

Abstract
Nanoparticles offer great potential in drug delivery and imaging, but shielding strategies are necessary to increase circulation time and performance. Structure-function studies are required to define the design rules to achieve effective shielding. With several formulations reaching clinical testing and approval, the ability to assess and detail nanoparticle formulations at the single particle level is becoming increasingly important. To address this need, we use cryo-electron tomography (cryo-ET) to investigate stealth-coated nanoparticles. As a model system, we studied the soft matter nanotubes formed by tobacco mosaic virus (TMV) coated with human serum albumin (SA) stealth proteins. Cryo-ET and subtomogram averaging allow for visualization of individual SA molecules and determination of their orientations relative to the TMV surface, and also for measurement of the surface coverage provided by added stealth proteins. This information fills a critical gap in the understanding of the structural morphology of stealth-coated nanoparticles, and therefore cryo-ET may play an important role in guiding the development of future nanoparticle-based therapeutics.
Graphical abstract
Introduction
Nanoparticles (NPs) offer promise as drug delivery vehicles to target drugs to diseased tissue, and they can be combined with contrast agents to integrate imaging to assess therapy success and disease progression. While the opportunities are wide ranging, challenges exist. When introduced into an organism, nanoparticles face biological hurdles such as immune recognition leading to clearance in non-target tissues. ‘Naked’ NPs can be recognized and tagged by antibodies and complement proteins and other components of the innate immune system, leading to clearance by the mononuclear phagocyte system.1,2 To circumnavigate this, stealth coatings have been developed to prevent carrier recognition and clearance, thereby enhancing the pharmacokinetic profiles and performance.
Two principal methods have been developed: stealthing with hydrophilic polymers and camouflage as ‘self’. Coating NPs with polyethylene glycol (PEG), termed PEGylation, is a popular strategy. The hydrophilic polymer reduces nanoparticle–protein interactions and therefore clearance3. However, PEG does not completely prevent immune recognition and its efficacy is dependent on the physical characteristics of the specific polymer selected.4,5 Furthermore, recent data suggests that PEG-specific antibodies are becoming more common in the human population (up to 25% of the population in 2012 compared to 0.2% of the population in 1984).6,7 Recent clinical trials have highlighted that the prevalence of anti-PEG antibodies can impair the functionality and safety of PEGylated therapies.8,9 Alternative polymer coatings and strategies are urgently needed to overcome this technological hurdle. Thus, more recent approaches make use of ‘self’ coatings, using proteins or peptides to mark the nanocarrier as self therefore overcoming immune clearance.10–12 Data indicate that these self-coatings may be superior to contemporary PEG coatings as demonstrated by reduced immune recognition along with enhanced circulation. In the present work, we set out to structurally characterize a nanocarrier coated with the ‘self’ protein serum albumin (SA), the most abundant plasma protein in humans.
While there are numerous methods to investigate NP stealth coatings, challenges still remain to understand the precise mechanism of enhancing the in vivo performance of nanocarriers. It is unclear why some stealth coatings perform better than others, including why different formulations of the same shielding strategy have varied effects. Thus, the need arises to characterize the physical and morphological properties of various stealthing strategies when applied to NP platforms. Multiple characterization techniques are usually combined to characterize stealth coatings. For example, the loading density of stealth polymers, proteins, or peptides can be determined through NMR, surface plasmon resonance, gel electrophoresis, and fluorescent modeling.13–16 Estimation of the size of coated nanoparticles can be obtained through techniques such as dynamic light scattering and chromatography techniques.17–20 Nevertheless, these techniques typically provide averaged information on a population of nanoparticles, rather than data on individual NPs. Techniques such as field emission scanning electron microscopy and negative stain transmission electron microscopy (TEM) can be used to visualize individual particles. But these techniques involve dried samples, whereas in the bloodstream the NPs would be in a hydrated state.21,22
To overcome these technological challenges, this work utilizes cryo-ET23 to visualize stealth coatings on the surface of a nanoparticle in a hydrated state. As proof-of-concept, we use tobacco mosaic virus (TMV) as a model NP platform coated with the stealth protein SA. TMV is a 300 nm by 18 nm rigid soft matter nanotube which has served as a model organism in structural biology studies, making it an ideal candidate platform for this study. It is a plant virus that we have engineered as a drug delivery vehicle and MRI contrast agent.24,25 However, as with other biologics, TMV has a short circulating half-life as a result of immune recognition. To overcome these properties, we recently developed SA-’camouflaged’ TMV NPs, which demonstrate reduced recognition by TMV-specific antibodies with no change in macrophage uptake compared to ‘naked’ and PEG-coated NPs.12 SA coating also extended the circulating half-life time more effectively than PEG-coatings. Here, we investigate the physical characteristics of SA-coated TMV NPs to understand the structural morphology of these camouflaged NPs and how the camouflage may influence their function.
Experimental
Materials and Methods
Virus propagation and purification
T158K mutant of TMV26 (TMV-lys) was propagated in Nicotiana benthamiana plants through mechanical inoculation using 5–10 μg of virus per leaf. Viruses were isolated and purified using established methods to yield approximately 1 mg of virus per gram of infected leaf material.27
TMV sCy5 labeling
TMVs were labeled with Cy5 at glutamic acid residues lining the central channel of TMV. Glutamic acids were first modified with alkynes by EDC coupling for 24 hours using 100 equivalents of propargylamine (Sigma Aldrich) per capsid protein with 50 equivalents of EDC in 100 mM HEPES buffer, pH 7.4. This was followed by an alkyne-azide click reaction for 30 minutes by adding 1 equivalent of sCy5-azide (Lumiprobe) per coat protein in the presence of 2 mM AMG (Fisher), 2 mM AsC (Fisher), 1 mM CuSO4 (Fisher) in 10 mM potassium phosphate buffer, pH 7.4 on ice. sCy5-labled TMV was purified by ultracentrifugation at 42,000 rpm for 3 hours on a 40% w/v sucrose cushion.
TMV conjugation
Human serum albumin (SA; Sigma Aldrich) was conjugated to the solvent-exposed exterior surface of TMV-lys through a PEG linker. SA was first conjugated using NHS-PEG4-SAT (ThermoFisher) at a 1-to-1 ratio in 10 mM potassium phosphate buffer, pH 7.4 containing 10% v/v DMSO overnight at RT. De-acetylation solution (0.5 M hydroxylamine, 25 mM EDTA in PBS, pH 7.2–7.5) was added to the reaction at a final concentration of 10% v/v to de-protect the thiol group and this de-protection reaction was carried for 2 hours at RT. TMV-lys was conjugated using maleimide-PEG4-NHS (ThermoFisher) at 10 equivalents PEG per TMV-lys coat protein in 10 mM potassium phosphate buffer, pH 7.4 containing 10% v/v DMSO for 2 hours at RT. The resulting SA-PEG4-SH conjugates and MAL-PEG4-TMV conjugates were purified through a PD MiniTrap G-25 desalting columns (GE). Purified SA-PEG4-SH was then reacted with purified MAL-PEG4-TMV at a ratio of 6 equivalents of SA-PEG4-SH per TMV coat protein overnight at room temperature. The reaction was quenched for 1 hour at RT by addition of excess glycine and l-cysteine. SA-PEG8-TMV (Fig. 1A) was then purified by ultracentrifugation at 55,000 rpm for 3 hours on a 40% w/v sucrose cushion. Conjugation was verified and quantified using SDS-PAGE analysis.
Fig. 1.

Production and characterization of SA-TMV. (A) Schematic representation of conjugation of TMV with SA. SA and TMV-lys are first separately conjugated with PEG linkers through NHS chemistry, followed by conjugation of SA-PEG4-SH and maleimide-PEG4-TMV to produce SA-PEG8-TMV. Analysis of particles before and after conjugation by (B) SDS-PAGE and (C) Western Blot (WB). Free SA was used as a reference for WB analysis. SA conjugation is indicated by the presence of multiple bands with MW > 64 kDa (MW of single TMV-lys CP is ~17 kDa; MW of single SA is ~67 kDa). WB immune recognition by anti-TMV antibodies shown in green, anti-SA antibodies shown in red. (D) Dot blot analysis of immune recognition of TMV and SA-TMV by anti-TMV and anti-cowpea mosaic virus (anti-CPMV, negative control). PBS is an additional negative control. (E) Quantitative densitometric analysis of the dot blots.
SDS-PAGE analysis
TMV samples were denatured by boiling at 100°C for 5 minutes in gel loading buffer (62.5 mM Tris–HCl pH 6.8, 2% w/v SDS, 10% v/v glycerol, 0.01% w/v bromophenol blue, 10% v/v 2-mercaptoethanol). 20 μg TMV were loaded on 4–12% NuPAGE gels in MOPS running buffer and separated at 200 V for 45 min. Gels were stained with Coomassie Blue and visualized using an AlphaImager imaging system (Biosciences) (Fig. 1B). Gels were analyzed by densitometry using ImageJ.28
Western blot analysis
Samples separated by SDS-PAGE (with 20 μg of loaded TMV) were transferred to nitrocellulose membranes at 100 V for 1 hour. Membranes were incubated in 5% w/v milk in TBS-Tween at RT for 1 hour, followed by incubation with either 0.5 μg/mL rabbit anti-TMV antibody (Pacific Immunology) or 0.5 μg/mL rabbit polyclonal anti-albumin antibody (Novus Biologicals) in 5% w/v milk in TBS-Tween for 1 hour at RT. Samples were then washed three times for 10 min each in TBS-Tween and incubated in 1 μg/mL alkaline phosphatase goat anti-rabbit antibody in 5% w/v milk in TBS-Tween at RT for 1 hour, followed by three washes for 10 min each in TBS-Tween and 1 min wash in Millipore water. Antibody binding was visualized using Novex AP Chromogenic Substrate (BCIP/NBT; Invitrogen) (Fig. 1C).
Immuno-dot blots
Dot blots were prepared by applying 1 μL spots of 150 μg/mL anti-TMV and 150 μg/mL anti-CPMV control antibodies (Pacific Immunology) on a nitrocellulose membrane after the membrane has been equilibrated in 10 mM PBS. Blots were incubated in 5% w/v milk in PBS at RT for 1 hour, washed three times for 5 min each in 10 mM PBS. The blots were then incubated in 40 μg/mL Cy5-labeled VNPs for 2.5 hours at RT, followed by three washes for 5 min each in PBS. Blots were then dried and imaged for fluorescence using a Maestro imaging system (Fig. 1D). Blots were analyzed for integrated density using ImageJ (Fig. 1E).
Negative stain transmission electron microscopy
3 μL SA-TMV or control naked TMV NPs at a concentration of 0.5 mg/mL were applied to glow-discharged carbon coated 200 mesh copper grids. After 1 min, excess sample was removed and grids were washed two times in dH2O for 1 min each. Grids were then stained two times for 30 sec each with 2% w/v uranyl acetate, followed by blotting until dry with Whatman 1 blotting paper. Grids were imaged in a FEI Tecnai G2 Spirit (120kV) transmission electron microscope with a 4k × 4k Gatan US4000 CCD camera (Fig. 2A,C).
Fig. 2.

Transmission electron microscopy of TMV and SA-TMV. Bare TMV-lys visualized by negative stain (A) and cryo (B) TEM. (C) Negative stain images of SA-conjugated particles show a rougher, uneven surface. (D) Cryo-EM images of SA-TMV samples reveal individual SA proteins on the TMV surface. Electron dense 10 nm gold fiducials are visible in the cryo micrographs. (Scale bars = 100 nm).
Cryo-electron microscopy and tomography
3 μL SA-TMV or control naked TMV NPs at a concentration of 0.5 mg/mL were mixed with 0.5 μL 10nm fiducial nanogold (Aurion). The solution was then applied to glow-discharged Quantifoil 2×2 200mesh holey carbon grids. Grids were blotted until nearly dry (approximately 2.5 sec blot) and rapidly plunged into liquid ethane cooled with liquid nitrogen using a manual plunger. Grids were imaged in a JEOL 2200FS transmission electron microscope equipped with an energy filter using a Tietz TVIPS 4k × 4k CMOS camera (for cryo-electron microscopy) (Fig. 2B,D) or a Direct Electron DE20 direct detector (for cryo-electron tomography) (Fig. 3). For tomography, SA-TMV or TMV grids were imaged as 0.5s long movies with four frames each, which were then motion-corrected using custom EMAN229 scripts to result in single images. Data was collected within a -70° to +70° tilt angle range or until a grid bar prevented further imaging. Tilt series were processed with the IMOD processing package30 to generate tomograms, which were visualized with UCSF Chimera.31
Fig. 3.

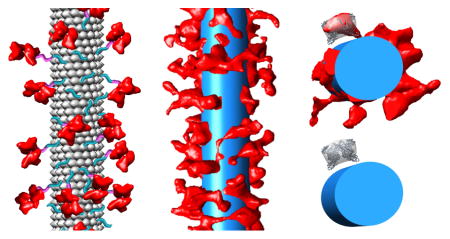
Cryo-electron tomography analysis of SA-TMV nanoparticles. (A) Central slice through processed tomogram of SA-TMV. A few electron dense fiducial markers are visible in this slice. (Scale bar = 100 nm). (B) Three-dimensional representation of SA-TMV tomogram. (Scale bar = 100 nm). (C) Single SA-TMV rod in blue, left, and in red with 300 nm × 18 nm cylinder shown in blue, right. (Scale bar = 25 nm).
Subtomogram averaging of SA
Selected individual densities attributed to SA were cropped from the tomogram density using UCSF Chimera. These densities were analyzed with the Jsubtomo software package32 using a 20 Å filtered map of the SA crystal structure33 (PDB: 1AO6) as a template map. Refined SA positions were selected for which Jsubtomo found dimer related orientations of the SA template in the cropped cryoEM density regions. The cryo-EM density from 10 refined SA positions, corresponding to five SA dimers, was averaged to generate an average SA density map. This average SA density map was positioned back into the tomogram using the refined SA positions to reveal the orientation of SA with respect to the TMV surface at multiple locations (Fig. 4).
Fig. 4.

Subtomogram averaging analysis of SA orientation on the TMV surface. (A) Averaged SA density (transparent gray) and the human SA crystal structure33 (PDB: 1AO6, gray ribbon) positioned with respect to the TMV cylinder (blue) at five locations along a single SA-TMV nanoparticle. These locations are shown both with (top) and without (bottom) the cryo-ET density (red). (B) SA crystal structure (PDB: 1AO6) with the C-alpha atoms of solvent-exposed lysine residues depicted as blue spheres.
Subtomogram averaging of SA-TMV segment
The tomogram of a whole SA-TMV particle was cropped into 21 segments, each corresponding to the length of six turns of the TMV helix using UCSF Chimera. These density segments were analyzed with the Jsubtomo software package.32 An SA-TMV template map was generated with an 8.8 nm diameter sphere, representing the same volume as SA, positioned 2.4 nm away from the surface of a 20 Å filtered map of six helical turns of TMV (PDB: 4UDV; Fig. 5A).34 Note that the average diameter of the PEG linker is estimated to be 2.4 nm. Refined SA-TMV segment positions were selected based on the highest cross-correlation score to the template map. The aligned cryo-EM density from all segments was averaged to generate an average SA-TMV segment map (Fig. 5B).
Fig. 5.
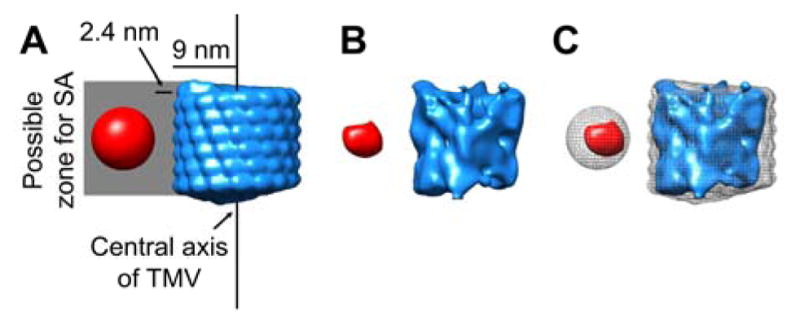
Subtomogram averaging analysis of SA-TMV segments. (A) Template map used for subtomogram averaging, with six turns of TMV density (PDB: 4UDV) shown in blue and a sphere representing SA shown in red. The sphere denoting SA is positioned 2.4 nm away from the TMV surface, as this distance is the average calculated length of an 8-mer PEG. The total possible zone for SA extends from the surface of TMV to 12.8 nm beyond the surface, or 21.8 nm from the central axis of TMV. (B) Results of averaging 21 aligned segments of SA-TMV with TMV density in blue and SA density in red. (C) Overlay of the template in grey mesh and the averaged density in solid blue (TMV) and red (SA).
Analysis of volume coverage
Individual SA-TMV or TMV rods were cropped from the SA-TMV tomogram or TMV tomogram, respectively, using UCSF Chimera. Rods were oriented along the Z-axis and clipped to one-pixel wide segments in UCSF Chimera. The integrated density of an oval containing only the TMV region of each segment was determined using the Measure tool in ImageJ. An oval was chosen because the density was skewed due to the missing wedge artifact of tomography data collection. The integrated density was also measured for a circle extending well beyond the TMV region to allow for SA. The TMV-only area was subtracted from the larger SA and TMV area to calculate the area filled by SA. This area was calculated for each one-pixel wide segment and plotted. The integration of this plot results in the total volume coverage around the TMV. By calculating the volume of a single SA in a similar fashion, the total number of SAs on an individual rod were determined (Fig. 6).
Fig. 6.
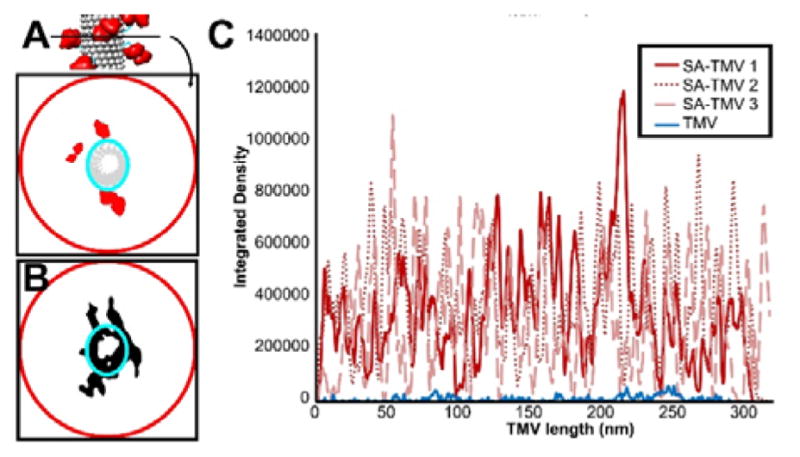
Analysis of SA volume coverage of TMV rod. (A) Schematic representation of coverage area analysis. Each TMV rod is broken up into one-pixel wide segments, and the areas filled with density within the larger TMV+SA circle (red) as well as within the smaller TMV-only oval (cyan) are measured. Subtracting the TMV-only area from TMV+SA area gives a measurement of the SA area at each segment along the TMV rod. (B) Experimental example of a one-pixel wide segment from an SA-TMV tomogram with the measurement circle and oval shown. Note that an oval is used to measure the TMV-only area since the tomograms are not corrected for the missing wedge, leading to apparent elongation of TMV along one axis of its cross section. (C) Plot of the coverage for three SA-TMV rods (red) compared to a naked control TMV rod (blue).
Analysis of surface coverage
Individual SA-TMV rods were cropped from the SA-TMV tomogram using UCSF Chimera. Rods were oriented along the Y-axis and pseudo-colored red, with a 300 nm by 18 nm blue cylinder also displayed using UCSF Chimera. At one degree rotations, images were saved over a total of 360 degrees. For each degree, the saved picture was opened in ImageJ and the amount of red and blue shown in a one-pixel wide line at the center of the SA-TMV was determined using the RGB Measure tool. The total amount of red in all 360 degrees divided by the total amount of red + blue yields a percent coverage for a single SA-TMV rod.
Results and Discussion
Particle preparation
To prepare SA-coated NPs, TMV containing a T158K mutation (termed TMV-lys)26 was incubated with MAL-PEG4-NHS yielding maleimide-functional MAL-PEG4-TMV. In a separate reaction, SA was reacted with NHS-PEG4-SH to produce thiol-activated SA-PEG4-SH. These two products were then reacted together via Michael addition to form SA-PEG8-TMV (in short SA-TMV, Fig. 1A). Conjugation and structural integrity of the SA-TMV formulation was confirmed first by SDS-polyacrylamide gel electrophoresis (SDS-PAGE; Fig. 1B). TMV coat proteins (CPs) have a molecular weight of 17 kDa, SA has a molecular weight of 66.5 kDa. SA-TMV shows the expected band pattern: free CP is detectable as a ~17 kDa band while conjugated SA-CP is detectable as high molecular weight bands (Fig. 1B). Western blotting against SA (Fig. 1C, red) and against TMV (Fig. 1C, green) indicate the higher order bands are in fact SA-conjugated CPs. According to densitometry analysis, ~0.25–0.35 mg SA were conjugated per 1 mg TMV particles yielding approximately 140–200 SAs per TMV.
TMV is comprised of 2,130 copies of an identical CP with the T158K mutation available for conjugation on the solvent-exposed exterior TMV surface. Each CP has an exposed exterior surface area of about 8 nm2. With the addition of the PEG linker, extending on average 2.4 nm from the TMV surface as calculated by the Flory dimension5, this generates an expanded surface area of ~10 nm2 per PEG-TMV CP. Based on the SA crystal structure, the footprint of the SA dimer would be 28–47 nm2, depending on its orientation. Since the footprint of SA is larger than the exposed surface area of a single PEG-TMV CP, SA dimers would be unable to bind to each PEG-TMV CP subunit. Steric constraints would limit SA dimers to binding to every 3–5 PEG-TMV CPs on average. The theoretical maximum coverage of TMV, if each SA dimer was oriented with its smallest footprint facing the TMV surface and with all PEG linkers fully extended (2.8 nm), is approximately 800 SA dimers per TMV. The practical maximum, however, would be much lower due to steric hindrance between SA molecules, random orientational preference on the TMV surface, and PEG linker flexibility.
To estimate the surface coverage based on the densitometry analysis, both the number of SAs per TMV and the orientation of each SA relative to the TMV rod must be considered. Assuming the exposed surface area of a TMV CP is approximately 8 nm2 and the footprint of SA is 28–47 nm2 depending on its orientation, the theoretical surface coverage of TMV is in the range of 20–55%. This underlines the importance to develop more quantitative experimental methods to determine coverage.
To confirm that SA-TMV maintains stealth properties, we investigated immune recognition from TMV-specific antibodies. Viral vectors are primarily eliminated from the body through antibody clearance, and TMV-specific antibodies have been previously found in blood collected from both smokers and non-smokers, presumably due to the prevalence of the virus in crops and tobacco cigarettes.35–38 Repeat administration of TMV-based therapeutics would also lead to development of TMV-specific antibodies, so it is critical to test the ability of stealth-coated particles to prevent immune recognition. Immuno dot blots with anti-TMV antibodies and fluorescent TMV particles were performed to test the shielding properties of the SA-TMV particles (Fig. 1D and 1E). To prepare the fluorescently labeled particles, Cy5 dyes were conjugated to the interior channel of TMV using click chemistry. Dot blots were performed by spotting polyclonal anti-TMV or control polyclonal anti-cowpea mosaic virus (CPMV) antibodies on a nitrocellulose membrane, which was then incubated with Cy5-labeled NPs. Recognition was measured using a Maestro Imaging system. As seen previously, SA-TMV particles have reduced antibody recognition compared to bare particles.12
Imaging and processing of naked and SA-coated particles
Visualization in 2D
The physical characteristics of the ‘naked’ TMV and SA-coated particles were obtained with a combination of negative stain and cryo-electron microscopy (cryo-EM), followed by a detailed analysis using cryo-ET. While cryo-samples are more technically challenging to prepare than negative stain samples, they offer the advantage of visualization of the sample in a frozen hydrated native-like state, albeit at lower contrast than observed with negative stain.
As expected, TMV displays much better contrast by negative stain (Fig. 2A) than by cryo-EM (Fig. 2B), while both methods reveal smooth rod-shaped particles. As we previously reported, the SA coating is detectable by negative stain TEM.12 SA-TMV exhibits a rough surface indicating presence of SA (Fig. 2C). Nevertheless, individual SAs cannot be identified in negative stain TEM images. In contrast, cryo-EM images of SA-TMV do reveal individual SA molecules as dots on the surface of the rod-shaped TMV (Fig. 2D). From a single two-dimensional cryo-micrograph of SA-TMV, it is apparent that there are numerous SAs per TMV nanoparticle and that the surface of TMV is fairly well covered.
Visualization in 3D by cryo-electron tomography
To investigate the three-dimensional architecture of SA on the surface of TMV, cryo-ET was performed. Data was collected using a JEOL 2200FS microscope equipped with an energy filter and a DE20 direct detector device. Images were motion corrected and processed using the IMOD software package30, resulting in an aligned tilt series (see ESI Movie S1) and reconstructed tomogram (see ESI Movie S2). A single slice from the reconstructed tomogram of SA-TMV is shown in Fig. 3A. As in the cryo-EM micrograph of SA-TMV (Fig. 2D), individual SA molecules are observed to dot the surface of the TMV rods. When the entire tomogram is displayed as a three-dimensional surface (Fig. 3B), it becomes apparent that SA molecules surround each TMV rod, leading to a relatively even coating of the NPs. An enlarged view of a single SA-TMV rod in Fig. 3C (left) shows a rough exterior with many protrusions. By displaying a cylinder with an 18 nm diameter (blue) centered on the SA-TMV density (red), it becomes possible to identify individual SA molecules positions relative to the TMV rod (Fig. 3C, right).
The radius of TMV is 9 nm and the site of attachment of the PEG linker (Lys-158) is on the external surface of TMV. The average diameter of the PEG linker is estimated to be 2.4 nm, as calculated from the Flory dimension of an 8-mer linear PEG polymer.5 Based on this information, on average most SA dimers are expected to be attached 2.4 nm from the surface of TMV or 11.4 nm from the central axis of TMV. Since the PEG linker is highly flexible, it is possible that the SA dimers could begin close to the TMV surface, or 9 nm from the central axis. Alternatively if a PEG linker were fully extended to its maximum length of 2.8 nm, then the distance between the SA dimer and TMV would be 2.8 nm, or 11.8 nm from the central axis. The dimensions of the SA dimer as calculated from the crystal structure are 6 nm × 6 nm × 10 nm.33 Taken together this means that the SA density in the SA-TMV NPs can range from 9 nm to 21.8 nm from the central axis of TMV. This agrees with the experimental data, in which SA density extends from the surface of the TMV NP in all directions for approximately 12 nm from the TMV surface (Fig. 3C, right).
Subtomogram averaging of SA
To determine the orientations of SA relative to the TMV surface, selected protrusions were cropped and aligned with the SA crystal structure, and averaged using the subtomogram averaging software Jsubtomo.32,33 The resulting average SA density was then placed into the refined SA positions within the tomogram to visualize how SA is oriented on the TMV surface. Fig. 4A shows the average SA density and docked SA crystal structure positioned with respect to the TMV rod at multiple sites. These results indicate that there is not one specific preferred orientation of SA relative to TMV. There are 120 lysine residues in the SA dimer, of which 79 are solvent-exposed (Fig. 4B). It is possible that a number of these lysine side chains are reactive and serve as anchors for the TMV linkage sites. Indeed, multiple lysine residues throughout the SA structure, including Lys 137, Lys 190, Lys 351, Lys 505, and Lys 525, have been found to be reactive groups in previous studies.39–42 The numerous possible SA attachment points, along with the flexibility of the 8-mer PEG linker would explain the heterogeneous display of SA observed for SA-TMV. Heterogeneous display may be advantageous for in vivo applications of these particles, to avoid innate immune recognition through pathogen-associated molecular patterns (PAMPs).
Subtomogram averaging of SA-TMV segment
To identify any effect a single SA might have on the positioning of its neighboring SA molecules, subtomogram averaging was performed on SA-TMV segments. The density map for SA-TMV was divided into 21 segments, each containing ~6 helical turns of TMV. Each segment was aligned to a generated template map of SA-TMV with a sphere representing SA positioned in the middle of the segment at the expected average distance from the TMV surface (Fig. 5A). A sphere with the same volume as the SA dimer was used in the template map to prevent bias in searching for a specific orientation of SA relative to the TMV central axis. The aligned segments were averaged using Jsubtomo (Fig. 5B). Through this analysis, any features that are repeated in all segments would show density in the average map and differences would be averaged away. In comparison to the template map, the average SA density is smaller than the sphere used in the template map and also smaller than expected for an SA dimer (Fig. 5C). This suggests that the subtomogram averaged segments may be aligned on one SA subunit, and that due to the multiple orientations of SA dimers relative to the TMV surface the other SA subunit in each dimer is effectively averaged away. Furthermore, there seems to be no pattern of neighboring SA molecules relative to the central SA dimer, as no density is apparent for additional SA dimers in the average SA-TMV segment map. Taken together, these results support the conclusion that although there is an average distance for SA dimers from the TMV surface (~2.4 nm), the SA orientations are random and there is no interaction between neighboring SA dimers.
Quantification of TMV coverage
To calculate the coverage of the volume surrounding TMV by the SA coating, three complete TMV rods were cut out from the three-dimensional tomogram, and each TMV was sliced into multiple one-pixel wide segments (Fig. 6A). At each segment, the integrated density of the image was measured to determine the area of the TMV portion alone, as well as the entire SA-TMV area (Fig. 6B). The TMV area was subtracted from the SA-TMV area to obtain a value for the SA area in each segment. These values are plotted for each TMV rod in Fig. 6C. The same was done for a single naked TMV rod extracted from a separate cryo tomogram of unmodified TMV (see ESI Fig. S1). The coverage plot shows that SA surrounds TMV in a relatively even fashion. The total integrated area under each SA curve can be quantified by comparing it to the area calculated for a single simulated SA molecule. This analysis indicates that each SA-TMV has a different number of conjugated SA molecules, ranging from approximately 180 to 210 SAs per TMV. This is in good agreement with the number calculated by the PAGE densitometry analysis (140–200 SAs per TMV, see Fig. 1). These results demonstrate that each SA-TMV has a different level of camouflage coating.
The cryo-tomogram also facilitates a rough calculation of the percent coverage of the TMV surface. The extent of uncoated and coated TMV surface can be quantified, analogous to the amounts of blue and red surface, respectively, as seen in Fig. 3C, right. Three SA-TMV rods were analyzed and found to have 30–45% surface coverage. These numbers are within the range calculated from the dot blot densitometric analysis (20–55%), further suggesting that the SAs tend to orient in a random manner.
To understand the biological relevance of this coverage, atomic models were built for SA-TMV segments and modeled with both intact IgG and Fab fragments. The structural modeling indicates that the spacing and density of SA on the TMV surface would likely prevent antibody-antigen interactions with the TMV surface – due to steric hindrance: i) SA is too densely packed to allow a Fab arm to easily find an opening large enough to access the TMV surface, and ii) the Fab arm is too short to penetrate between SAs at their average distance from the TMV surface to reach TMV epitopes. This is assuming an average Fab length of 6.5 nm.43 In rare cases, where the SA dimer is positioned next to the TMV surface and oriented such that its thinnest dimension (6 nm) is perpendicular to the TMV surface, then a Fab arm could reach an exposed TMV epitope. Mathematical calculations considering the average amount of exposed TMV surface area and the average antibody-antigen interaction patch size43 led to similar conclusions regarding SA shielding. The structural and mathematical modeling results correlate well with the experimental antibody binding data (Fig. 1D), indicating little antibody recognition for SA-TMV compared to strong recognition for TMV.
Conclusions
This work demonstrates the utility of cryo-ET as a characterization tool for stealth protein camouflaged NPs. When combined with subtomogram averaging, cryo-ET provides a way to visualize individual SA molecules heterogeneously displayed on the surface of TMV. While standard negative stain TEM can reveal the presence of a stealth protein coating on the surface of TMV, individual SAs are not resolved. Two dimensional cryo-TEM micrographs provide greater detail and individual SA molecules are observed as dots. Three-dimensional cryo-ET provides the possibility of quantitating surface coverage in a manner that is complementary to gel densitometric analysis. Furthermore, subtomogram averaging allows for analysis of the orientation of SA molecules relative to the TMV surface and potentially for the detection of interactions between neighboring SA molecules. With the rapid development of shielded nanoparticles for biomedical applications, this study serves as a proof-of-concept for using cryo-ET for visualization and quantitative structural analysis. It also opens the door for the development of further structure-function studies through correlation of structural morphology with in vivo efficacy.
Supplementary Material
Acknowledgments
We thank Dr. Sudheer Molugu for maintaining the TEM facility at the Cleveland Center for Membrane and Structural Biology, as well as the CWRU High Performance Computing staff for their assistance with campus computing resources. We thank Christina Wege and her team (University of Stuttgart) for providing TMV-lys. This work is supported by funding from the National Institutes of Health (R01-CA202814 to NFS and T32 GM008803 to NMG).
Footnotes
Electronic Supplementary Information (ESI) available: TMV-lys tomography results, and additional SA-TMV tomography movies and legends. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Lucas AT, Madden AJ, Zamboni WC. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:642–653. doi: 10.1002/wnan.1394. [DOI] [PubMed] [Google Scholar]
- 2.Pitek AS, Wen AM, Shukla S, Steinmetz NF. Small. 2016;12:1758–1769. doi: 10.1002/smll.201502458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owens DE, 3rd, Peppas NA. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, Napier M, Bear JE, DeSimone JM. Nano Lett. 2012;12:5304–5310. doi: 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee KL, Shukla S, Wu M, Ayat NR, El Sanadi CE, Wen AM, Edelbrock JF, Pokorski JK, Commandeur U, Dubyak GR, Steinmetz NF. Acta Biomater. 2015;19:166–179. doi: 10.1016/j.actbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garay RP, El-Gewely R, Armstrong JK, Garratty G, Richette P. Expert Opin Drug Deliv. 2012;9:1319–1323. doi: 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- 7.Richter AW, Akerblom E. Int Arch Allergy Appl Immunol. 1984;74:36–39. doi: 10.1159/000233512. [DOI] [PubMed] [Google Scholar]
- 8.Hershfield MS, Ganson NJ, Kelly SJ, Scarlett EL, Jaggers DA, Sundy JS. Arthritis Res Ther. 2014;16:R63. doi: 10.1186/ar4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganson NJ, Povsic TJ, Sullenger BA, Alexander JH, Zelenkofske SL, Sailstad JM, Rusconi CP, Hershfield MS. J Allergy Clin Immunol. 2016;137:1610–1613.e1617. doi: 10.1016/j.jaci.2015.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sosale NG, Spinler KR, Alvey C, Discher DE. Curr Opin Immunol. 2015;35:107–112. doi: 10.1016/j.coi.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitek AS, Jameson SA, Veliz FA, Shukla S, Steinmetz NF. Biomaterials. 2016;89:89–97. doi: 10.1016/j.biomaterials.2016.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Fuentes M, Torres D, Martin-Pastor M, Alonso MJ. Langmuir. 2004;20:8839–8845. doi: 10.1021/la049505j. [DOI] [PubMed] [Google Scholar]
- 14.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanomedicine (Lond) 2011;6:715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demers LM, Mirkin CA, Mucic RC, Reynolds RA, 3rd, Letsinger RL, Elghanian R, Viswanadham G. Anal Chem. 2000;72:5535–5541. doi: 10.1021/ac0006627. [DOI] [PubMed] [Google Scholar]
- 16.Steinmetz NF, Hong V, Spoerke ED, Lu P, Breitenkamp K, Finn MG, Manchester M. J Am Chem Soc. 2009;131:17093–17095. doi: 10.1021/ja902293w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanetti-Ramos BG, Fritzen-Garcia MB, Cristian SDOB, Andr APB, Soldi V, Borsali R, Creczynski-Pasa TB. Mat Sci Eng C-Bio S. 2009;29:638–640. [Google Scholar]
- 18.Seehuber A, Schmidt D, Dahint R. Langmuir. 2012;28:8700–8710. doi: 10.1021/la2050652. [DOI] [PubMed] [Google Scholar]
- 19.Cheng TL, Chuang KH, Chen BM, Roffler SR. Bioconjug Chem. 2012;23:881–899. doi: 10.1021/bc200478w. [DOI] [PubMed] [Google Scholar]
- 20.Li SD, Huang L. Biochim Biophys Acta. 2009;1788:2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duncanson WJ, Figa MA, Hallock K, Zalipsky S, Hamilton JA, Wong JY. Biomaterials. 2007;28:4991–4999. doi: 10.1016/j.biomaterials.2007.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gref R, Quellec P, Sanchez A, Calvo P, Dellacherie E, Alonso MJ. Eur J Pharm Biopharm. 2001;51:111–118. doi: 10.1016/s0939-6411(00)00143-0. [DOI] [PubMed] [Google Scholar]
- 23.Stewart PL. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016 doi: 10.1002/wnan.1417. [DOI] [PubMed] [Google Scholar]
- 24.Bruckman MA, Jiang K, Simpson EJ, Randolph LN, Luyt LG, Yu X, Steinmetz NF. Nano Lett. 2014;14:1551–1558. doi: 10.1021/nl404816m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czapar AE, Zheng YR, Riddell IA, Shukla S, Awuah SG, Lippard SJ, Steinmetz NF. ACS Nano. 2016;10:4119–4126. doi: 10.1021/acsnano.5b07360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geiger FC, Eber FJ, Eiben S, Mueller A, Jeske H, Spatz JP, Wege C. Nanoscale. 2013;5:3808–3816. doi: 10.1039/c3nr33724c. [DOI] [PubMed] [Google Scholar]
- 27.Bruckman MA, Steinmetz NF. Methods Mol Biol. 2014;1108:173–185. doi: 10.1007/978-1-62703-751-8_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Kremer JR, Mastronarde DN, McIntosh JR. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Huiskonen JT, Parsy ML, Li S, Bitto D, Renner M, Bowden TA. J Vis Exp. 2014:e51714. doi: 10.3791/51714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K. Protein Eng. 1999;12:439–446. doi: 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- 34.Fromm SA, Bharat TA, Jakobi AJ, Hagen WJ, Sachse C. J Struct Biol. 2015;189:87–97. doi: 10.1016/j.jsb.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.da Silva RM, de Souto ER, Pedroso JC, Arakava R, Almeida AMR, Barboza AAL, Vida JB. Braz Arch Biol Techn. 2008;51:903–909. [Google Scholar]
- 36.Hu Q, Niu Y, Zhang K, Liu Y, Zhou X. Virol J. 2011;8:41. doi: 10.1186/1743-422X-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wetter C. Naturwissenschaften. 1975;62:533. doi: 10.1007/BF00609072. [DOI] [PubMed] [Google Scholar]
- 38.Balique F, Colson P, Raoult D. J Clin Virol. 2012;55:374–376. doi: 10.1016/j.jcv.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Krantz A, Hanel AM, Strug I, Wilczynski A, Wolff JJ, Huang W, Huang LH, Settineri T, Holmes DL, Hardy MC, Bridon DP. Comput Struct Biotechnol J. 2014;9:e201403001. doi: 10.5936/csbj.201403001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shuck SC, Wauchope OR, Rose KL, Kingsley PJ, Rouzer CA, Shell SM, Sugitani N, Chazin WJ, Zagol-Ikapitte I, Boutaud O, Oates JA, Galligan JJ, Beavers WN, Marnett LJ. Chem Res Toxicol. 2014;27:1732–1742. doi: 10.1021/tx500218g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng X, Maggs JL, Usui T, Whitaker P, French NS, Naisbitt DJ, Park BK. Chem Res Toxicol. 2015;28:51–58. doi: 10.1021/tx500285k. [DOI] [PubMed] [Google Scholar]
- 42.Anraku M, Shintomo R, Taguchi K, Kragh-Hansen U, Kai T, Maruyama T, Otagiri M. Life Sci. 2015;134:36–41. doi: 10.1016/j.lfs.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Stanfield RL, Wilson IA. Microbiol Spectr. 2014:2. doi: 10.1128/microbiolspec.AID-0012-2013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.