward from the basal body, the foundation of the cilium generated by the mother centriole of the centrosome (62) (Figure 3). Sheathed in plasma membrane and decorated with numerous receptors and ion channels, the primary cilium serves as a major signaling center, particularly for the Hedgehog and WNT signaling pathways (147). IHH signaling is a major regulator of skeletal growth, so perturbation of the primary cilium has the potential to disrupt IHH signaling and growth of long bones.
Figure 3. Primary cilium proteins implicated in skeletal dysplasias.
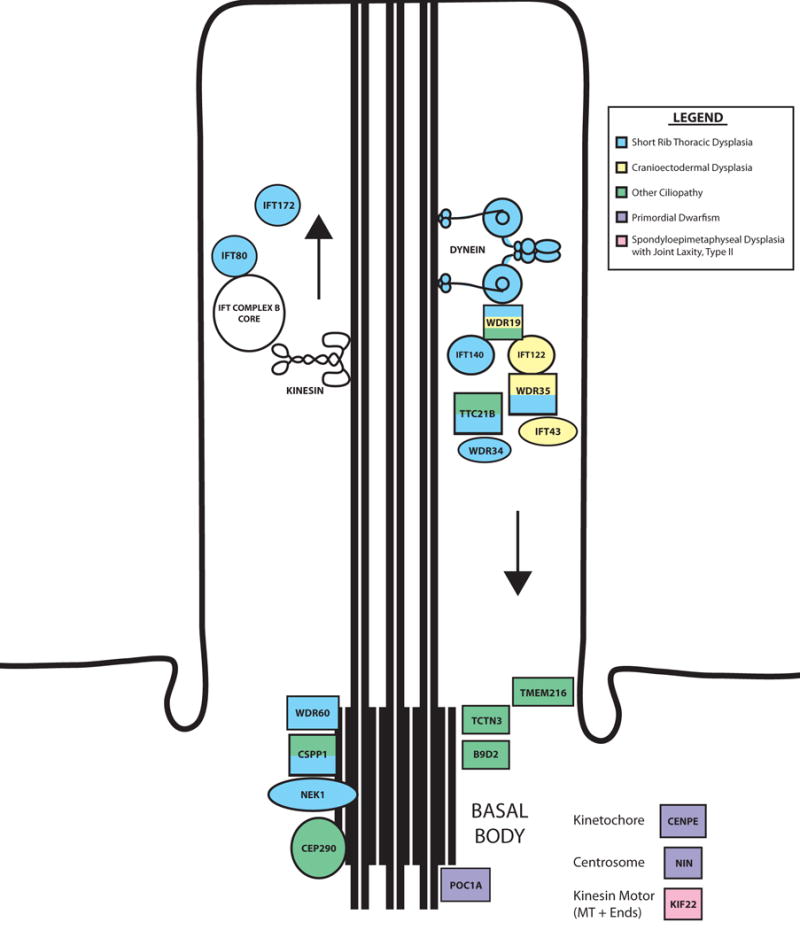
Components of the primary cilium are disrupted in many forms of skeletal dysplasia. These components include genes that encode parts of motor proteins (e.g., dynein) and parts of both the IFT-A and IFT-B complexes, which regulate retrograde and anterograde ciliary trafficking, respectively. The colors of the cilium components signify the types of disorders with which they are associated.
The primary cilium maintains growth plate architecture and organization (132). Chondrocytes are normally organized in columns within the proliferative zone. As the chondrocytes divide, the daughter cells divide perpendicular to the column and spread over one another to align within the column (84, 118a). This process, called chondrocyte rotation (132), is regulated by WNT signaling through the planar-cell polarity pathway (84, 119). Mouse models of skeletal ciliopathies have disorganized growth plates that underlie the growth insufficiency. The disorganized growth plate may arise from perturbation of cell polarity (WNT) and/or the rate of growth plate chondrocyte proliferation and hypertrophic differentiation (IHH), or through some other means. Mouse models of skeletal ciliopathies should help clarify the role of the primary cilium in the development of growth insufficiency.
Many genes implicated in skeletal ciliopathies regulate intraflagellar transport (IFT), the mechanism responsible for trafficking components into (IFT complex B) or out of (IFT complex A) the cilium (Figure 3). Kinesin motors carry IFT-B components, whereas dynein motors carry IFT-A components. This trafficking system ensures that the cilium is maintained and lengthened and that receptors and downstream effectors of signaling pathways are targeted properly to the cilium. Mutation of basal body–associated genes (Figure 3) could cause disruptions in primary cilium formation and maintenance or affect mitotic spindle formation and orientation.
Mutations in NEK1, a basal body–associated component of the cilium, cause short-rib polydactyly syndrome type Majewski (135). The never-in-mitosis gene A–related kinases (NEKs) constitute a large gene family composed of 11 members (NEK1–NEK11) (43). NEKs regulate microtubule dynamics, cell cycle progression, and ciliogenesis. Mutations in other NEK genes may cause skeletal dysplasia (including but not limited to primordial dwarfisms), classical ciliopathies, or cancer. Several other gene families, including WDR and CEP, are associated largely with the primary cilium and are excellent candidates for human disease genes and worthy of functional analysis in model organisms.
The newest potential member of the skeletal ciliopathies is a novel form of primordial dwarfism caused by mutation of POC1A, encoding protein of centriole 1A. Patients are smaller than average size in utero and remain so throughout their lives (121, 124). Two homozygous mutations were found in highly consanguineous families: a premature stop codon (R81X) (124) and a missense mutation (L171P) (121). Surprisingly, the premature stop codon undergoes efficient read-through in patient fibroblast cultures, and POC1A protein levels are reduced by only 15–50% (124). The fibroblasts have abnormal mitotic spindle orientation, form multipolar spindles, and have multiple centrosomes in noncycling cells. The frequency of primary cilium detection is only approximately half of the control, and the cilia that do form are approximately two-thirds normal size. The missense mutation is a nonconservative change in a region conserved from mammals to algae. Skin fibroblasts from these patients exhibit centrosome amplification (124) and alterations in cholera toxin trafficking (121). There are shared and distinct clinical features between the two families.
The effects both mutations have on cilia and centrosomes may be sufficient to produce the growth insufficiency. Two lines of evidence support this idea. First, knockdown of both POC1A and the related POC1B gene in HeLa cells alters the orientation of the mitotic spindle, causing unequal bipolar, monopolar, or even multipolar spindles (143). These knockdowns cause failure of daughter centrioles to mature into new mother centrioles. The lack of mother centrioles could affect the cells’ abilities to form primary cilia, as the mother centriole is responsible for the formation of the basal body and the extension of the ciliary axoneme (62). This could affect growth plate organization and signaling (132, 147), leading to severe short stature. The defects noted in mitotic spindles could affect orientation of chondrocyte cell division, leading to improper chondrocyte placement and faulty rotation. Additionally, mutations in POC1A, ORC1, CENPJ, and PCNT cause very similar growth deficiencies and microcephaly, and all of these genes encode proteins associated with the centrosome (3, 47, 57, 117, 133, 143).
It will be interesting to determine whether defects in the other primordial dwarfism genes cause the cilium defects similar to those observed in cells from POC1A patients, or whether, like ORC1 mutations, the cells form relatively normal cilia but at a slower rate (133). Histology of the growth plates in skeletal ciliopathy mouse models could determine which genes cause disorganization of the growth plate, and primary fibroblasts could be used to study the primary cilium. Both approaches will clarify the roles of these genes in the development of postnatal growth retardation in skeletal ciliopathy patients, a class that may include some forms of primordial dwarfism. These analyses could also shed light on the potential connection between the primary cilium, planar-cell polarity, and components of the cell cycle such as ORC1 that are linked to the centrosome/primary cilium (53, 57, 133). Although there is clearly a role for both the planar-cell polarity pathway (43a, 84, 119, 147a) and the primary cilium (132) in skeletal development and growth, their functional relationship is intensely debated (12a, 147). Clearly, there is much work to be done to clarify these associations, but the data collected will certainly be of clinical and basic significance.
NOVEL SKELETAL DISORDERS OF UNKNOWN ETIOLOGY
Although genetic causes of the >450 forms of skeletal disorders (149) are being uncovered quite rapidly, distinct skeletal disorders are still being discovered with no known genetic etiology. We do not have the ability to cover all of these disorders here, but the best strategy for identifying the genetic etiology of these new disorders may be exome sequencing or genome sequencing. In sporadic cases, analysis of trios may reveal de novo mutations. In familial cases, a combination of exome sequencing and either genome sequencing or SNP typing could yield linkage information for prioritization of rare variants. Current trends indicate that the causative mutations for these novel disorders will be uncovered very soon, with mechanistic details and therapeutic avenues likely to follow closely behind.
SKELETAL DYSPLASIA GENETICS AND HUMAN HEIGHT VARIATION
Do mutations in skeletal dysplasia patients provide information relevant to height variation in the human population? The answer is probably yes, because some skeletal dysplasia–associated genes have already been implicated in human height. Two genome-wide association studies (GWAS) conducted on human height variation (77a, 152c) have revealed height-associated loci that partially overlap with genes known to cause skeletal disorders. A GWAS from 2010 reported that 21 of 241 genes (8.7%) are known to cause a human growth disorder in their height-associated genomic loci, including genes influencing the endocrine system and skeleton (77a). The more recent GWAS, published in 2014, found 697 variants that explain one-fifth of the height heritability in individuals of European ancestry (152c). Of the genes that are specifically mentioned in the article, 20% of them are associated with a human skeletal disorder, and the majority of them are in some way connected to the signaling pathways and/or cellular processes discussed elsewhere in this review (152c). Approximately 8% of these genes had no previous association with skeletal growth, meaning that GWAS could provide new loci for consideration in the genetic analysis of skeletal disorders (152c). An important next step is to identify functional variants in the GWAS-associated candidate genes and determine how genetic interactions between these loci contribute to human height variation.
FUTURE DIRECTIONS: HUMAN STUDIES, MOUSE MODELS, AND IN VITRO TECHNOLOGIES IMPACT SKELETAL DYSPLASIA RESEARCH
The contributions of genetics to understanding skeletal dysplasia begin by assigning a gene to a disorder and extend to understanding the pathophysiology of the disease and development of treatments. Receiving a molecular diagnosis often gives patients and their families relief because they can understand the cause, the progression of disease over a lifetime, and the risk for future pregnancies. Human disease gene discovery is a starting point for understanding the normal regulation of skeletal growth and for studying development in model systems. Mouse models of human skeletal dysplasias and development of chondrocyte cell lines have been invaluable for mechanistic insight that explains the clinical features at the molecular level. These data provide a foundation for developing and testing therapeutic interventions, completing the circle from bedside to bench and back to bedside.
Studies in mice have contributed to understanding the genetic basis of skeletal dysplasias, disease pathophysiology, and therapeutic testing (6, 7, 19, 35, 66, 81, 100, 104, 109, 131, 134, 144). The data collected from skeletal dysplasia patients are usually limited by the necessity for minimal invasiveness, and include radiographs; photographs; measurements of height, weight, and hormone levels; and isolation of skin fibroblasts, blood leukocytes, and genomic DNA. By contrast, mouse models provide researchers with accessible tissue throughout development, the potential to assess genetic interactions underlying digenic or oligogenic disease using complementation studies, and piloting treatments (44, 63, 64, 68, 87, 101, 130, 153, 158).
In many of these studies, the description of the mouse model was followed by reports of mutations in the orthologous human gene that cause similar skeletal abnormalities. For example, Smits et al. (131) recently found that an ethylnitrosourea (ENU)–induced mutation in mice is a model of achondrogenesis type 1A in humans. This autosomal recessive mutation caused perinatal lethality, severely disrupted endochondral ossification, greatly reduced the mineralization of skeletal elements, and disturbed the trafficking of extracellular matrix proteins, resulting in acute intracellular stress in affected chondrocytes. The mutant locus was mapped to mouse chromosome 12 using a SNP array and subsequently narrowed to a 3.7-Mb interval. Candidate gene screening led to the discovery of a nonsense mutation (c.5003T→A, p.L1668X) in Trip11, which encodes Golgi microtubule-associated protein 210 (GMAP-210). TRIP11 was evaluated as a candidate gene for achondrogenesis type 1A because of similarities in the mouse and human phenotypes. Indeed, 10 out of 10 achondrogenesis type 1A patients screened had mutations in TRIP11. Thus, the discovery of mouse mutations contributes significantly to understanding the genetic basis for skeletal dysplasias in humans.
In addition to mouse models, induced pluripotent stem cells (iPS cells) generated from skeletal dysplasia patients have the potential to serve the research community as a tool for drug discovery and design (107a, 154a). Successful generation of chondrocytes from dermal fibroblasts from mice and humans has been reported (53a, 109a, 109b), but more importantly, the analysis of these cells provides valuable insight into possible treatment regimes. In the case of type 2 collagen disorders, patient-derived dermal fibroblasts that either were stimulated to form chondrocytes directly or differentiated following an induction of pluripotency revealed that matrix-promoting treatments such as ascorbate worsen the cellular pathology (107a). However, treatment with the chemical chaperone trimethylamine N-oxide (TMAO) resulted in an amelioration of the cellular phenotype, making it an attractive therapeutic candidate (107a). Another study, in which iPS cells derived from a patient with achondroplasia and thanatophoric dysplasia type I were differentiated into chondrocytes, revealed a safe, effective therapy for these two forms of skeletal dysplasia: the statins (154a). Statin therapy in vitro rescued the cellular phenotypes, which included a reduced level of collagen 2 and other known chondrocyte markers. Remarkably, statin treatment also increased the longitudinal growth of Fgfr3ach mice. Although there are major caveats, such as the lack of true growth plate architecture in iPS-chondrocyte cultures, iPS cell–based strategies could provide a wealth of new information, including new therapeutic avenues, through large-scale drug screens.
Developing a therapy for achondroplasia is a top priority, given that it is the most common form (71), and it is one of the best examples of the progression from gene discovery to potential therapies in less than ~20 years. Mutations in FGFR3 cause a number of additional disorders (9), meaning that a treatment for achondroplasia could be an appropriate therapy for these other FGFR3-related disorders or vice versa. Interestingly, the discovery of mutations in NPR2 in acromesomelic dysplasia type Maroteaux (AMDM) patients (12) has helped shape the trajectory of this area of research, as work in cell culture and mouse models has revealed that the genetic and molecular etiologies of the two disorders are intertwined. Activation of NPR2 inhibits FGFR3-mediated activation of the MEK/ERK mitogen-activated protein kinase (MAPK) pathway (72), which allows the growth plate to promote chondrocyte differentiation. Loss of CNP-NPR2-mediated inhibition of FGF signaling in the mouse model of AMDM leads to overactivation of the MEK/ERK MAPK pathway (44), and treatment of chondrocytes with CNP leads to reduced ERK activation (72). Treating achondroplasia patients with CNP, the ligand for NPR2, was a rational therapy based on knowledge of the effect of the FGF mutation on MEK/ERK and the oppositional effects of CNP signaling. This therapy is somewhat successful in mouse models with various methods of continuous administration (156–158). Statins (154a), soluble “decoy” human FGFR3 (43c), PTH (153), FGFR3 inhibitors (63, 64), CNP (152a, 156–158), CNP analogs (87), and meclozine (87d) are all being explored as potential treatments for achondroplasia. The administration of MEK/ERK inhibitors has been postulated as an intervention in mouse models of Apert syndrome (gain-of-function mutation in FGFR2) (130) and AMDM (44). Although these therapies are still under development, it is promising that targeting these pathways has improved growth in vitro and even in vivo in some cases. Future research may involve identifying additional compounds that modulate this interconnected signaling network with greater specificity, and improving delivery of therapeutics to the growth plate. The progression from mutation discovery to therapies in achondroplasia should serve as a model for research on other forms of skeletal dysplasia.
CONCLUDING REMARKS
There has been remarkable progress recently in understanding the genetic and molecular etiologies of skeletal dysplasias. Genetic causes of fifty-six disorders have been uncovered (Table 1). The speed and volume of these discoveries lead us to propose the construction of a Database of Nosology and Classification of Genetic Skeletal Disorders, where new information can be submitted as it is published. Such a database could be expanded to other types of growth disorders, providing a valuable resource for clinical, basic, and translational research. Finally, the progress made in therapeutics through in vitro and in vivo work represents a spectacularly promising start toward developing therapies for all skeletal disorders.
Acknowledgments
We acknowledge support from a Regents’ Fellowship for Cellular and Molecular Biology (2010) and a University of Michigan Reproductive Sciences Program Fellowship (2012) (to K.A.G.) along with National Institutes of Health grants R01 HD030428-20 and HD034283-16 (to S.A.C.). We thank Matt Warman (Harvard) and Miriam Meisler and Gregory Dressler (both University of Michigan) for advice on the manuscript.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- 1a.Ahmad NN, Ala-Kokko L, Knowlton RG, Jimenez SA, Weaver EJ, et al. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy) PNAS. 1991;88:6624–27. doi: 10.1073/pnas.88.15.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akizu N, Silhavy JL, Rosti RO, Scott E, Fenstermaker AG, et al. Mutations in CSPP1 lead to classical Joubert syndrome. Am J Hum Genet. 2014;94:80–86. doi: 10.1016/j.ajhg.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS. Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47:411–14. doi: 10.1136/jmg.2009.076646. [DOI] [PubMed] [Google Scholar]
- 3a.Allen DB, Cuttler L. Clinical practice. Short stature in childhood—challenges and choices. N Engl J Med. 2013;368:1220–1228. doi: 10.1056/NEJMcp1213178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. 2012;44:788–92. doi: 10.1038/ng.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–58. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 7.Arikawa-Hirasawa E, Wilcox WR, Le AH, Silverman N, Govindraj P, et al. Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat Genet. 2001;27:431–34. doi: 10.1038/86941. [DOI] [PubMed] [Google Scholar]
- 7a.Asharani PV, Keupp K, Semler O, Wang W, Li Y, et al. Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. Am J Hum Genet. 2012;90:661–74. doi: 10.1016/j.ajhg.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker S, Booth C, Fillman C, Shapiro M, Blair MP, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A. 2011;155A:1668–72. doi: 10.1002/ajmg.a.34071. [DOI] [PubMed] [Google Scholar]
- 9.Baldridge D, Shchelochkov O, Kelley B, Lee B. Signaling pathways in human skeletal dysplasias. Annu Rev Genomics Hum Genet. 2010;11:189–217. doi: 10.1146/annurev-genom-082908-150158. [DOI] [PubMed] [Google Scholar]
- 10.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 10a.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b.Baple EL, Chambers H, Cross HE, Fawcett H, Nakazawa Y, et al. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J Clin Investig. 2014;124:3137–46. doi: 10.1172/JCI74593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartels CF, Bukulmez H, Padayatti P, Rhee DK, van Ravenswaaij-Arts C, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet. 2004;75:27–34. doi: 10.1086/422013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.Basten SG, Giles RH. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia. 2013;2:6. doi: 10.1186/2046-2530-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82. doi: 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beier F, Loeser RF. Biology and pathology of Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in chondrocytes. J Cell Biochem. 2010;110:573–80. doi: 10.1002/jcb.22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Below JE, Earl DL, Shively KM, McMillin MJ, Smith JD, et al. Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am J Hum Genet. 2013;92:137–43. doi: 10.1016/j.ajhg.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15a.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nature structural & molecular biology. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 16.Bicknell LS, Bongers EM, Leitch A, Brown S, Schoots J, et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet. 2011;43:356–59. doi: 10.1038/ng.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bicknell LS, Walker S, Klingseisen A, Stiff T, Leitch A, et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet. 2011;43:350–55. doi: 10.1038/ng.776. [DOI] [PubMed] [Google Scholar]
- 18.Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151–57. doi: 10.1038/sj.ejhg.5201638. [DOI] [PubMed] [Google Scholar]
- 19.Bonadio J, Jepsen KJ, Mansoura MK, Jaenisch R, Kuhn JL, Goldstein SA. A murine skeletal adaptation that significantly increases cortical bone mechanical properties. Implications for human skeletal fragility. J Clin Investig. 1993;92:1697–705. doi: 10.1172/JCI116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bongers EM, Opitz JM, Fryer A, Sarda P, Hennekam RC, et al. Meier-Gorlin syndrome: report of eight additional cases and review. Am J Med Genet. 2001;102:115–24. doi: 10.1002/ajmg.1452. [DOI] [PubMed] [Google Scholar]
- 21.Boyden ED, Campos-Xavier AB, Kalamajski S, Cameron TL, Suarez P, et al. Recurrent dominant mutations affecting two adjacent residues in the motor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity. Am J Hum Genet. 2011;89:767–72. doi: 10.1016/j.ajhg.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89:634–43. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22a.Bui C, Huber C, Tuysuz B, Alanay Y, Bole-Feysot C, et al. XYLT1 mutations in Desbuquois dysplasia type 2. Am J Hum Genet. 2014;94:405–14. doi: 10.1016/j.ajhg.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campeau PM, Kim JC, Lu JT, Schwartzentruber JA, Abdul-Rahman OA, et al. Mutations in KAT6B, encoding a histone acetyltransferase, cause genitopatellar syndrome. Am J Hum Genet. 2012;90:282–89. doi: 10.1016/j.ajhg.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campeau PM, Lenk GM, Lu JT, Bae Y, Burrage L, et al. Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet. 2013;92:781–91. doi: 10.1016/j.ajhg.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caux F, Plauchu H, Chibon F, Faivre L, Fain O, et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet. 2007;15:767–73. doi: 10.1038/sj.ejhg.5201823. [DOI] [PubMed] [Google Scholar]
- 26.Celli J, van Bokhoven H, Brunner HG. Feingold syndrome: clinical review and genetic mapping. Am J Med Genet A. 2003;122A:294–300. doi: 10.1002/ajmg.a.20471. [DOI] [PubMed] [Google Scholar]
- 26a.Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91:343–48. doi: 10.1016/j.ajhg.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clayton-Smith J, O’Sullivan J, Daly S, Bhaskar S, Day R, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89:675–81. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Courtens W, Levi S, Verbelen F, Verloes A, Vamos E. Feingold syndrome: report of a new family and review. Am J Med Genet. 1997;73:55–60. [PubMed] [Google Scholar]
- 29.Dauber A, Lafranchi SH, Maliga Z, Lui JC, Moon JE, et al. Novel microcephalic primordial dwarfism disorder associated with variants in the centrosomal protein ninein. J Clin Endocrinol Metab. 2012;97:E2140–51. doi: 10.1210/jc.2012-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43(3):189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Munnik SA, Bicknell LS, Aftimos S, Al-Aama JY, van Bever Y, et al. Meier-Gorlin syndrome genotype-phenotype studies: 35 individuals with pre-replication complex gene mutations and 10 without molecular diagnosis. Eur J Hum Genet. 2012;20:598–606. doi: 10.1038/ejhg.2011.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Pontual L, Yao E, Callier P, Faivre L, Drouin V, et al. Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat Genet. 2011;43:1026–30. doi: 10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–21. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 33.Dickson LA, Pihlajaniemi T, Deak S, Pope FM, Nicholls A, et al. Nuclease S1 mapping of a homozygous mutation in the carboxyl-propeptide-coding region of the pro alpha 2(I) collagen gene in a patient with osteogenesis imperfecta. PNAS. 1984;81:4524–28. doi: 10.1073/pnas.81.14.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dighe M, Fligner C, Cheng E, Warren B, Dubinsky T. Fetal skeletal dysplasia: an approach to diagnosis with illustrative cases. Radiographics. 2008;28:1061–77. doi: 10.1148/rg.284075122. [DOI] [PubMed] [Google Scholar]
- 35.Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SG, et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet. 2011;89:94–110. doi: 10.1016/j.ajhg.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 37.Edery P, Marcaillou C, Sahbatou M, Labalme A, Chastang J, et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science. 2011;332:240–43. doi: 10.1126/science.1202205. [DOI] [PubMed] [Google Scholar]
- 38.Favaro FP, Alvizi L, Zechi-Ceide RM, Bertola D, Felix TM, et al. A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am J Hum Genet. 2014;94:120–28. doi: 10.1016/j.ajhg.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feingold M, Hall BD, Lacassie Y, Martinez-Frias ML. Syndrome of microcephaly, facial and hand abnormalities, tracheoesophageal fistula, duodenal atresia, and developmental delay. Am J Med Genet. 1997;69:245–49. [PubMed] [Google Scholar]
- 40.Ford-Hutchinson AF, Ali Z, Lines SE, Hallgrimsson B, Boyd SK, Jirik FR. Inactivation of Pten in osteo-chondroprogenitor cells leads to epiphyseal growth plate abnormalities and skeletal overgrowth. J Bone Miner Res. 2007;22:1245–59. doi: 10.1359/jbmr.070420. [DOI] [PubMed] [Google Scholar]
- 41.Francomano CA, Liberfarb RM, Hirose T, Maumenee IH, Streeten EA, et al. The Stickler syndrome: evidence for close linkage to the structural gene for type II collagen. Genomics. 1987;1:293–96. doi: 10.1016/0888-7543(87)90027-9. [DOI] [PubMed] [Google Scholar]
- 42.Francomano CA, Ortiz de Luna RI, Hefferon TW, Bellus GA, Turner CE, et al. Localization of the achondroplasia gene to the distal 2.5 Mb of human chromosome 4p. Hum Mol Genet. 1994;3:787–92. doi: 10.1093/hmg/3.5.787. [DOI] [PubMed] [Google Scholar]
- 43.Fry AM, O’Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci. 2012;125:4423–33. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43a.Gao B, Song H, Bishop K, Elliot G, Garrett L, et al. Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev Cell. 2011;20:163–76. doi: 10.1016/j.devcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43b.Garbes L, Kim K, Riess A, Hoyer-Kuhn H, Beleggia F, et al. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet. 2015;96:432–39. doi: 10.1016/j.ajhg.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43c.Garcia S, Dirat B, Tognacci T, Rochet N, Mouska X, et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci Transl Med. 2013;5:203ra124. doi: 10.1126/scitranslmed.3006247. [DOI] [PubMed] [Google Scholar]
- 44.Geister KA, Brinkmeier ML, Hsieh M, Faust SM, Karolyi IJ, et al. A novel loss-of-function mutation in Npr2 clarifies primary role in female reproduction and reveals a potential therapy for acromesomelic dysplasia, Maroteaux type. Hum Mol Genet. 2013;22:345–57. doi: 10.1093/hmg/dds432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glazov EA, Zankl A, Donskoi M, Kenna TJ, Thomas GP, et al. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLOS Genet. 2011;7:e1002027. doi: 10.1371/journal.pgen.1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45a.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–44. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet. 2012;90:175–200. doi: 10.1016/j.ajhg.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet. 2008;40:232–36. doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47a.Grosch M, Gruner B, Spranger S, Stutz A, Rausch T, et al. Identification of a Ninein (NIN) mutation in a family with spondyloepimetaphyseal dysplasia with joint laxity (leptodactylic type)-like phenotype. Mat Bio. 2013;32:387–392. doi: 10.1016/j.matbio.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 47b.Growth Hormone Research, S. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab. 2000;85:3990–3993. doi: 10.1210/jcem.85.11.6984. [DOI] [PubMed] [Google Scholar]
- 49.Guernsey DL, Matsuoka M, Jiang H, Evans S, Macgillivray C, et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat Genet. 2011;43:360–64. doi: 10.1038/ng.777. [DOI] [PubMed] [Google Scholar]
- 50.Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93:915–25. doi: 10.1016/j.ajhg.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 52.He H, Liyanarachchi S, Akagi K, Nagy R, Li J, et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science. 2011;332:238–40. doi: 10.1126/science.1200587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hemerly AS, Prasanth SG, Siddiqui K, Stillman B. Orc1 controls centriole and centrosome copy number in human cells. Science. 2009;323:789–93. doi: 10.1126/science.1166745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53a.Hiramatsu K, Sasagawa S, Outani H, Nakagawa K, Yoshikawa H, Tsumaki N. Generation of hyaline cartilaginous tissue from mouse adult dermal fibroblast culture by defined factors. J Clin Investig. 2011;121:640–57. doi: 10.1172/JCI44605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirata M, Kugimiya F, Fukai A, Ohba S, Kawamura N, et al. C/EBPβ promotes transition from proliferation to hypertrophic differentiation of chondrocytes through transactivation of p57Kip2. PLOS ONE. 2009;4:e4543. doi: 10.1371/journal.pone.0004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoover-Fong J, Sobreira N, Jurgens J, Modaff P, Blout C, et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94:105–12. doi: 10.1016/j.ajhg.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162–72. doi: 10.1016/S0140-6736(07)61090-3. [DOI] [PubMed] [Google Scholar]
- 57.Hossain M, Stillman B. Meier-Gorlin syndrome mutations disrupt an Orc1 CDK inhibitory domain and cause centrosome reduplication. Genes Dev. 2012;26:1797–810. doi: 10.1101/gad.197178.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsieh SC, Chen NT, Lo SH. Conditional loss of PTEN leads to skeletal abnormalities and lipoma formation. Mol Carcinog. 2009;48:545–52. doi: 10.1002/mc.20491. [DOI] [PubMed] [Google Scholar]
- 59.Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C. 2012;160C:165–74. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 60.Huber C, Faqeih EA, Bartholdi D, Bole-Feysot C, Borochowitz Z, et al. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am J Hum Genet. 2013;92:144–49. doi: 10.1016/j.ajhg.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huber C, Wu S, Kim AS, Sigaudy S, Sarukhanov A, et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia. Am J Hum Genet. 2013;93:926–31. doi: 10.1016/j.ajhg.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61a.Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, et al. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development. 2011;138:1507–19. doi: 10.1242/dev.057802. [DOI] [PubMed] [Google Scholar]
- 62.Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol. 2011;12:222–34. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- 63.Jin M, Yu Y, Qi H, Xie Y, Su N, et al. A novel FGFR3-binding peptide inhibits FGFR3 signaling and reverses the lethal phenotype of mice mimicking human thanatophoric dysplasia. Hum Mol Genet. 2012;21:5443–55. doi: 10.1093/hmg/dds390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jonquoy A, Mugniery E, Benoist-Lasselin C, Kaci N, Le Corre L, et al. A novel tyrosine kinase inhibitor restores chondrocyte differentiation and promotes bone growth in a gain-of-function Fgfr3 mouse model. Hum Mol Genet. 2012;21:841–51. doi: 10.1093/hmg/ddr514. [DOI] [PubMed] [Google Scholar]
- 65.Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43:23–26. doi: 10.1038/ng.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–89. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 67.Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu Rev Cell Dev Biol. 2009;25:629–48. doi: 10.1146/annurev.cellbio.042308.113308. [DOI] [PubMed] [Google Scholar]
- 68.Kawasaki Y, Kugimiya F, Chikuda H, Kamekura S, Ikeda T, et al. Phosphorylation of GSK-3β by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes. J Clin Investig. 2008;118:2506–15. doi: 10.1172/JCI35243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68a.Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92:565–74. doi: 10.1016/j.ajhg.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klingseisen A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 2011;25:2011–24. doi: 10.1101/gad.169037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–60. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med. 2010;12:327–41. doi: 10.1097/GIM.0b013e3181daae9b. [DOI] [PubMed] [Google Scholar]
- 72.Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, et al. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci. 2005;118:5089–100. doi: 10.1242/jcs.02618. [DOI] [PubMed] [Google Scholar]
- 73.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–36. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 74.Kronenberg HM. PTHrP and skeletal development. Ann NY Acad Sci. 2006;1068:1–13. doi: 10.1196/annals.1346.002. [DOI] [PubMed] [Google Scholar]
- 75.Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012;484:115–19. doi: 10.1038/nature10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lachman RS, Tiller GE, Graham JM, Jr, Rimoin DL. Collagen, genes and the skeletal dysplasias on the edge of a new era: a review and update. Eur J Radiol. 1992;14:1–10. doi: 10.1016/0720-048x(92)90052-b. [DOI] [PubMed] [Google Scholar]
- 77a.Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–38. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2012;44:85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 79.Le Goff C, Mahaut C, Wang LW, Allali S, Abhyankar A, et al. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. doi: 10.1016/j.ajhg.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Le Merrer M, Rousseau F, Legeai-Mallet L, Landais JC, Pelet A, et al. A gene for achondroplasia-hypochondroplasia maps to chromosome 4p. Nat Genet. 1994;6:318–21. doi: 10.1038/ng0394-318. [DOI] [PubMed] [Google Scholar]
- 81.Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–10. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 82.Lee B, Vissing H, Ramirez F, Rogers D, Rimoin D. Identification of the molecular defect in a family with spondyloepiphyseal dysplasia. Science. 1989;244:978–80. doi: 10.1126/science.2543071. [DOI] [PubMed] [Google Scholar]
- 83.Lee H, Graham JM, Jr, Rimoin DL, Lachman RS, Krejci P, et al. Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet. 2012;90:746–51. doi: 10.1016/j.ajhg.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y, Dudley AT. Noncanonical frizzled signaling regulates cell polarity of growth plate chondrocytes. Development. 2009;136:1083–92. doi: 10.1242/dev.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–19. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, et al. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med. 2011;364:2218–26. doi: 10.1056/NEJMoa1012717. [DOI] [PubMed] [Google Scholar]
- 87.Lorget F, Kaci N, Peng J, Benoist-Lasselin C, Mugniery E, et al. Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia. Am J Hum Genet. 2012;91:1108–14. doi: 10.1016/j.ajhg.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87a.Marini JC, Blissett AR. New genes in bone development: what’s new in osteogenesis imperfecta. J Clin Endocrinol Metab. 2013;98:3095–103. doi: 10.1210/jc.2013-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87b.Martin CA, Ahmad I, Klingseisen A, Hussain MS, Bicknell LS, et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet. 2014;46:1283–92. doi: 10.1038/ng.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87c.Martinez-Glez V, Valencia M, Caparros-Martin JA, Aglan M, Temtamy S, et al. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33:343–50. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87d.Matsushita M, Kitoh H, Ohkawara B, Mishima K, Kaneko H, et al. Meclozine facilitates proliferation and differentiation of chondrocytes by attenuating abnormally activated FGFR3 signaling in achondroplasia. PLOS ONE. 2013;8:e81569. doi: 10.1371/journal.pone.0081569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McInerney-Leo AM, Schmidts M, Cortes CR, Leo PJ, Gener B, et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet. 2013;93:515–23. doi: 10.1016/j.ajhg.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mehawej C, Delahodde A, Legeai-Mallet L, Delague V, Kaci N, et al. The impairment of MAGMAS function in human is responsible for a severe skeletal dysplasia. PLOS Genet. 2014;10:e1004311. doi: 10.1371/journal.pgen.1004311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Merrill AE, Sarukhanov A, Krejci P, Idoni B, Camacho N, et al. Bent bone dysplasia-FGFR2 type, a distinct skeletal disorder, has deficient canonical FGF signaling. Am J Hum Genet. 2012;90:550–57. doi: 10.1016/j.ajhg.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 93.Michot C, Le Goff C, Goldenberg A, Abhyankar A, Klein C, et al. Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet. 2012;90:740–45. doi: 10.1016/j.ajhg.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet. 2011;88:508–15. doi: 10.1016/j.ajhg.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Min BJ, Kim N, Chung T, Kim OH, Nishimura G, et al. Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am J Hum Genet. 2011;89:760–66. doi: 10.1016/j.ajhg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mirzaa GM, Vitre B, Carpenter G, Abramowicz I, Gleeson JG, et al. Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum Genet. 2014;133:1023–39. doi: 10.1007/s00439-014-1443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moens CB, Auerbach AB, Conlon RA, Joyner AL, Rossant J. A targeted mutation reveals a role for N-myc in branching morphogenesis in the embryonic mouse lung. Genes Dev. 1992;6:691–704. doi: 10.1101/gad.6.5.691. [DOI] [PubMed] [Google Scholar]
- 98.Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA, Alkuraya FS. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am J Hum Genet. 2013;92:157–61. doi: 10.1016/j.ajhg.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111:63–72. doi: 10.1016/j.ymgme.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 100.Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–79. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 101.Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102a.Murray JE, van der Burg M, H IJ, Carroll P, Wu Q, et al. Mutations in the NHEJ component XRCC4 cause primordial dwarfism. Am J Hum Genet. 2015;96:412–24. doi: 10.1016/j.ajhg.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nagy A, Moens C, Ivanyi E, Pawling J, Gertsenstein M, et al. Dissecting the role of N-myc in development using a single targeting vector to generate a series of alleles. Curr Biol. 1998;8:661–64. doi: 10.1016/s0960-9822(98)70254-4. [DOI] [PubMed] [Google Scholar]
- 104.Nicole S, Davoine CS, Topaloglu H, Cattolico L, Barral D, et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia) Nat Genet. 2000;26:480–83. doi: 10.1038/82638. [DOI] [PubMed] [Google Scholar]
- 105.Nigg EA, Stearns T. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–60. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105a.Nundlall S, Rajpar MH, Bell PA, Clowes C, Zeeff LA, et al. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones. 2010;15:835–49. doi: 10.1007/s12192-010-0193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 107.Ogi T, Walker S, Stiff T, Hobson E, Limsirichaikul S, et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLOS Genet. 2012;8:e1002945. doi: 10.1371/journal.pgen.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107a.Okada M, Ikegawa S, Morioka M, Yamashita A, Saito A, et al. Modeling type II collagenopathy skeletal dysplasia by directed conversion and induced pluripotent stem cells. Hum Mol Genet. 2015;24:299–313. doi: 10.1093/hmg/ddu444. [DOI] [PubMed] [Google Scholar]
- 108.Ota S, Zhou ZQ, Keene DR, Knoepfler P, Hurlin PJ. Activities of N-Myc in the developing limb link control of skeletal size with digit separation. Development. 2007;134:1583–92. doi: 10.1242/dev.000703. [DOI] [PubMed] [Google Scholar]
- 109.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 109a.Outani H, Okada M, Hiramatsu K, Yoshikawa H, Tsumaki N. Induction of chondrogenic cells from dermal fibroblast culture by defined factors does not involve a pluripotent state. Biochem Biophys Res Commun. 2011;411:607–12. doi: 10.1016/j.bbrc.2011.06.194. [DOI] [PubMed] [Google Scholar]
- 109b.Outani H, Okada M, Yamashita A, Nakagawa K, Yoshikawa H, Tsumaki N. Direct induction of chondrogenic cells from human dermal fibroblast culture by defined factors. PLOS ONE. 2013;8:e77365. doi: 10.1371/journal.pone.0077365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pansuriya TC, van Eijk R, d’Adamo P, van Ruler MA, Kuijjer ML, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–61. doi: 10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110a.Payne F, Colnaghi R, Rocha N, Seth A, Harris J, et al. Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J Clin Investig. 2014;124:4028–38. doi: 10.1172/JCI73264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, et al. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet. 2012;90:864–70. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M. The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol. 2000;149:317–30. doi: 10.1083/jcb.149.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pihlajaniemi T, Dickson LA, Pope FM, Korhonen VR, Nicholls A, et al. Osteogenesis imperfecta: cloning of a pro-alpha 2(I) collagen gene with a frameshift mutation. J Biol Chem. 1984;259:12941–44. [PubMed] [Google Scholar]
- 116.Qvist P, Huertas P, Jimeno S, Nyegaard M, Hassan MJ, et al. CtIP mutations cause Seckel and Jawad syndromes. PLOS Genet. 2011;7:e1002310. doi: 10.1371/journal.pgen.1002310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116a.Rajpar MH, McDermott B, Kung L, Eardley R, Knowles L, et al. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLOS Genet. 2009;5:e1000691. doi: 10.1371/journal.pgen.1000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–19. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 118.Rios JJ, Paria N, Burns DK, Israel BA, Cornelia R, et al. Somatic gain-of-function mutations in PIK3CA in patients with macrodactyly. Hum Mol Genet. 2013;22:444–51. doi: 10.1093/hmg/dds440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118a.Romereim SM, Conoan NH, Chen B, Dudley AT. A dynamic cell adhesion surface regulates tissue architecture in growth plate cartilage. Development. 2014;141:2085–95. doi: 10.1242/dev.105452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Romereim SM, Dudley AT. Cell polarity: the missing link in skeletal morphogenesis? Organogenesis. 2011;7:217–28. doi: 10.4161/org.7.3.18583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–54. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 121.Sarig O, Nahum S, Rapaport D, Ishida-Yamamoto A, Fuchs-Telem D, et al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. Am J Hum Genet. 2012;91:337–42. doi: 10.1016/j.ajhg.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Savarirayan R, Rimoin DL. The skeletal dysplasias. Best Pract Res Clin Endocrinol Metab. 2002;16:547–60. doi: 10.1053/beem.2002.0210. [DOI] [PubMed] [Google Scholar]
- 123.Schmidts M, Vodopiutz J, Christou-Savina S, Cortes CR, McInerney-Leo AM, et al. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2013;93:932–44. doi: 10.1016/j.ajhg.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123a.Schreml J, Durmaz B, Cogulu O, Keupp K, Beleggia F, et al. The missing “link”: an autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum Genet. 2014;133:29–39. doi: 10.1007/s00439-013-1351-y. [DOI] [PubMed] [Google Scholar]
- 123b.Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, et al. A mutation in the 5´-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91:349–57. doi: 10.1016/j.ajhg.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123c.Shaheen R, Al Tala S, Almoisheer A, Alkuraya FS. Mutation in PLK4, encoding a master regulator of centriole formation, defines a novel locus for primordial dwarfism. J Med Genet. 2014;51:814–16. doi: 10.1136/jmedgenet-2014-102790. [DOI] [PubMed] [Google Scholar]
- 123d.Shaheen R, Alazami AM, Alshammari MJ, Faqeih E, Alhashmi N, et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet. 2012;49:630–35. doi: 10.1136/jmedgenet-2012-101142. [DOI] [PubMed] [Google Scholar]
- 124.Shaheen R, Faqeih E, Shamseldin HE, Noche RR, Sunker A, et al. POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am J Hum Genet. 2012;91:330–36. doi: 10.1016/j.ajhg.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124a.Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet. 2015;24:1410–19. doi: 10.1093/hmg/ddu555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, et al. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet. 2014;94:73–79. doi: 10.1016/j.ajhg.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shamseldin HE, Rajab A, Alhashem A, Shaheen R, Al-Shidi T, et al. Mutations in DDX59 implicate RNA helicase in the pathogenesis of orofaciodigital syndrome. Am J Hum Genet. 2013;93:555–60. doi: 10.1016/j.ajhg.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–42. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 130.Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39:1145–50. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- 131.Smits P, Bolton AD, Funari V, Hong M, Boyden ED, et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N Engl J Med. 2010;362:206–16. doi: 10.1056/NEJMoa0900158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Song B, Haycraft CJ, Seo HS, Yoder BK, Serra R. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev Biol. 2007;305:202–16. doi: 10.1016/j.ydbio.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stiff T, Alagoz M, Alcantara D, Outwin E, Brunner HG, et al. Deficiency in origin licensing proteins impairs cilia formation: implications for the aetiology of Meier-Gorlin syndrome. PLOS Genet. 2013;9:e1003360. doi: 10.1371/journal.pgen.1003360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Storm EE, Huynh TV, Copeland NG, Jenkins NA, Kingsley DM, Lee SJ. Limb alterations in brachypodism mice due to mutations in a new member of the TGFβ-superfamily. Nature. 1994;368:639–43. doi: 10.1038/368639a0. [DOI] [PubMed] [Google Scholar]
- 134a.Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014;95:96–107. doi: 10.1016/j.ajhg.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134b.Symoens S, Malfait F, D’Hondt S, Callewaert B, Dheedene A, et al. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis. 2013;8:154. doi: 10.1186/1750-1172-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, et al. NEK1 mutations cause short-rib polydactyly syndrome type Majewski. Am J Hum Genet. 2011;88:106–14. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thomas S, Legendre M, Saunier S, Bessieres B, Alby C, et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am J Hum Genet. 2012;91:372–78. doi: 10.1016/j.ajhg.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136a.Tsang KY, Chan D, Cheslett D, Chan WC, So CL, et al. Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLOS Biol. 2007;5:e44. doi: 10.1371/journal.pbio.0050044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsipouras P, Myers JC, Ramirez F, Prockop DJ. Restriction fragment length polymorphism associated with the pro alpha 2(I) gene of human type I procollagen. Application to a family with an autosomal dominant form of osteogenesis imperfecta. J Clin Investig. 1983;72:1262–67. doi: 10.1172/JCI111082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tunkel D, Alade Y, Kerbavaz R, Smith B, Rose-Hardison D, Hoover-Fong J. Hearing loss in skeletal dysplasia patients. Am J Med Genet A. 2012;158A:1551–55. doi: 10.1002/ajmg.a.35373. [DOI] [PubMed] [Google Scholar]
- 139.Tuz K, Bachmann-Gagescu R, O’Day DR, Hua K, Isabella CR, et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2014;94:62–72. doi: 10.1016/j.ajhg.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139a.Ulici V, James CG, Hoenselaar KD, Beier F. Regulation of gene expression by PI3K in mouse growth plate chondrocytes. PLOS ONE. 2010;5:e8866. doi: 10.1371/journal.pone.0008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139b.Unlu G, Levic DS, Melville DB, Knapik EW. Trafficking mechanisms of extracellular matrix macromolecules: insights from vertebrate development and human diseases. Int J Biochem Cell Biol. 2014;47:57–67. doi: 10.1016/j.biocel.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–25. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van Bokhoven H, Celli J, van Reeuwijk J, Rinne T, Glaudemans B, et al. MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat Genet. 2005;37:465–67. doi: 10.1038/ng1546. [DOI] [PubMed] [Google Scholar]
- 142.Velinov M, Slaugenhaupt SA, Stoilov I, Scott CI, Jr, Gusella JF, Tsipouras P. The gene for achondroplasia maps to the telomeric region of chromosome 4p. Nat Genet. 1994;6:314–17. doi: 10.1038/ng0394-314. [DOI] [PubMed] [Google Scholar]
- 143.Venoux M, Tait X, Hames RS, Straatman KR, Woodland HR, Fry AM. Poc1A and Poc1B act together in human cells to ensure centriole integrity. J Cell Sci. 2013;126:163–75. doi: 10.1242/jcs.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vilain E, Le Merrer M, Lecointre C, Desangles F, Kay MA, et al. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J Clin Endocrinol Metab. 1999;84:4335–40. doi: 10.1210/jcem.84.12.6186. [DOI] [PubMed] [Google Scholar]
- 146.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–22. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 147.Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–13. doi: 10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147a.Wang B, Sinha T, Jiao K, Serra R, Wang J. Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Hum Mol Genet. 2011;20:271–85. doi: 10.1093/hmg/ddq462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang X, Tsai JW, Imai JH, Lian WN, Vallee RB, Shi SH. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 2009;461:947–55. doi: 10.1038/nature08435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148a.Wang Y, Wysocka J, Perlin JR, Leonelli L, Allis CD, Coonrod SA. Linking covalent histone modifications to epigenetics: the rigidity and plasticity of the marks. Cold Spring Harbor symposia on quantitative biology. 2004;69:161–169. doi: 10.1101/sqb.2004.69.161. [DOI] [PubMed] [Google Scholar]
- 149.Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–68. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Webster MK, D’Avis PY, Robertson SC, Donoghue DJ. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol Cell Biol. 1996;16:4081–87. doi: 10.1128/mcb.16.8.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 1996;15:520–27. [PMC free article] [PubMed] [Google Scholar]
- 152.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–95. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 152a.Wendt DJ, Dvorak-Ewell M, Bullens S, Lorget F, Bell SM, et al. Neutral endopeptidase-resistant C-type natriuretic peptide variant represents a new therapeutic approach for treatment of fibroblast growth factor receptor 3-related dwarfism. J Pharmacol Exp Ther. 2015;353:132–49. doi: 10.1124/jpet.114.218560. [DOI] [PubMed] [Google Scholar]
- 152b.Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, et al. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152c.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xie Y, Su N, Jin M, Qi H, Yang J, et al. Intermittent PTH (1–34) injection rescues the retarded skeletal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Hum Mol Genet. 2012;21:3941–55. doi: 10.1093/hmg/dds181. [DOI] [PubMed] [Google Scholar]
- 154.Yamamoto GL, Baratela WA, Almeida TF, Lazar M, Afonso CL, et al. Mutations in PCYT1A cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94:113–19. doi: 10.1016/j.ajhg.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154a.Yamashita A, Morioka M, Kishi H, Kimura T, Yahara Y, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature. 2014;513:507–11. doi: 10.1038/nature13775. [DOI] [PubMed] [Google Scholar]
- 155.Yang G, Sun Q, Teng Y, Li F, Weng T, Yang X. PTEN deficiency causes dyschondroplasia in mice by enhanced hypoxia-inducible factor 1α signaling and endoplasmic reticulum stress. Development. 2008;135:3587–97. doi: 10.1242/dev.028118. [DOI] [PubMed] [Google Scholar]
- 155a.Yang G, Zhu L, Hou N, Lan Y, Wu XM, et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014;24:1266–69. doi: 10.1038/cr.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155b.Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. PNAS. 2014;111:12097–102. doi: 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yasoda A, Kitamura H, Fujii T, Kondo E, Murao N, et al. Systemic administration of C-type natriuretic peptide as a novel therapeutic strategy for skeletal dysplasias. Endocrinology. 2009;150:3138–44. doi: 10.1210/en.2008-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yasoda A, Komatsu Y, Chusho H, Miyazawa T, Ozasa A, et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10:80–86. doi: 10.1038/nm971. [DOI] [PubMed] [Google Scholar]
- 158.Yasoda A, Nakao K. Translational research of C-type natriuretic peptide (CNP) into skeletal dysplasias. Endocr J. 2010;57:659–66. doi: 10.1507/endocrj.k10e-164. [DOI] [PubMed] [Google Scholar]
- 158a.Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLOS Genet. 2014;10:e1004820. doi: 10.1371/journal.pgen.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]