

Abstract
Mutant allele specific imbalance (MASI) was initially coined to describe copy number alterations associated with the mutant allele of an oncogene. The copy number gain (CNG) specific to the mutant allele can be readily observed in electropherograms. With the development of genome-wide analyses at base-pair resolution with copy number counts, we can now further differentiate MASI into those with CNG, with copy neutral alteration (also termed acquired uniparental disomy; UPD), or with loss of heterozygosity (LOH) due to the loss of the wild-type (WT) allele. Here we summarize the occurrence of MASI with CNG, aUPD, or MASI with LOH in some major oncogenes (such as EGFR, KRAS, PIK3CA, and BRAF). We also discuss how these various classifications of MASI have been demonstrated to impact tumorigenesis, progression, metastasis, prognosis, and potentially therapeutic responses in cancer, notably in lung, colorectal, and pancreatic cancers.
Keywords: MASI, UPD, aUPD, LOH, Loss of wild-type allele, CNG, KRAS, EGFR, PDAC, lung cancer, colorectal cancer, oncogenes, tumor-suppressor gene
1. INTRODUCTION
An oncogene is a gene that, when activated by mutation, increases the selective growth advantages of the cell in which it resides. Oncogenes often encode proteins that control cell processes such as proliferation and survival. These proteins include transcription factors, chromatin remodelers, growth factor receptors, signal transducers, and apoptosis regulators. Mutation, structural rearrangement (chromosomal rearrangement, gene fusion), and gene amplification (or copy number gains, CNGs) are common mechanisms that activate an oncogene, and result in an increased or a deregulated expression and/or function of the gene. Therefore, cells with such alterations in oncogenes possess a growth advantage or an increased survival rate. Translocations and mutations typically occur early in tumorigenesis, whereas copy number changes usually occur during late tumor stages.
Until recently somatic mutation and gene amplification were considered two independent and largely mutually exclusive mechanisms of oncogene activation. In general, a single copy of the mutated allele is sufficient to convert a proto-oncogene into an activated oncogene, while amplification often involves the non-mutated, wild-type (WT) allele. For example, KRAS is activated via point-mutations while HER-2/neu and MYC family genes gain their oncogenic properties largely through chromosomal aberrations and amplification [1; 2; 3; 4]. With the advances in genome-wide analysis, through single nucleotide polymorphism (SNP)-based array techniques and sequencing of the exome or whole genome, identifying genes altered in cancer at base-pair resolution with copy number counts has become routine. These advances have led to growing recognition of mutant allele specific imbalance (MASI) with CNG [5], acquired uniparental disomy (aUPD) (equivalent to MASI without CNG) [6; 7; 8], and MASI with loss of the WT allele in oncogenes [9], and their significance in tumorigenesis. While it remains true that activating somatic mutations in one allele of an oncogene is sufficient to confer a selective growth advantage on the cell, MASI with CNG, aUPD, and MASI with loss of the WT allele in oncogenes have now been reported to contribute to enhanced malignancies of tumor cells and therapeutic susceptibilities (Figure 1 and Table 1).
Figure 1. Mutant allele specific imbalance (MASI) with or without copy number alteration.
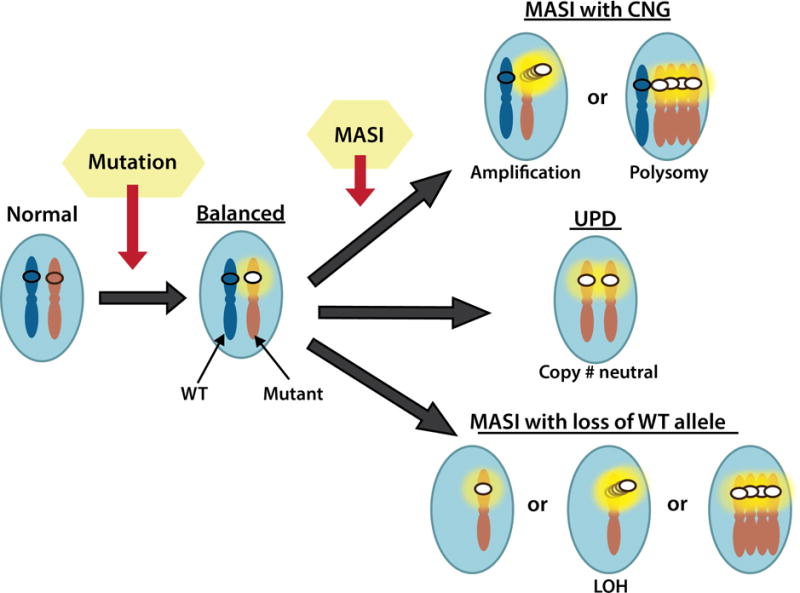
The diagram depicts three commonly observed MASI classifications that involve copy number gain (CNG), copy number neutral alteration (equivalent to uniparental disomy; UPD), or loss of heterozygosity (LOH) due to the loss of the wild-type (WT) allele.
Table 1.
Copy number alterations in oncogene activation
| Classifications | Mechanisms |
|---|---|
| Amplification | CNG of WT, no Mutation |
| Balanced | Mut: WT = 1: 1, no CNG |
| MASI or MASI with CNG | Relative increase of the Mut allele; Mut: WT > 1 due to CNG of the mutant allele |
| aUPD, MASI without CNG, or copy-neutral LOH | MASI with copy neutral changes, Mut: WT= 2:0 |
| Reverse MASI | Relative increase of the WT allele; Mut: WT < 1 due to CNG of the WT allele |
| MASI with LOH or MASI with loss of the WT allele | Relative increase of the Mut allele; Mut: WT > 1 due to the loss of the WT allele |
The EGFR pathway contains numerous well-investigated oncogenes, such as the EGFR, KRAS, BRAF, and PIK3CA genes, with activating alterations in various tumors including lung, colorectal (CRC), and pancreatic ductal adenocarcinoma (PDAC) [10; 11; 12; 13; 14; 15]. These three cancer types are responsible for >410,000 cases (~24%) of cancer incidence and ~250,000 (42%) cancer deaths in the USA estimated for 2016 [16]. KRAS is a major oncogene that is frequently activated in PDAC (>90%) [17; 18], CRC (>40%) [19; 20], and lung cancer (predominantly in lung adenocarcinoma where it is the most commonly mutated oncogene at >20%) [21; 22; 23]. BRAF and PIK3CA are also often activated by mutations in CRC (18% and 32% respectively) [11; 12; 24; 25], occasionally in lung cancers (3% and 4%) [11; 25], and in a subtype of PDAC (those associated with IPMN; intraductal papillary mucinous neoplasm) [13; 26]. Activating mutations of the EGFR gene are present in 15–30% of NSCLC (non-small cell lung cancer), more frequently in adenocarcinomas (>30%), women (~37%), and non-smokers (~50%), while they have been rarely detected in other types of human cancers [27; 28; 29]. EGFR CNGs have also been reported in NSCLC and may play a role in survival and in response to tyrosine kinase inhibitor therapy [10; 30; 31], while KRAS CNGs have not been investigated in-depth in clinical tumors including NSCLCs. Here we discuss how activating alterations of these oncogenes via mutation, mitotic error, and/or copy number alteration, resulting in MASI with CNG, aUPD, or MASI with loss of the WT allele, may impact tumorigenesis, progression, and potentially therapeutic responses in cancers.
2. MUTANT ALLELE SPECIFIC IMBALANCE (MASI) WITH CNG
Activating somatic mutations with nucleotide changes (Figure 1 & Table 1, balanced) and gene CNGs due to focal amplification or chromosomal polysomy (Figure 1 & Table1, amplification) are two major categories of oncogene activation, which occur via independent mechanisms, and are largely but not completely mutually exclusive in tumor cells [32].
EGFR is a tyrosine kinase (TK) receptor of the ErbB family that is commonly altered in epithelial tumors. The EGFR can induce cancer via at least three major mechanisms: overexpression of EGFR ligands, amplification of EGFR, and mutational activation of EGFR. Mutations that have been described to activate EGFR include many variants of small mutations, insertions, and deletions, leading to enhanced dimerization/enhanced ATP binding/pathway activation [27; 28; 29]. Notably, mutations in the tyrosine kinase (TK) domain of the EGFR are unique to NSCLC and rarely detected in other tumor types including CRC and PDAC [27; 28; 29]. While most tumors are heterozygous for EGFR mutations, studies suggest that these genetic alterations are often coupled with gene amplification [27; 33; 34]. For example, analysis of electropherograms discovered ~40% of tumors with mutations have the mutant allele equal to or greater than the WT allele, indicating gene amplification of the mutant allele [27]. In some instances, the mutations were presented as homozygous on the electropherograms, with no detectable WT sequence, which implied the amplification of the mutant allele or the loss of the WT allele [33; 34].
KRAS is a member of the Ras family GTPases that regulates cell growth, differentiation, and survival [35]. KRAS is a major oncogene for PDAC, CRC, and lung cancer and is activated mainly via somatic mutations [17; 18; 19; 20; 21; 22; 23]. KRAS amplification is rare in comparison, although it has been detected in gastric, lung, and uterine cancers [32]. Conversely, unlike in other cancer types, 11% of ovarian cancers harbor KRAS amplification, a much higher rate than KRAS mutations in this cancer type [32], suggesting that amplification of WT KRAS may be an independent cancer driver in disease subtypes [36]. KRAS mutation and WT KRAS amplifications are largely mutually exclusive in these cancer types [32]. Amplifications of the mutant KRAS allele have also been reported decades ago [37; 38; 39; 40], but the term mutant allele specific imbalance (MASI) was only recently coined by Soh et al. to describe these preferential copy number alterations specific to the mutant allele in oncogenes [5] (Table 1).
Using both SNP array analysis and gene specific assays, Soh et al demonstrated that complete MASI (homozygous mutations, with or without CNG) is a frequent occurrence in KRAS (36%) and EGFR (29%), rare for PIK3CA (6%), and intermediate at the BRAF locus (17%) in a panel of 833 cell lines of 12 tumor types [5]. Among the cancer cell lines with mutations, MASI is a frequent event in mutant EGFR (75%) and was due mainly to CNGs, while MASI in mutant KRAS (58%) was due to UPD [5]. MASI is less frequent in cancer cells with mutant BRAF (38%) and was more associated with CNGs although not conclusively. PIK3CA mutation is rare and MASI frequency among those with mutant PIK3CA is equally rare (8%). For PIK3CA, CNGs without mutation were the most frequent change [5]. The cause for the observed variations in MASI frequency among the oncogenes is unknown.
Subsequent studies confirmed that KRAS MASI is associated with selective amplification of the KRAS mutant allele at 4–18% lung cancer cases examined and correlates with poor prognosis [41; 42; 43]. Recently it has been demonstrated in vivo that mutant Kras copy gain can result in the metabolic reprogramming and increased malignancy in lung tumor cells [44]. KRAS MASI has also been reported in 18.4% of PDAC and is associated with the progression to undifferentiated carcinoma of the pancreas [45]. In CRC, KRAS MASI has been detected at 12.8–55%, is more frequently associated with G13D mutation [46; 47], and has been demonstrated to be an independent adverse prognostic factor in one study [47]. In in vitro studies, simulated KRAS MASI (with increasing amount of the mutant KRAS plasmid vector) was shown to reduced treatment responses to cetuximab treatment [46].
Preferential amplification of the mutant EGFR allele in lung cancer has also been confirmed by independent studies [48; 49]. In a report on lung adenocarcinoma, EGFR MASI was detected in 26% of the cases examined, more commonly associated with exon 19 mutations than with exon 21 mutation, and also associated with poor disease-specific survival [48]. In another study on NSCLC, EGFR MASI was detected in 37% of the cases and associated with exon 19 deletion [49]. The different techniques employed to evaluate MASI, intertumor and intratumor heterogeneity, and variations in sample collection (such as the portion of early stage vs. advanced tumors in a given study) may all account for the disparities among the reported EGFR or KRAS MASI frequencies. However, EGFR MASI does not appear to play a role in therapeutic responses to first-generation EGFR small molecular inhibitors [49], TTP (time to progress), or OS (overall survival) [50]. It’s possible that such a difference is difficult to be discerned statistically because either EGFR mutation or EGFR CNG (most studies refer to toal CNG without making a distinction between WT and mutant EGFR) alone is already associated significantly with better clinical outcomes to treatment of EGFR inhibitors [30; 50; 51; 52]. Given the rarity of the EGFR mutation, non-exclusivity of KRAS and EGFR CNGs, the lack of genomic data from large clinical trials, to thoroughly unravel the predictive value of EGFR MASI on therapeutic responsiveness and patient prognosis may require a global collaboration.
BRAF mutation is not a frequent event for lung, colorectal, or pancreatic cancers, therefore there is no BRAF MASI-oriented report in these cancer types to-date. BRAF is a major oncogene for melanoma. Recently Pfarr et al. reported that even though BRAF was mutated at high frequency in the melanoma examined (36%), only 3% harbored co-occurring mutations and amplifications of BRAF [43]. This is consistent with the previous report of Soh et al that BRAF MASI frequency is far lower comparing to KRAS and EGFR MASI, and may not be a significant mechanism for regulating BRAF signaling in cancer.
It’s important to note that in the majority of the studies on MASI, there were no vested efforts made to differentiate those MASI with CNGs from MASI without CNGs (also called aUPD) or MASI with loss of the WT allele, which will be discussed below. Given that MASI may correlate with prognosis and/or treatment responses, extra care to discern various types of MASI in future research will be highly desirable to appreciate the significance of MASI in clinical correlative studies.
3. ACQUIRED UNIPARENTAL DISOMY (aUPD) or MASI WITHOUT CNG
The development of genome-wide approaches, especially high resolution SNP arrays, enables evaluation of dynamic chromosomal as well as focal changes of copy number alterations and loss of heterozygosity (LOH) with high resolution, and thus allowing the identification of aUPD in cancer cells. Whereas LOH has been most commonly referred to gross deletions of chromosomal material encountered in cancer, aUPD is equivalent to copy-neutral LOH or MASI without CNG (Figure 1 and Table 1). Both are somatic events, with LOH due to deletion results in hemizygosity, while aUPD results in homozygosity.
UPD was initially defined to describe the inheritance of a pair of chromosomes/segments from only one parent with regard to development and developmental disorders. UPD was first reported by Engels et al in 1980 proposing that constitutional UPD was likely to occur due to the high rates of meiotic error [53]. Recent advances in molecular genetics have permitted the precise mapping and frequency of UPD to be assessed. It is now known that the extent of UPD can range from a small chromosomal segment to an entire chromosome, as proposed by Engel. The severity of the resulting phenotype of each germline UPD varies greatly. UPDs that are expected to lead to early lethality in zygotic development would go undetected. UPDs without discernable phenotypes will also go undetected. As a result, constitutional UPDs are often identified through studies of inherited diseases.
Acquired UPDs have been revealed in a variety of cancer types comparing constitutional and tumor DNA genotypes, with most studies being performed on hematological malignancies [6; 7; 8], but also in some solid tumors including CRC [54; 55], lung cancer [5], PDAC [56], pancreatic endocrine tumors [57], breast cancer [58; 59; 60], basal cell carcinoma [61], retinoblastoma [62], neuroblastomas [63], and clear cell renal carcinomas [64]. These studies have revealed that aUPD can occur in almost any chromosome, but it is becoming evident that aUPDs are non-randomly distributed with cooperation occurring between the aUPD and gene mutations. These genes can be homozygous for mutations, and mutated genes can be inactivated tumor-suppressor genes or activated oncogenes. Affected genes in regions of aUPD to-date include but not restricted to KRAS, EGFR, BRAF, MPL, MSH2, MAP2, MLH1, FHIT, TET2, APC, HLA-A/B/C, A20, EZH2, JAK2, CDKN2A, PTCH, WT1, H19, IGF2, H19, HRAS, CDKN1C, CBL, FLT3, miR-15a, miR-16-1, RB1, BRCA2, p53, NF1, BRCA1, CEBPA, RUNX1,JAK, IRF8,TNFRSF14, VPS39,PPM1D, PPM1E, C17orf71, SLCA3R1, TRIM37, PIK3CA, PTEN, CDH1, TPM3, MUC1, THBS3, CBLB, MAF, and FBXW7 [5; 6; 7; 8; 59; 60; 65; 66; 67; 68].
The underlying mechanism for aUPD is not completely understood. It has been proposed that aUPD can result from mitotic homologous recombination events, or it may represent an attempt to correct for the unbalanced loss of chromosomal material by using the remaining allele as a template. It is thought that if aUPD occurs in only a segment of the chromosome, it does so probably through mitotic recombination between identical low copy repeats in the G2 phase of the cell cycle. If aUPD involves the entire chromosome, the aUPD probably arises from a chromosomal segregation error in mitosis, in which one allele is lost in anaphase lag and the remaining allele is reduplicated [7; 8].
Correlations between somatic UPDs and clinical outcomes have been reported. In breast cancer, aUPD regions are often associated with poor overall survival, although they are not consistent adverse predictors for metastasis-free survival [60]. Higher frequency of aUPD has also been reported in triple-negative breast cancer than Her2/neu-positive and/or ER or PR-positive cases [59]. In serous ovarian cancer, aUPD is a common event and some recurrent loci are associated with a poor outcome [69]. Both better and inferior clinical outcomes have been reported to be associated with different regions of aUPD in acute myeloid leukemia [70; 71].
Despite the recognition of aUPD in oncogenes, aUPD in the EGFR pathway has been scarcely described to-date. In the study of lung cancer, CRC, and PDAC by Soh et al [5], it was demonstrated that KRAS aUPD contributed to 55% of all KRAS MASI observed, while MASI at the EGFR, BRAF, and PIK3CA were largely caused by CNG (78%, 67%, and 100%, respectively). The cause for the disparities among the oncogenes remains unexplored. Chiosea et al also reported that 47% of KRAS MASI in lung adenocarcinoma was due to CNGs, implying that the remaining cases (53%) might have harbored KRAS aUPD [41] and therefore is consistent with the previous finding. In juvenile myelomonocytic leukemia (JMML), KRAS aUPD was thought to facilitate aggressive transformation from an indolent course to fatal malignancy in a case study [67]. aUPD at the PIK3CA, FGFR3, and CDKN2A genes were thought to contribute to bladder tumorigenesis [72]. As mentioned previously, although MASI at KRAS or EGFR have been reported to be adverse prognostic markers in lung, CRC, and PDAC [41; 42; 45; 47; 48; 49], those studies failed to make a distinction between MASI with or without CNG, therefore the clinical significance of aUPD at KRAS and EGFR remains to be determined in those cancer types. Evaluation of oncogene aUPD frequency and their potential impacts on prognosis, survival, and treatment responses is much anticipated and should be addressed in future studies.
4. MASI with loss of wild-type allele or MASI with LOH
We have recently detected loss of the WT Kras allele in a genetically-engineered mouse model (GEMM) that we have developed for PDAC [9]. The GEMM, with the genotype of p16lox/lox;LSL-KrasG12D;Pdx1-Cre (referred to as PKP mice), uniformly develops precursor lesions (pancreatic intraepithelial neoplasia, PanIN) that progress to PDAC and eventually metastasis, mimicking human PanIN/PDA development and progression to metastasis at both genetic and histologic levels. From the clonal cell lines derived from both primary pancreatic tumors and metastases developed in the PKP GEMM, we detected loss of the WT Kras allele (MASI with LOH at Kras). Intriguingly the frequency of MASI with LOH at Kras is higher among those cancer cell lines derived from metastases then those from primary pancreatic tumors [9]. MASI with LOH at Kras did not appear to be a random event because it resulted in discernible functional advantages shown by colony formation, cell proliferation, and motility assays [9]. Using real-time PCR, we eliminated the possibility that differential amplification of the mutant or WT Kras allele contributed to the differences observed [9]. We further confirmed the functional advantages resulted from the loss of WT KRAS allele in isogenic colorectal cancer cell lines [73; 74] (KRASG12D/WT vs. KRASG12D/−genotypes) using colony formation, cell proliferation, and motility assays (data not shown). To ascertain that these results are not artifacts of tissue culture adaption, we microdissected liver metastases from both our mouse models and human specimens to confirm that MASI with LOH at Kras indeed occurred in vivo [9].
Non-biased LOH profiling comparing cancer cell lines derived from human primary pancreatic tumors (n=19) and metastases (n=10) was performed using SNP chip analyses. LOH at chromosome 12p, which contains KRAS, was observed in 37% of primary and 80% of metastatic cancer cell lines (p<0.02) and is the singular chromosomal arm that showed statistical difference between the two LOH profiles in this whole-genome scanning study [9]. The lack of significant difference in allelic loss on other chromosomes, indicates that this event at chromosome 12p likely occurs selectively, and is not a random manifestation of increased genomic stability during progression [9]. These data corroborate the observations made in our PKP mice and indicate that MASI with LOH at Kras is a selective event that occurs in vivo, confers growth advantage to tumor cells, and may promote metastasis.
Prior to our discovery, LOH at chromosome 12p (where KRAS resides) had been reported to correlate with the presence of KRAS mutations in human lung cancer, CRC, PDAC, and prostate cancer [75; 76; 77; 78; 79; 80]. Loss of the WT Kras allele was also found associated with Kras activation in a spontaneous lung cancer mouse model [81; 82]. Increased loss of the WT Kras allele and elevated Ras signaling also correlated with high-grade tumors in a lung cancer mouse model [83], suggesting that the loss of the WT Kras allele may facilitate tumor progression. Heterozygous deletion of the WT Kras allele was shown to be sufficient to promote tumorigenesis in vivo [84; 85]. In vitro, the reintroduction of the WT KRAS allele to colonic cancer cells harboring oncogenic KRAS resulted in growth inhibition and altered expression profiles in cell proliferation, metabolism, and transcriptional control [86; 87]. Recently, spontaneous loss of the WT Kras allele has also been reported in a T-ALL GEMM, in which the authors also demonstrated that restoration of the WT Kras protein expression in vivo has tumor-suppressive effects [88]. These results are independently verified by another related study on oncogenic Kras-driven leukemia, which demonstrated that genetic or epigenetic loss of WT Kras expression promoted tumor growth and shortened survival [89]. Our data echo these observations, but take them a step further and offer the first potential explanation for the selective loss of the WT allele- the loss of the WT KRAS allele in the context of mutant KRAS, which is akin to MASI with LOH (Table 1, Figure 1), may promote metastasis in mice and humans [9]. Our data also is supported by the findings of Martins and colleagues that the loss of the WT Kras is a selected event in tumorigenesis and that mutant Kras lung tumors are not a single disease but rather a heterogeneous group comprising two classes of tumors based on their mutant Kras allelic counts (or the WT Kras status) which contribute to their distinct metabolic profiles, prognosis, and therapeutic susceptibility [44; 83]. Together, these reports provide evidence in favor of the tumor-suppressive role of WT KRAS.
Over the decades, there has been a considerable amount of speculations on the mechanisms underlying the tumor-suppressive function of WT KRAS [90; 91; 92]. One potential mechanism is that WT RAS can bring about growth suppression by contesting for the same targets as oncogenic Ras or by interacting with an unexplored downstream target. Alternatively, normal RAS may compete with the oncogenic form, either for unknown regulatory events, or for downstream effectors. Presently, there is no consensus supporting a particular mechanism or downstream pathway from published literatures. For example, an inverse correlation between WT Kras expression and ERK activity was reported in one study and was offered as a possible molecular mechanism for the inhibitory effect of WT Kras on cellular transformation [84]. Other studies revealed that the disruption of WT RAS isoforms not cognate to the mutant RAS would compromise MAP/ERK signaling, suggesting WT RAS isoforms serve as tumor promoters in this scenario [93; 94; 95]. It has also been proposed that WT RAS may exert growth inhibition by binding to tumor suppressive RASSF1A-Nore heterodimers to activate downstream pro-apoptotic genes [96]. However, conflicting data revealed that WT KRAS can antagonize mutant KRAS-induced apoptosis via the RASSF1A-MST2-LATS1 pathway [97; 98]. This area of research remains to be further elucidated and is essential to secure the tumor-suppressive role for WT KRAS.
As discussed previously, it has been reported that MASI at KRAS is associated with worse prognosis in pancreatic and lung cancers and the observed MASI is presumably due to amplification of the mutant allele [42; 45]. Acquired UPD has been considered to be associated with tumor-suppressor genes, and until recently, rarely with oncogenes [5; 6; 99; 100]. However, we now have evidence indicating that in addition to MASI with CNGs and aUPD, MASI with LOH at KRAS also contribute to the observed MASI at the KRAS locus. Using a combination of direct genomic sequencing (mutation and MASI are determined by electrophergram), Q-PCR (provides total copy number of KRAS alleles), and Taqman Mutation Analysis Assay (provides ratio of mutant to WT KRAS alleles), we are able to differentiate between MASI resulting from amplification of the mutant allele alone (MASI with CNG) and MASI with LOH at KRAS (loss of the WT KRAS allele with or without amplification of the mutant allele) (Figure 2A). Applying this two-step analysis in a pilot study to 64 lung cancer samples with known KRAS mutations at the Columbia University Medical Center, we found that 29.4% of primary tumors and 76.9% of metastases displayed MASI with LOH at KRAS (p=0.003) (Figure 2B), further confirming that MASI with LOH at KRAS is a significant event in tumor progression and is associated with metastasis. The impacts of MASI with LOH at KRAS on other clinical parameters, such as survival or therapeutic responses, remain to be investigated.
Figure 2. Copy number analyses for mutant vs. wild-type alleles of KRAS at hot-spot G12D.
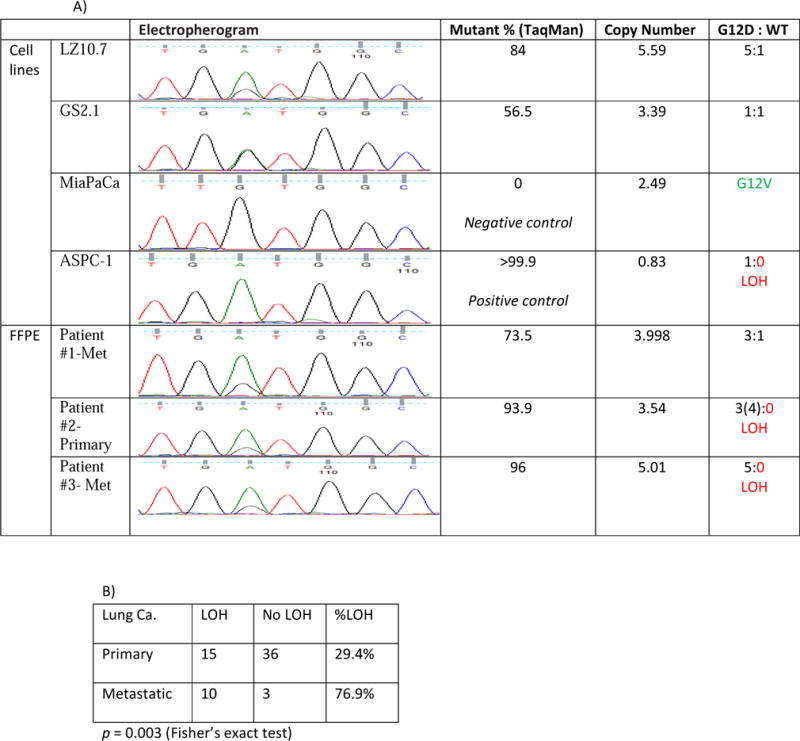
MASI with LOH at KRAS was determined by a combination of the direct genomic sequencing (electropherogram), Taqman mutation analysis assay, and Q-PCR. A) Pancreatic cancer cell lines with G12D mutation with no LOH, G12V mutation (negative control), and G12D mutation with LOH (positive control) were used to develop the assay. The LOH status for each formalin-fixed, paraffin-embedded (FFPE) patient sample was then assessed using this combinative assay. Three representative cases are presented here: patient #1 had MASI with CNG, and #2 and #3 had MASI with LOH. B) Breakdowns of MASI with or without LOH present in primary lung tumors vs. metastases. MASI with LOH at KRAS is a significant event in lung cancer and is associated with metastasis (p=0.003).
5. CONCLUSION
The recognition of MASI and its classifications allows a common language to describe genomic changes pertaining to the allele specific (mutant and/or WT) alterations in oncogenes. While it is now indisputable that MASI, aUPD, and MASI with loss of the WT allele are common occurrence in cancers, their functional or clinical implications are less certain. In general, MASI is an adverse prognostic marker in numerous cancer types examined. We and colleagues have demonstrated that MASI with loss of the WT allele can enhance or promote malignant growth. We have also demonstrated that MASI with LOH at KRAS is associated with metastasis. aUPD is not unique to oncogenes; in fact, it is more consistently offered as a mechanism to inactivate tumor-suppressor genes, such as p16, p53, Rb. Therefore, the frequency of aUPD in oncogenes and their implication in tumorigenesis are scarcely described. Overall, most of the MASI studies to-date do not make a distinction on MASI with CNGs, MASI without CNGs/aUPD, or MASI with loss of the WT allele. The value of discerning various types of MASI in clinicopathological correlative studies may seem uncertain, however, given that genetic mutation profiles alone do not always predict treatment responses of target therapies based on genetic mutations, and MASI has been shown to associate with treatment responses in some early studies, additional efforts to distinguish various types of MASI may be warranted to generate more precise data in future clinical correlative analyses.
Investigations on the mechanisms and functional impacts of MASI would be equally anticipated in the future research. Whereas it may be conceivable to image amplification of the mutant allele of an oncogene would promote tumorigenesis and correlate to worse prognosis, it has been more challenging to convince skeptics that the WT allele of an oncogene may possess tumor-suppressive function in the presence of the mutant allele. The identification of the underlying mechanism responsible for the profound effect of the WT KRAS allele on mutant KRAS-driven tumorigenesis would undoubtedly further cement its putative role in tumor suppression. The finding that WT KRAS does function as a tumor suppressor may come with significant implications for the development of therapeutics that target KRAS activity- it may be essential to design future target therapies to inactivate mutant form of KRAS specifically without affecting WT KRAS functions, because KRAS target therapies that do not distinguish between WT and mutant KRAS may inadvertently promote tumor progression and/or metastasis, and would have unintended devastating impacts on cancer patients.
Over the past decade, comprehensive sequencing efforts have revealed the genomic landscapes of common forms of human cancer. To date, these studies have revealed ~140 genes that, when altered by intragenic mutations can promote or “drive” tumorigenesis. As these efforts have matured, it would only be appropriate to consider other mechanisms of gene regulation that may contribute to tumor progression and metastasis in addition to intragenic mutation. While the concept of gene amplification is not new, the significance of secondary copy number alterations that could further regulate an activated oncogene and thus provide growth advantages to tumor cells has not yet been fully realized. The profound impacts of MASI on tumor progression and metastasis, patient prognosis and survival, therapeutic responses, and tumor recurrence would be worthy in-depth examinations in the near future.
HIGHLIGHTS.
This review describes and explains various classes of mutant allele specific imbalance (MASI) pertaining to oncogenes and their significance in tumorigenesis.
This review highlights the occurrence of MASI at the EGFR and KRAS loci and their impacts on tumor growth, progression, and metastasis, as well as their implications in patient prognosis and treatment responses.
We review and discuss the putative tumor-suppressive role of the wild-type KRAS allele in the context of the activated mutant KRAS allele in cancer, notably in pancreatic and lung cancers.
Acknowledgments
Grant Support: This study was supported by NIH/NCI R01 CA109525 and R01 CA178445
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: No potential conflicts of interest were disclosed.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grutzmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA, I. Australian Pancreatic Cancer Genome. Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV, Grimmond SM. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levens D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harb Perspect Med. 2013;3 doi: 10.1101/cshperspect.a014233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacot W, Fiche M, Zaman K, Wolfer A, Lamy PJ. The HER2 amplicon in breast cancer: Topoisomerase IIA and beyond. Biochim Biophys Acta. 2013;1836:146–157. doi: 10.1016/j.bbcan.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Soh J, Okumura N, Lockwood WW, Yamamoto H, Shigematsu H, Zhang W, Chari R, Shames DS, Tang X, MacAulay C, Varella-Garcia M, Vooder T, Wistuba, Lam S, Brekken R, Toyooka S, Minna JD, Lam WL, Gazdar AF. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4:e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med. 2009;15:120–128. doi: 10.1016/j.molmed.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Lapunzina P, Monk D. The consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer. Biol Cell. 2011;103:303–317. doi: 10.1042/BC20110013. [DOI] [PubMed] [Google Scholar]
- 8.Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:3913–3923. doi: 10.1158/1078-0432.CCR-10-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, Chen D, Arking DE, Lowy AM, Hruban RH, Remotti HE, Su GH. Disruption of p16 and Activation of Kras in Pancreas Increases Ductal Adenocarcinoma Formation and Metastasis in vivo. Oncotarget. 2011;2:862–873. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandhi J, Zhang J, Xie Y, Soh J, Shigematsu H, Zhang W, Yamamoto H, Peyton M, Girard L, Lockwood WW, Lam WL, Varella-Garcia M, Minna JD, Gazdar AF. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4:e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 12.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 13.Schonleben F, Qiu W, Ciau NT, Ho DJ, Li X, Allendorf JD, Remotti HE, Su GH. PIK3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:3851–3855. doi: 10.1158/1078-0432.CCR-06-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984;223:661–664. doi: 10.1126/science.6695174. [DOI] [PubMed] [Google Scholar]
- 15.Bos JL. Ras oncogenes in human cancer: A review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 16.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a Cancer Journal for Clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 17.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 18.Smit VT, Boot AJM, Smits AMM, Fleuren GJ, Cornelisse CJ, Bos JL. K-ras codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobunai T, Watanabe T, Yamamoto Y, Eshima K. The frequency of KRAS mutation detection in human colon carcinoma is influenced by the sensitivity of assay methodology: a comparison between direct sequencing and real-time PCR. Biochem Biophys Res Commun. 2010;395:158–162. doi: 10.1016/j.bbrc.2010.03.167. [DOI] [PubMed] [Google Scholar]
- 20.Cejas P, Lopez-Gomez M, Aguayo C, Madero R, de Castro Carpeno J, Belda-Iniesta C, Barriuso J, Moreno Garcia V, Larrauri J, Lopez R, Casado E, Gonzalez-Baron M, Feliu J. KRAS mutations in primary colorectal cancer tumors and related metastases: a potential role in prediction of lung metastasis. PLoS One. 2009;4:e8199. doi: 10.1371/journal.pone.0008199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, van Bodegom PC, Bos JL. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res. 1988;48:5738–5741. [PubMed] [Google Scholar]
- 22.Suzuki Y, Orita M, Shiraishi M, Hayashi K, Sekiya T. Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene. 1990;5:1037–1043. [PubMed] [Google Scholar]
- 23.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, Stratton MR. Cosmic 2005. Br J Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, Futreal PA, Stratton MR, Wooster R, Leung SY. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002;62:6451–6455. [PubMed] [Google Scholar]
- 25.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 26.Schonleben F, Qiu W, Bruckman KC, Ciau NT, Li X, Lauerman MH, Frucht H, Chabot JA, Allendorf JD, Remotti HE, Su GH. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Lett. 2006;8:8. doi: 10.1016/j.canlet.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2005 doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 28.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 29.Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:2556–2568. doi: 10.1200/JCO.2005.07.799. [DOI] [PubMed] [Google Scholar]
- 30.Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA, Jr, Varella-Garcia M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–655. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch FR, Varella-Garcia M, Cappuzzo F, McCoy J, Bemis L, Xavier AC, Dziadziuszko R, Gumerlock P, Chansky K, West H, Gazdar AF, Crino L, Gandara DR, Franklin WA, Bunn PA., Jr Combination of EGFR gene copy number and protein expression predicts outcome for advanced non-small-cell lung cancer patients treated with gefitinib. Ann Oncol. 2007;18:752–760. doi: 10.1093/annonc/mdm003. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, Buchanan S, Peng SB, Staschke KA, Yadav V, Yue Y, Kouros-Mehr H. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014;9:e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sankaranarayanan P, Schomay TE, Aiello KA, Alter O. Tensor GSVD of patient- and platform-matched tumor and normal DNA copy-number profiles uncovers chromosome arm-wide patterns of tumor-exclusive platform-consistent alterations encoding for cell transformation and predicting ovarian cancer survival. PLoS One. 2015;10:e0121396. doi: 10.1371/journal.pone.0121396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winter E, Perucho M. Oncogene amplification during tumorigenesis of established rat fibroblasts reversibly transformed by activated human ras oncogenes. Mol Cell Biol. 1986;6:2562–2570. doi: 10.1128/mcb.6.7.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winter E, Yamamoto F, Almoguera C, Perucho M. A method to detect and characterize point mutations in transcribed genes: amplification and overexpression of the mutant c-Ki-ras allele in human tumor cells. Proc Natl Acad Sci U S A. 1985;82:7575–7579. doi: 10.1073/pnas.82.22.7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6:1353–1362. [PubMed] [Google Scholar]
- 40.Modrek B, Ge L, Pandita A, Lin E, Mohan S, Yue P, Guerrero S, Lin WM, Pham T, Modrusan Z, Seshagiri S, Stern HM, Waring P, Garraway LA, Chant J, Stokoe D, Cavet G. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244–1252. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- 41.Chiosea SI, Sherer CK, Jelic T, Dacic S. KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod Pathol. 2011;24:1571–1577. doi: 10.1038/modpathol.2011.109. [DOI] [PubMed] [Google Scholar]
- 42.Villaruz LC, Socinski MA, Cunningham DE, Chiosea SI, Burns TF, Siegfried JM, Dacic S. The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer. 2013;119:2268–2274. doi: 10.1002/cncr.28039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfarr N, Penzel R, Klauschen F, Heim D, Brandt R, Kazdal D, Jesinghaus M, Herpel E, Schirmacher P, Warth A, Weichert W, Endris V, Stenzinger A. Copy number changes of clinically actionable genes in melanoma, non-small cell lung cancer and colorectal cancer-A survey across 822 routine diagnostic cases. Genes Chromosomes Cancer. 2016;55:821–833. doi: 10.1002/gcc.22378. [DOI] [PubMed] [Google Scholar]
- 44.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krasinskas AM, Moser AJ, Saka B, Adsay NV, Chiosea SI. KRAS mutant allele-specific imbalance is associated with worse prognosis in pancreatic cancer and progression to undifferentiated carcinoma of the pancreas. Mod Pathol. 2013 doi: 10.1038/modpathol.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malapelle U, Sgariglia R, De Stefano A, Bellevicine C, Vigliar E, de Biase D, Sepe R, Pallante P, Carlomagno C, Tallini G, Troncone G. KRAS mutant allele-specific imbalance (MASI) assessment in routine samples of patients with metastatic colorectal cancer. J Clin Pathol. 2015;68:265–269. doi: 10.1136/jclinpath-2014-202761. [DOI] [PubMed] [Google Scholar]
- 47.Hartman DJ, Davison JM, Foxwell TJ, Nikiforova MN, Chiosea SI. Mutant allele-specific imbalance modulates prognostic impact of KRAS mutations in colorectal adenocarcinoma and is associated with worse overall survival. Int J Cancer. 2012;131:1810–1817. doi: 10.1002/ijc.27461. [DOI] [PubMed] [Google Scholar]
- 48.Oakley GJ, 3rd, Chiosea SI. Higher dosage of the epidermal growth factor receptor mutant allele in lung adenocarcinoma correlates with younger age, stage IV at presentation, and poorer survival. J Thorac Oncol. 2011;6:1407–1412. doi: 10.1097/JTO.0b013e31821d41af. [DOI] [PubMed] [Google Scholar]
- 49.Malapelle U, Vatrano S, Russo S, Bellevicine C, de Luca C, Sgariglia R, Rocco D, de Pietro L, Riccardi F, Gobbini E, Righi L, Troncone G. EGFR mutant allelic-specific imbalance assessment in routine samples of non-small cell lung cancer. J Clin Pathol. 2015;68:739–741. doi: 10.1136/jclinpath-2015-203101. [DOI] [PubMed] [Google Scholar]
- 50.Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Shibata T, Sakiyama T, Yoshida T, Tamura T. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:6829–6837. doi: 10.1200/JCO.2005.01.0793. [DOI] [PubMed] [Google Scholar]
- 51.Li AR, Chitale D, Riely GJ, Pao W, Miller VA, Zakowski MF, Rusch V, Kris MG, Ladanyi M. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. The Journal of molecular diagnostics : JMD. 2008;10:242–248. doi: 10.2353/jmoldx.2008.070178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dahabreh IJ, Linardou H, Siannis F, Kosmidis P, Bafaloukos D, Murray S. Somatic EGFR mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:291–303. doi: 10.1158/1078-0432.CCR-09-1660. [DOI] [PubMed] [Google Scholar]
- 53.Engel E. A new genetic concept: the uniparental disomy and its potential effect, the isodisomy (author’s transl) J Genet Hum. 1980;28:11–22. [PubMed] [Google Scholar]
- 54.Andersen CL, Wiuf C, Kruhoffer M, Korsgaard M, Laurberg S, Orntoft TF. Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis. 2007;28:38–48. doi: 10.1093/carcin/bgl086. [DOI] [PubMed] [Google Scholar]
- 55.Melcher R, Al-Taie O, Kudlich T, Hartmann E, Maisch S, Steinlein C, Schmid M, Rosenwald A, Menzel T, Scheppach W, Luhrs H. SNP-Array genotyping and spectral karyotyping reveal uniparental disomy as early mutational event in MSS- and MSI-colorectal cancer cell lines. Cytogenet Genome Res. 2007;118:214–221. doi: 10.1159/000108303. [DOI] [PubMed] [Google Scholar]
- 56.Harada T, Chelala C, Bhakta V, Chaplin T, Caulee K, Baril P, Young BD, Lemoine NR. Genome-wide DNA copy number analysis in pancreatic cancer using high-density single nucleotide polymorphism arrays. Oncogene. 2008;27:1951–1960. doi: 10.1038/sj.onc.1210832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagano Y, Kim DH, Zhang L, White JA, Yao JC, Hamilton SR, Rashid A. Allelic alterations in pancreatic endocrine tumors identified by genome-wide single nucleotide polymorphism analysis. Endocr Relat Cancer. 2007;14:483–492. doi: 10.1677/ERC-06-0090. [DOI] [PubMed] [Google Scholar]
- 58.Murthy SK, DiFrancesco LM, Ogilvie RT, Demetrick DJ. Loss of heterozygosity associated with uniparental disomy in breast carcinoma. Mod Pathol. 2002;15:1241–1250. doi: 10.1097/01.MP.0000032535.62750.D1. [DOI] [PubMed] [Google Scholar]
- 59.Tuna M, Smid M, Zhu D, Martens JW, Amos CI. Association between acquired uniparental disomy and homozygous mutations and HER2/ER/PR status in breast cancer. PLoS One. 2010;5:e15094. doi: 10.1371/journal.pone.0015094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tuna M, Smid M, Martens JW, Foekens JA. Prognostic value of acquired uniparental disomy (aUPD) in primary breast cancer. Breast Cancer Res Treat. 2012;132:189–196. doi: 10.1007/s10549-011-1579-y. [DOI] [PubMed] [Google Scholar]
- 61.Teh MT, Blaydon D, Chaplin T, Foot NJ, Skoulakis S, Raghavan M, Harwood CA, Proby CM, Philpott MP, Young BD, Kelsell DP. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Res. 2005;65:8597–8603. doi: 10.1158/0008-5472.CAN-05-0842. [DOI] [PubMed] [Google Scholar]
- 62.Hagstrom SA, Dryja TP. Mitotic recombination map of 13cen-13q14 derived from an investigation of loss of heterozygosity in retinoblastomas. Proc Natl Acad Sci U S A. 1999;96:2952–2957. doi: 10.1073/pnas.96.6.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caren H, Erichsen J, Olsson L, Enerback C, Sjoberg RM, Abrahamsson J, Kogner P, Martinsson T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: four cases of homozygous deletions of the CDKN2A gene. BMC Genomics. 2008;9:353. doi: 10.1186/1471-2164-9-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toma MI, Grosser M, Herr A, Aust DE, Meye A, Hoefling C, Fuessel S, Wuttig D, Wirth MP, Baretton GB. Loss of heterozygosity and copy number abnormality in clear cell renal cell carcinoma discovered by high-density affymetrix 10K single nucleotide polymorphism mapping array. Neoplasia. 2008;10:634–642. doi: 10.1593/neo.08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawashima-Goto S, Imamura T, Seki M, Kato M, Yoshida K, Sugimoto A, Kaneda D, Fujiki A, Miyachi M, Nakatani T, Osone S, Ishida H, Taki T, Takita J, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Ogawa S, Hosoi H. Identification of a homozygous JAK3 V674A mutation caused by acquired uniparental disomy in a relapsed early T-cell precursor ALL patient. Int J Hematol. 2015;101:411–416. doi: 10.1007/s12185-014-1711-y. [DOI] [PubMed] [Google Scholar]
- 66.Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, Fox H, Jacobi K, Saiya-Cork K, Bixby D, Lebovic D, Roulston D, Shedden K, Sabel M, Marentette L, Cimmino V, Chang AE, Malek SN. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123:1487–1498. doi: 10.1182/blood-2013-05-500264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kato M, Yasui N, Seki M, Kishimoto H, Sato-Otsubo A, Hasegawa D, Kiyokawa N, Hanada R, Ogawa S, Manabe A, Takita J, Koh K. Aggressive transformation of juvenile myelomonocytic leukemia associated with duplication of oncogenic KRAS due to acquired uniparental disomy. J Pediatr. 2013;162:1285–1288. 1288 e1281. doi: 10.1016/j.jpeds.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 68.Eldai H, Periyasamy S, Qarni S Al, Rodayyan M Al, Mustafa SM, Deeb A, Sheikh E Al, Khan MA, Johani M, Yousef Z, Aziz MA. Novel Genes Associated with Colorectal Cancer Are Revealed by High Resolution Cytogenetic Analysis in a Patient Specific Manner. PLoS One. 2013;8 doi: 10.1371/journal.pone.0076251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tuna M, Ju Z, Smid M, Amos CI, Mills GB. Prognostic relevance of acquired uniparental disomy in serous ovarian cancer. Mol Cancer. 2015;14:29. doi: 10.1186/s12943-015-0289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bullinger L, Kronke J, Schon C, Radtke I, Urlbauer K, Botzenhardt U, Gaidzik V, Cario A, Senger C, Schlenk RF, Downing JR, Holzmann K, Dohner K, Dohner H. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2010;24:438–449. doi: 10.1038/leu.2009.263. [DOI] [PubMed] [Google Scholar]
- 71.Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, Lillington D, Lister TA, Young BD. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65:9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- 72.Schepeler T, Lamy P, Hvidberg V, Laurberg JR, Fristrup N, Reinert T, Bartkova J, Tropia L, Bartek J, Halazonetis TD, Pan CC, Borre M, Dyrskjot L, Orntoft TF. A high resolution genomic portrait of bladder cancer: correlation between genomic aberrations and the DNA damage response. Oncogene. 2013;32:3577–3586. doi: 10.1038/onc.2012.381. [DOI] [PubMed] [Google Scholar]
- 73.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2001;19:940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 74.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA, Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J, Zhang Z, Dai Z, Plass C, Morrison C, Wang Y, Wiest JS, Anderson MW, You M. LOH of chromosome 12p correlates with Kras2 mutation in non-small cell lung cancer. Oncogene. 2003;22:1243–1246. doi: 10.1038/sj.onc.1206192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wan J, Li H, Li Y, Zhu ML, Zhao P. Loss of heterozygosity of Kras2 gene on 12p12-13 in Chinese colon carcinoma patients. World Journal of Gastroenterology. 2006;12:1033–1037. doi: 10.3748/wjg.v12.i7.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takeuchi S, Mori N, Koike M, Slater J, Park S, Miller CW, Miyoshi I, Koeffler HP. Frequent loss of heterozygosity in region of the KIP1 locus in non-small cell lung cancer: evidence for a new tumor suppressor gene on the short arm of chromosome 12. Cancer Res. 1996;56:738–740. [PubMed] [Google Scholar]
- 78.Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, Rosell R, Monzo M, Wu L, Westra WH, Yang SC, Jen J, Sidransky D. Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res. 2001;61:1309–1313. [PubMed] [Google Scholar]
- 79.Kibel AS, Schutte M, Kern SE, Isaacs WB, Bova GS. Identification of 12p as a region of frequent deletion in advanced prostate cancer. Cancer Res. 1998;58:5652–5655. [PubMed] [Google Scholar]
- 80.Mahlamaki EH, Hoglund M, Gorunova L, Karhu R, Dawiskiba S, Andren-Sandberg A, Kallioniemi OP, Johansson B. Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Genes Chromosomes Cancer. 1997;20:383–391. doi: 10.1002/(sici)1098-2264(199712)20:4<383::aid-gcc10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 81.Hegi ME, Devereux TR, Dietrich WF, Cochran CJ, Lander ES, Foley JF, Maronpot RR, Anderson MW, Wiseman RW. Allelotype analysis of mouse lung carcinomas reveals frequent allelic losses on chromosome 4 and an association between allelic imbalances on chromosome 6 and K-ras activation. Cancer Res. 1994;54:6257–6264. [PubMed] [Google Scholar]
- 82.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 83.Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GI, Martins CP. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z, Wang Y, Vikis H, Johnson L, Liu G, Li J, Anderson M, Sills R, Hong H, Devereux T, Jacks T, Guan K, You M. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 85.James RM, Arends MJ, Plowman SJ, Brooks DG, Miles CG, West JD, Patek CE. K-ras proto-oncogene exhibits tumor suppressor activity as its absence promotes tumorigenesis in murine teratomas. Mol Cancer Res. 2003;1:820–825. [PubMed] [Google Scholar]
- 86.Li H, Cao HF, Wan J, Li Y, Zhu ML, Zhao P. Growth inhibitory effect of wild-type Kras2 gene on a colonic adenocarcinoma cell line. World Journal of Gastroenterology. 2007;13:934–938. doi: 10.3748/wjg.v13.i6.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H, Cao HF, Li Y, Zhu ML, Wan J. Changes in gene-expression profiles of colon carcinoma cells induced by wild type K-ras2. World Journal of Gastroenterology. 2007;13:4620–4625. doi: 10.3748/wjg.v13.i34.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Staffas A, Karlsson C, Persson M, Palmqvist L, Bergo MO. Wild-type KRAS inhibits oncogenic KRAS-induced T-ALL in mice. Leukemia. 2014 doi: 10.1038/leu.2014.315. [DOI] [PubMed] [Google Scholar]
- 89.Kong G, Chang YI, Damnernsawad A, You X, Du J, Ranheim EA, Lee W, Ryu MJ, Zhou Y, Xing Y, Chang Q, Burd CE, Zhang J. Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia. 2016;30:1542–1551. doi: 10.1038/leu.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh A, Sowjanya AP, Ramakrishna G. The wild-type Ras: road ahead. FASEB J. 2005;19:161–169. doi: 10.1096/fj.04-2584hyp. [DOI] [PubMed] [Google Scholar]
- 91.Pfeifer GP. A new verdict for an old convict. Nat Genet. 2001;29:3–4. doi: 10.1038/ng0901-3. [DOI] [PubMed] [Google Scholar]
- 92.Zhou B, Der CJ, Cox AD. The role of wild type RAS isoforms in cancer. Semin Cell Dev Biol. 2016 doi: 10.1016/j.semcdb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bentley C, Jurinka SS, Kljavin NM, Vartanian S, Ramani SR, Gonzalez LC, Yu K, Modrusan Z, Du P, Bourgon R, Neve RM, Stokoe D. A requirement for wild-type Ras isoforms in mutant KRas-driven signalling and transformation. Biochem J. 2013;452:313–320. doi: 10.1042/BJ20121578. [DOI] [PubMed] [Google Scholar]
- 94.Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH, Bar-Sagi D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discovery. 2013;3:112–123. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- 96.Ortiz-Vega S, Khokhlatchev A, Nedwidek M, Zhang XF, Dammann R, Pfeifer GP, Avruch J. The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene. 2002;21:1381–1390. doi: 10.1038/sj.onc.1205192. [DOI] [PubMed] [Google Scholar]
- 97.Matallanas D, Romano D, Al-Mulla F, O’Neill E, Al-Ali W, Crespo P, Doyle B, Nixon C, Sansom O, Drosten M, Barbacid M, Kolch W. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol Cell. 2011;44:893–906. doi: 10.1016/j.molcel.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 98.Romano D, Maccario H, Doherty C, Quinn NP, Kolch W, Matallanas D. The differential effects of wild-type and mutated K-Ras on MST2 signaling are determined by K-Ras activation kinetics. Mol Cell Biol. 2013;33:1859–1868. doi: 10.1128/MCB.01414-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Walsh CS, Ogawa S, Scoles DR, Miller CW, Kawamata N, Narod SA, Koeffler HP, Karlan BY. Genome-wide loss of heterozygosity and uniparental disomy in BRCA1/2-associated ovarian carcinomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:7645–7651. doi: 10.1158/1078-0432.CCR-08-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bacolod MD, Schemmann GS, Giardina SF, Paty P, Notterman DA, Barany F. Emerging paradigms in cancer genetics: some important findings from high-density single nucleotide polymorphism array studies. Cancer Res. 2009;69:723–727. doi: 10.1158/0008-5472.CAN-08-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]