

Abstract
Digoxin is a drug widely used to treat heart failure and studies have demonstrated its potential as anticancer agent. In addition, digoxin presents the potential to interact with a series of other compounds used in medicine. The aim of the present study was to evaluate in vitro the cytotoxicity, genotoxicity and mutagenicity of digoxin and its potential to interact with the mutagen Mitomycin C (MMC). The cytotoxicity of digoxin was assessed by employing the MTT method and the comet assay was performed to assess the genotoxicity of this medicine in CHO-K1 and HeLa cell lines. Besides, the cytokinesis-block micronucleus assay was performed to assess the mutagenicity and the antimutagenicity of this drug. The Ames assay was also performed with TA98 and TA100 strains of S. typhimurium. Results showed that digoxin was cytotoxic, genotoxic and mutagenic for HeLa and CHO-K1 cell lines at concentrations many times higher than those observed in human therapeutic conditions. Nevertheless, an antimutagenic effect against the mutagen MMC was observed on both cell lines in concentrations near those used therapeutically in humans. This chemoprotective effect observed is an interesting finding that should be better explored regarding its impact in anticancer chemotherapy.
Keywords: Mutagenicity, Antimutagenicity, Genotoxicity, Desmutagen
Introduction
Cardiac glycosides are common substances in the plant kingdom and different molecules of this group, such as digoxin, digitoxin and ouabain, have been used for the treatment of heart failure and atrial arrhythmia (Rahimtoola and Tak 1996). They consist of three structures: a steroidal nucleus, an unsaturated lactone (jointly called “aglycone”) and a carbohydrate (Cerella et al. 2013). The main pharmacological effect of cardiac glycosides in therapeutic doses is the inhibition of Na, K-ATPase. In the myocardium, these agents bind reversibly to the α subunit of Na, K-ATPase, leading to raised intracellular sodium levels that result in an increase of calcium ions in the myocytes (Hallböök et al. 2011).
Nevertheless, besides the effects described in myocytes, epidemiological studies developed in the middle of the 1960s indicated that oncology patients receiving cardiac glycosides presented significantly lower mortality rates than patients not treated with such drugs (Xue et al. 2015). For instance, Stenkvist et al. (1979) reported that the tumor cell populations from breast cancer patients on digitalis medication for cardiac problems appeared to have a lower proliferative rate than tumor cells from patients who were not on digitalis treatment, principally digoxin. Moreover, Stenkvist et al. (1982) also described that the recurrence rate of breast cancer in patients not on digitalis was almost ten times higher than that in breast cancer patients treated with digitalis. However, later studies have shown that the use of digoxin by women increased the risks of breast cancer (Ahern et al. 2008; Biggar et al. 2011; Ahern et al. 2014). Complementarily, Boursi et al. (2014) affirm that digoxin use is associated with increased human colorectal cancer risk. Despite the well-characterized effects of digoxin on Na, K-ATPase, especially in cardiac tissue, its effects in other biological contexts are unclear and, in some cases, controversial.
In vitro studies have shown that cardiac glycosides induce different effects against a variety of cancer cells, including apoptotic and antimigratory action (Özdemir et al. 2016), autophagic death induction (Wang et al. 2012) and reduction of proliferative rates (Rascón-Valenzuela et al. 2015). Bearing in mind the examples of effects observed in cancer cells exposed to cardiac glycosides, exploring their anticancer effect is an interesting approach, since they have been on the market for decades. In this sense, due to the high cost and long period required for research & development (R&D) of new drugs as anticancer agents, the discovery of such potential drugs that are already known and used clinically can be considered a relevant short cut, which can contribute much in this area of the pharmaceutical industry (Hung and Liu 2012).
In the present work, the mutagenicity of digoxin was evaluated by employing different biological systems to assess gene and chromosomal mutations in vitro. In addition, the interactions of digoxin with a known anticancer chemotherapeutic agent were also evaluated in different protocols in human and rodent cell lines and an antimutagenic effect was identified. The term “mutagenicity” refers to the induction of heritable changes in the genetic material of living systems, which may result in changes in the phenotype. On the other hand, antimutagens are compounds, natural or synthetic, that are able to inactivate the effects of mutagenic agents (Sloczyska et al. 2014). Since the 70s, chemoprevention has been considered a potential strategy to lower cancer incidence and, consequently, cancer-related death (Sporn et al. 1976).
In this way, the experimental information described in this study can provide a better understanding of the genetic effects associated with the use of digoxin and help to elucidate the biological results of the action of this molecule at cellular level.
Materials and methods
Mammalian cell cultures
HeLa (Human Cervix Carcinoma—ATCC CCL-2) and CHO-K1 (Chinese Hamster Ovary—ATCC CCL-61) cells were used in the present study. HeLa was cultured in DMEM (Sigma, St. Louis, MO, USA) and CHO-K1 was maintained in HAM F-10 medium (Sigma, St. Louis, MO, USA). For all cultures, media were supplemented with 10% fetal bovine serum (FBS), 60 µg/ml streptomycin and 100 U/ml penicillin; the cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
Cell viability assay
The effects of digoxin on cell viability in the CHO-K1 cell line were assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma, St. Louis, MO, USA) colorimetric method. Briefly, the cells were plated in 96-well plates (1 × 105 cells/well) and incubated for 24 h at 37 °C in a humid atmosphere with 5% CO2. After this period, the wells were washed with culture medium and incubated with digoxin at different concentrations (3.9–500 μM, diluted in dimethyl sulfoxide (DMSO)/water for 24 or 48 h). The final concentration of DMSO in cell culture medium during the treatments did not exceed 1%. After incubation, the plates were treated with MTT and the formazan formed was dissolved with DMSO. The readings were performed using a microplate reader (Biotek Powerwave XS2) at 550 nm. Cytotoxicity was scored as the percentage reduction in absorbance in treatments with digoxin versus the solvent control group (Gomes et al. 2011; Gonçalves et al. 2014). All experiments were performed in triplicate.
Genotoxicity assays
Cytokinesis-block micronucleus assay
In this study, to assess the induction of chromosomal mutations in vitro the cytokinesis-block micronucleus assay (CBMN) was performed as described by Fenech (2000, 2007), with some adaptations. Cells (HeLa and CHO-K1) were seeded in 24-well plates (2.5 × 105 cells/well) and maintained at 37 °C in a humid atmosphere with 5% CO2. After 24 h, cells were washed twice with PBS (phosphate buffer—0.06 M Na2HPO4 and 0.06 M KH2PO4, pH 6.8) and the treatments were performed in culture medium without serum for three hours. The negative control group was treated with PBS and a positive control group was established with the treatment of the cells with methyl methanesulfonate (MMS—400 µM).
Different concentrations of digoxin were assessed. For the HeLa cell line, the selection of concentrations employed was based on a previous study of our group (Rocha et al. 2014). For the CHO-K1 cell line, the selection of concentrations employed was based on the results of the MTT assay described above. To complement the results, both cell lines were also treated with noncytotoxic concentrations of digoxin, near those observed in the plasma of patients that use this drug in cardiac treatments (0.7, 1.5 and 3.0 nM).
After completing the treatments with digoxin (3 h), cells were washed twice with PBS and fresh complete medium containing Cytochalasin-B (3.0 µg/mL) was added. Cytochalasin-B prevented polymerization of the actin filaments and blocked cytokinesis, resulting in the formation of binucleated cells. After 15 h (CHO-K1) or 24 h (HeLa), cells were washed twice with PBS, trypsinized and centrifuged for 5 min at 400 g. The pellet was then resuspended in chilled hypotonic solution (1% sodium citrate) together with 10 µl of 1% formaldehyde and carefully homogenized with a Pasteur pipette. The cell suspension was centrifuged for 5 min at 400 g and resuspended in 5 mL methanol/acetic acid (3:1 v/v). Next, the tubes were centrifuged for 5 min, the supernatant discarded, and the cell suspension poured onto slides previously cleaned and covered with a film of chilled ultrapure water.
At the time of cytogenetic analysis, slides were stained with DAPI (4′,6-diamidino-2-phenylindole) diluted in PBS for 2 min, washed with distilled water and analyzed in a fluorescent microscope (Zeiss, Axioscope—excitation filter of 365 nm and barrier filter of 445/450 nm) (Gontijo et al. 2015).
One thousand binucleated cells were analyzed for each treatment and the frequencies of micronucleated cells were obtained. In a blind test, cells containing 1–4 micronuclei were scored. The criteria for the identification of MN followed the recommendations of Fenech (2000) and Titenko-Holland et al. (1997). Statistical analysis was performed applying ANOVA followed by a Student–Newman–Keuls test.
Nuclear Division Index (NDI)
The influence of digoxin on cell proliferation was assessed by calculating the Nuclear Division Index (NDI) for HeLa and CHO-K1 cells. For calculating the NDI, the slides prepared for the CBMN assay were used and 300 cells with a well-preserved cytoplasm were counted using fluorescence microscopy. The NDI was calculated according to Eastmond and Tucker (1989) using the formula: NDI = (M1 + 2(M2) + 3(M3) + 4(M4))/N, where M1–M4 is the number of cells with 1, 2, 3 and 4 nuclei, respectively, and N is the total number of viable cells counted.
Single-cell gel electrophoresis assay
The single-cell gel electrophoresis assay (Comet assay) was performed in the alkaline version according to Olive and Banáth (2006) with adaptations. Briefly, CHO-K1 and HeLa cells were seeded in 24-well plates (2 × 105 cells/well) in complete medium. Twenty-four hours after this, cells were washed twice with PBS and incubated with the different treatments for 3 h in culture medium without serum. The CHO-K1 cell line was treated with 0.7, 1.5, 3.0 nM, 20, 35 and 50 µM of digoxin. The HeLa cell line was treated with 0.7, 1.5, 3.0, 25, 50 and 100 nM of digoxin. The positive control group was exposed to MMS (120 µM) and the negative control group was treated with 10 µL of PBS.
After the treatments, the cells were washed twice with PBS and detached using a trypsin–EDTA solution (0.25%) and the cell suspension obtained was centrifuged (5 min, 400 g). The pellet was resuspended in 500 µL of PBS and 30 µL aliquots of the cell suspensions were mixed with 120 µL of low-melting-point agarose (0.75% in PBS). These mixtures were poured onto slides precoated with normal-melting-point agarose (1.5% in PBS), covered with coverslips and maintained for 5 min at 4 °C. After this, the coverslips were removed and the slides were immersed in the lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10; 1% Triton X-100 and 10% DMSO) for at least 1 h (4 °C).
After the lysis process, the slides were washed with PBS and maintained in a horizontal electrophoresis box filled with cold alkaline buffer (1 mM EDTA, 300 mM NaOH, pH ≥ 13) for 40 min. Electrophoresis was conducted at approximately 1 V/cm and 300 mA for 20 min and the slides were then neutralized in a Tris solution (0.4 M, pH 7.5), fixed with methanol, air dried and stained with ethidium bromide (2 µg/mL). The analysis was carried out under fluorescence microscopy (Zeiss Axioscope A1—excitation 510–560 nm; emission 590–650 nm). The quantification of DNA breakages was achieved by visual scoring with the comets being classified from 0 (no damage) to 4 (maximum damage) (Collins 2004). For each treatment, 100 comets were analyzed and the level of DNA damage for each experimental condition was based on the score, which was calculated employing the equation:
where C0–C4 are the numbers of comets in each classification of damage.
The means of the score values were calculated for each treatment based on independent experiments. For statistical analysis, ANOVA was performed followed by the Student–Newman–Keuls post-test with a significance level of 0.05.
Ames mutagenicity assay
The Ames mutagenicity assay was performed employing the Kado microsuspension method with the Salmonella typhimurium strains TA100 and TA98, with and without metabolic activation (Mortelmans and Zeiger 2000). Five different concentrations of digoxin (diluted in DMSO) were evaluated in this assay (200–1000 µM). The selection of concentrations was based on the bacterial toxicity identified in preliminary tests. Each concentration of digoxin (in a maximum volume of 10 µL) was added to 50 µL of PBS (pH 7.4) and 10 µL of bacterial culture (1–2 × 109 CFU) and then incubated at 37 °C for 90 min. After this time, 2 mL of top agar was added to the mixture and poured on to a plate containing minimal agar. The plates were incubated at 37 °C for 48 h, and his + revertant colonies were manually counted. The influence of metabolic activation was tested by adding 50 µL of S9 mixture (4%). The S9 mix was freshly prepared before each test from an Aroclor-1254-induced rat liver fraction (lyophilized—Moltox Molecular Toxicology Inc, Boone, NC, USA). All experiments were performed in triplicate.
The standard mutagens 4-nitro-o-phenylenediamine (1 µg/plate) and sodium azide (0.125 µg/plate) were used as positive controls in experiments without S9 mix for TA98 and TA100, respectively. In the experiments with metabolic activation, 2-aminoanthracene (0.125 µg/plate) was used for both strains. DMSO served as negative control (10 µL/plate).
Statistical analysis was performed with Salanal software (Integrated Laboratory Systems, Research Triangle Park, NC, USA) applying Bernstein’s model. The Mutagenicity Index (MI) was calculated applying the equation MI = Rev Trat/Rev Control for each concentration assessed, where “Rev Trat” is the frequency of revertants per plate in treated groups and “Rev Control” is the frequency of revertants in the negative control group. Only conditions that induced an MI ≥ 2 were considered mutagenic in this study (Thome et al. 2012; Santos et al. 2013).
Antimutagenicity assays
Cytokinesis-block micronucleus assay and Nuclear Division Index
The same procedures used in the CBMN assay to evaluate the mutagenicity were used in the evaluation of antimutagenicity. However, the treatment with digoxin (0.7, 1.5 and 3.0 nM) was performed in association with the known mutagen Mitomycin C (MMC—4.8 µM) in different treatment protocols: pre-complexation treatment, simultaneous treatment, pre-treatment and post-treatment (Fig. 1).
Fig. 1.

Schema of treatments in CBMN assay for evaluation of the antimutagenicity in HeLa and CHO-K1 cell lines
The antimutagenic activity was defined by the percentage of reduction of mutagenicity (%Red) calculated for each digoxin concentration associated with MMC following the equation %Red = ((MN A − MN B)/(MN A − MN C)) × 100, where MN A is the group of cells treated with the mutagen (MMC—4.8 µM), MN B is the group of cells treated with digoxin plus the mutagen, and MN C is the negative control group (Manoharan and Banerjee 1985; Waters et al. 1990).
Results
The results presented in Fig. 2 show that digoxin was cytotoxic to the CHO-K1 cell line but only concentrations ≥125 µM reduced significantly the cell viability (24 and 48 h). In a previous work by our research group (Rocha et al. 2014), digoxin reduced the cell viability of HeLa cells treated with concentrations ≥150 nM, in 24 and 48 h of exposition.
Fig. 2.

Cell viability obtained from the MTT assay performed with the CHO-K1 cell line after treatment with different concentrations of digoxin in 24–48 h of exposition. *(p < 0.05)
The CBMN assay was realized with both cell lines employing noncytotoxic concentrations and, as verified in Tables 1, 2 and 3, digoxin induced chromosomal mutations just in higher concentrations. The micronuclei frequency was higher than in the control group when CHO-K1 cells were treated with 20, 35 and 50 µM of digoxin. The same was observed in HeLa cells at concentrations of 25, 50 and 100 nM. Besides the mutagenicity described above, the treatment with digoxin reduced the mitotic index in the CHO-K1 cell line, in all concentrations assessed. However, when concentrations near those observed in the blood plasma of patients in digoxin medical treatments were evaluated, no mutagenic effects were observed.
Table 1.
Frequency of binucleated cells with micronucleus and Nuclear Division Index (NDI) in CBMN assay performed in three independent experiments with higher concentrations of digoxin in the CHO-K1 cell line
| Treatments (µM) | BNMN | NDI | |||||
|---|---|---|---|---|---|---|---|
| Experiments | Mean | SD | Mean | SD | |||
| I | II | III | |||||
| 0.0 | 4 | 6 | 7 | 5.7 | 1.2 | 1.56 | 0.01 |
| 20 | 9 | 13 | 14 | 12.0* | 2.2 | 1.31* | 0.02 |
| 35 | 13 | 16 | 15 | 14.7* | 1.2 | 1.38* | 0.02 |
| 50 | 12 | 14 | 15 | 13.7* | 1.2 | 1.39* | 0.03 |
| MMS | 36 | 39 | 40 | 38.0* | 1.7 | 1.31* | 0.01 |
BNMN binucleated cells with micronucleus; SD standard deviation; MMS methyl methanesulfonate (400 µM)
* Statistically different (p < 0.05) from negative control (0.0 µM)
Table 2.
Frequency of binucleated cells with micronucleus and Nuclear Division Index (NDI) in CBMN assay performed in three independent experiments with higher concentrations of digoxin in the HeLa cell line
| Treatments (nM) | BNMN | NDI | |||||
|---|---|---|---|---|---|---|---|
| Experiments | Mean | SD | Mean | SD | |||
| I | II | III | |||||
| 0.0 | 8 | 7 | 6 | 7.00 | 0.8 | 1.45 | 0.01 |
| 25 | 10 | 9 | 8 | 9.00 | 0.8 | 1.43 | 0.02 |
| 50 | 10 | 12 | 10 | 10.7* | 0.9 | 1.43 | 0.01 |
| 100 | 14 | 15 | 17 | 15.3* | 1.2 | 1.40* | 0.01 |
| MMS | 36 | 32 | 31 | 33.0* | 2.2 | 1.32* | 0.01 |
BNMN binucleated cells with micronucleus, SD standard deviation, MMS methyl methanesulfonate (400 µM)
* Statistically different (p < 0.05) from negative control (0.0 µM)
Table 3.
Frequency of binucleated cells with micronucleus and Nuclear Division Index (NDI) in CBMN assay performed in three independent experiments with lower concentrations of digoxin in CHO-K1 and HeLa cell lines
| Treatments (nM) | CHO-K1 | HeLa | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BNMN | NDI | BNMN | NDI | |||||||||||
| Experiments | Mean | SD | Mean | SD | Experiments | Mean | SD | Mean | SD | |||||
| I | II | III | I | II | III | |||||||||
| 0.0 | 11 | 11 | 9 | 10.3 | 0.9 | 1.54 | 0.01 | 6 | 4 | 4 | 4.57 | 0.9 | 1.47 | 0.02 |
| 0.7 | 10 | 12 | 8 | 10.0 | 1.6 | 1.54 | 0.01 | 4 | 5 | 3 | 4.00 | 0.8 | 1.43 | 0.01 |
| 1.5 | 13 | 11 | 14 | 12.7 | 1.2 | 1.53 | 0.01 | 4 | 6 | 6 | 5.33 | 0.9 | 1.43 | 0.02 |
| 3.0 | 14 | 15 | 12 | 13.7 | 1.2 | 1.53 | 0.01 | 7 | 7 | 6 | 6.67 | 0.5 | 1.43 | 0.01 |
| MMS | 31 | 33 | 29 | 31.0 | 1.6 | 1.48 | 0.01 | 36 | 32 | 31 | 30.7 | 2.9 | 1.38 | 0.01 |
BNMN binucleated cells with micronucleus, SD standard deviation, MMS methyl methanesulfonate (400 µM)
The results obtained in the Alkaline Comet assay show that digoxin induced chromosomal breaks in CHO-K1 cells at higher concentrations (20, 35 and 50 µM). Similar results were observed for the HeLa cell line, which presented scores statistically higher than those observed in negative control at 50 and 100 nM of digoxin (Fig. 3). Nevertheless, in the Ames assay, digoxin did not present mutagenicity. None of the concentrations evaluated induced gene mutations without or with metabolic activation in either of the S. typhimurium strains employed (Fig. 4).
Fig. 3.
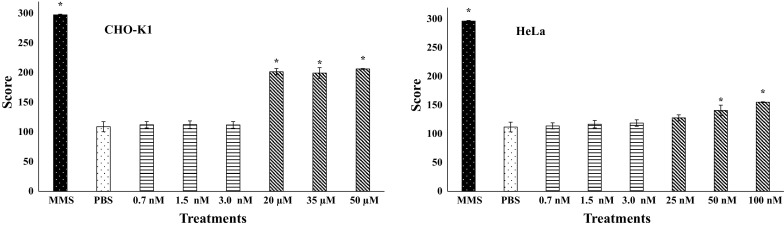
Mean of the scores and standard deviation obtained in Alkaline Comet assay performed with different concentrations of digoxin in CHO-K1 and HeLa cell lines. MMS: methyl methanesulfonate (120 µM); *statistically different (p < 0.05) from negative control (PBS)
Fig. 4.
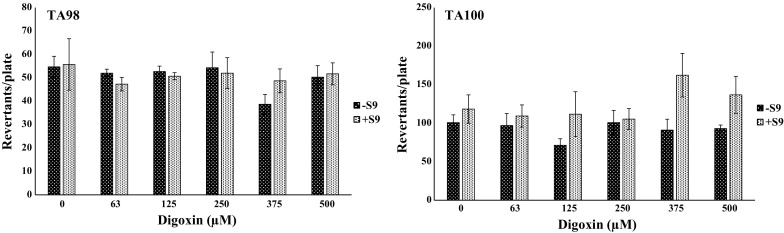
Mean of the frequency of revertants per plate and standard deviation in Ames assay performed with the strains TA98 and TA100 of Salmonella typhimurium to assess different concentrations of digoxin, with (+S9) or without (−S9) metabolic activation
The antimutagenicity studies showed that digoxin (1.5 and 3.0 nM) reduced the frequency of chromosomal mutations induced by MMC in CHO-K1 in CBMN in all treatment protocols, except in post-treatment (Table 4). For the HeLa cell line, digoxin was antimutagenic in the pre-treatment protocol at the two highest concentrations tested (1.5 and 3.0 nM). In the pre-complexation and simultaneous treatments, only the highest concentration assessed (3.0 nM) was antimutagenic, while in the post-treatment this effect was not observed (Table 5).
Table 4.
Frequency of binucleated cells with micronucleus, Nuclear Division Index (NDI) and percentage of reduction in mutagenicity (%Red) obtained in CBMN assay performed with digoxin associated with MMC in different protocols of treatment on CHO-K1 cell line
| Treatments | Protocols | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-complexation | Pre-treatment | Simultaneous treatment | Post-treatment | |||||||||
| MNBN ( | NDI ( | %Red (%) | MNBN ( | NDI ( | %Red (%) | MNBN ( | NDI ( | %Red (%) | MNBN ( | NDI ( | %Red (%) | |
| PBS | 6.3 ± 1.2* | 1.5 ± 0.01 | 7.3 ± 0.9* | 1.5 ± 0.01 | 7.0 ± 0.8* | 1.5 ± 0.01 | 11.0 ± 0.8* | 1.5 ± 0.01 | ||||
| MMC | 30.7 ± 1.7 | 1.5 ± 0.01 | 30.7 ± 1.7 | 1.5 ± 0.01 | 30.0 ± 1.6 | 1.5 ± 0.01 | 29.7 ± 1.2 | 1.5 ± 0.01 | ||||
| 0.7 + MMC | 29.0 ± 0.8 | 1.5 ± 0.01 | – | 28.0 ± 0.8 | 1.5 ± 0.01 | – | 26.7 ± 1.2 | 1.5 ± 0.01 | – | 30.3 ± 1.2 | 1.5 ± 0.01 | – |
| 1.5 + MMC | 24.7 ± 1.2* | 1.5 ± 0.01 | 24.4 | 21.3 ± 1.2* | 1.5 ± 0.01 | 40.2 | 23.3 ± 0.47* | 1.5 ± 0.01 | 28.8 | 28.7 ± 0.9 | 1.5 ± 0.01 | – |
| 3.0 + MMC | 17.7 ± 1.2* | 1.5 ± 0.01 | 53.1 | 18.0 ± 0.8* | 1.5 ± 0.01 | 54.3 | 15.0 ± 0.82* | 1.5 ± 0.01 | 65.2 | 27.3 ± 0.9 | 1.5 ± 0.01 | – |
MNBN binucleated cells with micronucleus, mean ± standard deviation, MMC Mitomycin C (4.8 µM)
* Statistically different from group treated with mutagen MMC (p < 0.05)
#Statistically different from negative control (PBS) (p < 0.05)
Table 5.
Frequency of binucleated cells with micronucleus, Nuclear Division Index (NDI) and percentage of reduction in mutagenicity (%Red) obtained in CBMN assay performed with digoxin associated with MMC in different protocols of treatment on the HeLa cell line
| Treatments | Protocols | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-complexation | Pre-treatment | Simultaneous treatment | Post-treatment | |||||||||
| MNBN ( | NDI ( | %Red (%) | MNBN ( | NDI ( | %Red (%) | MNBN ( | NDI ( | %Red (%) | MNBN ( | NDI ( | %Red (%) | |
| PBS | 8.0 ± 0.8* | 1.5 ± 0.01 | 8.7 ± 1.2* | 1.5 ± 0.02 | 6.7 ± 1.2* | 1.5 ± 0.03 | 8.0 ± 1.6* | 1.4 ± 0.01 | ||||
| MMC | 39.7 ± 1.2 | 1.4 ± 0.01# | 36.3 ± 1.2 | 1.4 ± 0.01# | 39.0 ± 1.6 | 1.3 ± 0.01# | 35.0 ± 1.4 | 1.4 ± 0.00# | ||||
| 0.7 + MMC | 37.3 ± 0.9 | 1.3 ± 0.01# | – | 38.0 ± 2.2 | 1.2 ± 0.03# | – | 35.0 ± 1.4 | 1.2 ± 0.02# | – | 34.7 ± 1.7 | 1.4 ± 0.01 | – |
| 1.5 + MMC | 38.7 ± 1.2 | 1.4 ± 0.01# | – | 27.7 ± 1.2* | 1.4 ± 0.01 | 31.3 | 38.3 ± 1.2 | 1.5 ± 0.02 | – | 31.3 ± 2.0 | 1.4 ± 0.01 | – |
| 3.0 + MMC | 23.0 ± 1.6* | 1.5 ± 0.01 | 52.6 | 31.7 ± 1.7* | 1.4 ± 0.01 | 34.0 | 22.7 ± 1.2* | 1.5 ± 0.02 | 50.3 | 27.3 ± 0.9 | 1.4 ± 0.01 | – |
MNBN binucleated cells with micronucleus, mean ± standard deviation, MMC Mitomycin C (4.8 µM)
* Statistically different from group treated with mutagen MMC (p < 0.05)
#Statistically different from negative control (PBS) (p < 0.05)
Discussion
The genetic toxicological effects associated with exposure to chemical substances, including medications, have been identified in previous studies (Schmitt et al. 2001; Brambilla et al. 2013), revealing the relevance of the analysis of these biological parameters. In addition, in light of the potential of cardiac glycosides, including digoxin, as antitumor agents, a better understanding of the genotoxic potential of these compounds can help to elucidate the possible mechanisms related to cell death induced on tumor cells.
Reviews performed by Snyder and Green (2001) and Brambilla and Martelli (2009) revealed that until the 2000s, studies about the mutagenicity/genotoxicity of digoxin were scarce. More recently, studies by Sedigh-Ardekani et al. (2013) and Lu et al. (2014) demonstrated that digoxin induced chromosomal damages and cell death in vitro and data obtained in the present study corroborate these works.
As observed in Fig. 2, digoxin reduced the cell viability of the CHO-K1 cell line in the MTT assay. The cell viability for this rodent cell line was only affected after treatment with concentrations of digoxin of between 125 and 500 µM. When HeLa cells were exposed to digoxin for 24 and 48 h, the cell viability decreased significantly at concentrations ≥150 nM (Rocha et al. 2014), showing that the response of CHO-K1 cells (rodent origin) to treatment with digoxin was very different to that observed in the human cell line.
Our results are in agreement with other data found in the scientific literature. According to Hallböök et al. (2011), digoxin was cytotoxic to normal peripheral blood cells and to leukemia cell lines at concentrations of between 0.02 and 1 µM. Similar cytotoxic concentrations were observed in another study that used the HeLa cell line (Lu et al. 2014) and in colon cancer cells (Felth et al. 2009). Also, the difference between the sensibility of human cells in comparison to rodent cells to digoxin in vitro is remarkable, as observed in our results. Schoonen et al. (2005) showed that concentrations of digoxin capable of inducing cell damage and cell death in human lineages have no effect on CHO-K1 cell lines. Moreover, Gupta et al. (1986) described the difference in the sensibility of the alpha 1 subunit of Na, K-ATPase, with humans and monkeys presenting high affinity and rodents (mice, Chinese hamsters and Syrian hamsters) showing low affinity for cardiotonic steroids. The differences observed between human and rodent cells were also observed for other cardiac glycosides. Oleandrin, for instance, presents high cytotoxicity to human tumor cells but mouse pancreatic cancer cells are nonresponsive to exposure to this chemical agent (Newman et al. 2008).
Different possible cytotoxic mechanisms of action have been proposed for cardiac glycosides, including digoxin. Cardiotonic steroids, like digoxin and digitalis, may influence cell proliferation, differentiation and, eventually, cell death via the Na, K-ATPase signalosome pathways (Schoner and Scheiner-Bobis 2007). However, other mechanisms can be related to cell responses after exposure to cardiac glycosides. For instance, bufalin, a hydrophobic cardiotonic steroid, downregulates the expression of Cyclin A, Bcl-2 and Bcl-XL, and upregulates the expression of p21 and Bax in ovarian endometrial cyst stromal cells, affecting cell cycle progression and inducing apoptosis (Nasu et al. 2005). Digoxin at low concentrations (<10 nM) prevents apoptosis in HeLa cells, but at higher concentrations (>10 nM) the apoptotic process is triggered in these cells in a cytochrome-c release-dependent pathway (Ramirez-Ortega et al. 2006).
In the Ames assay, digoxin did not induce gene mutations by substitution, addition or deletion of bases in DNA structure, without or with metabolic activation (Fig. 4). Nevertheless, as shown in Table 1, digoxin induced chromosomal mutations in CHO-K1 at higher assessed concentrations (20, 35 and 50 µM). In HeLa cell lines, mutagenicity was also observed, but at much lower concentrations (Table 2). The concentrations tested near that observed in the blood plasma of patients in digoxin medical treatments were not able to cause significant chromosomal damages or reduce the NDI values for either cell line (Table 3).
The results observed in the Comet assay corroborate those observed in the micronucleus assay. The score values obtained with this methodology were different from negative control for the three highest concentrations assessed with the CKO-K1 cell line and for the two highest concentrations evaluated in HeLa (Fig. 3). These complementary results enable us to understand that the micronucleus detected in CBMN resulted from chromosomal breaks caused by the exposition to digoxin at higher concentrations.
DNA damage can be caused by agents that interact directly or indirectly with DNA and some authors have described the potential of cardiac glycosides to produce reactive oxygen species (ROS), which are recognized as genotoxic agents (Huang et al. 2004; Schoner and Scheiner-Bobis 2007). Also, one study showed that different cardiac glycosides, including digoxin, inhibit the activity of topoisomerase II (Bielawski et al. 2006) and, consequently, exposition to this kind of agent can result in single-strand and double-strand breaks in the DNA structure (Godard et al. 2002; Hajji et al. 2005). In addition, according to Lu et al. (2014), digoxin modulated G2/M arrest, DNA damage and apoptosis through the p53-dependent pathway in HeLa cells.
As described above, the genotoxicity of digoxin shown in the present study should not cause immediate concern because the concentrations necessary to induce DNA damage in human or rodent cells were much higher than the plasmatic levels of digoxin observed in human users of this drug. In this context, the potential interaction of digoxin with the DNA-damaging agent Mitomycin C was evaluated at the three lowest concentrations assessed in the CBMN and Comet assays. These concentrations did not induce chromosomal damages and were selected because they are near that observed in the blood of human users of this medicament to treat cardiac diseases.
The results presented in Tables 4 and 5 show that digoxin reduced the mutagenicity induced by MMC in different treatment protocols (pre-complexation, simultaneous and pre-treatment) in CHO-K1 and HeLa cell lines. In addition, the NDI in the treatments where this effect was observed was not affected, indicating an effective chemoprotective effect of digoxin.
The profile of action identified in this study indicates that digoxin acts as a desmutagen as the protective effect was only observed in treatments where the DNA damages were induced after or simultaneously to the exposition to this drug. Desmutagens act directly on the compounds that induce mutations in DNA, inactivating them chemically or enzymatically, inhibiting the metabolic activation of pro-mutagens or sequestering reactive molecules, thereby preventing their mutagenic effects (Sloczyska et al. 2014).
These results are very interesting because they demonstrate that co-treatment with digoxin has the potential to be used in a chemopreventive approach in very low concentrations, near those already employed. Chemopreventive agents can be used not only to prevent cancer, but also in therapy, since many of them can be associated with chemotherapeutic agents to enhance the effect with the use of lower doses and thereby minimize toxicity induced by them (Dorai and Aggarwal 2004).
Therefore, this study contributes to the elucidation of mutagenic and antimutagenic effects of digoxin and how they interfere in cell death. Complementary studies should be performed to assist comprehension of the action mechanisms of digoxin and the interaction with the other genotoxic agents. However, the relevance of these results is clear because they show that digoxin can have a drug–drug interaction with mutagenic agents, however, with a protective effect against DNA damages.
Conclusions
The results presented herein show that digoxin was mutagenic at higher concentrations both in rodent and human cell lines. However, in concentrations near those used in the therapy of cardiac diseases, this drug did not induce chromosomal mutations. In addition, in these same conditions, digoxin reduced the frequency of chromosomal damages induced by Mitomycin C in human and rodent cell lines. This finding should be better explored, along with the interaction of digoxin with other chemotherapeutic anticancer agents, because this interesting effect can affect the efficacy of treatments or may be used in an approach to reduce the collateral effects of chemotherapy.
Acknowledgements
The authors would like to acknowledge the financial support from the Fundação de Amparo à Pesquisa de Minas Gerais—FAPEMIG (APQ-01254-15; CBB-PPM-00560-13), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (478629/2013-3).
Compliance with ethical standards
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Ahern TP, Lash TL, Sørensen HT, Pedersen L. Digoxin treatment is associated with an increased incidence of breast cancer: a population-based case-control study. Breast Cancer Res. 2008;10:R102. doi: 10.1186/bcr2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern TP, Tamimi RM, Rosner BA, Hankinson SE. Digoxin use and risk of invasive breast cancer: evidence from the Nurses’ Health Study and meta-analysis. Breast Cancer Res Treat. 2014;144:427–435. doi: 10.1007/s10549-014-2886-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielawski K, Winnicka K, Bielawska A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol Pharm Bull. 2006;29:1493–1497. doi: 10.1248/bpb.29.1493. [DOI] [PubMed] [Google Scholar]
- Biggar RJ, Wohlfahrt J, Oudin A, et al. Digoxin use and the risk of breast cancer in women. J Clin Oncol. 2011;29:2165–2170. doi: 10.1200/JCO.2010.32.8146. [DOI] [PubMed] [Google Scholar]
- Boursi B, Haynes K, Mamtani R, Yang Y-X. Digoxin use and the risk for colorectal cancer. Pharmacoepidemiol Drug Saf. 2014;23:1147–1153. doi: 10.1002/pds.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla G, Martelli A. Update on genotoxicity and carcinogenicity testing of 472 marketed pharmaceuticals. Mutat Res Mutat Res. 2009;681:209–229. doi: 10.1016/j.mrrev.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Mattioli F, Robbiano L, Martelli A. Genotoxicity and carcinogenicity studies of bronchodilators and antiasthma drugs. Basic Clin Pharmacol Toxicol. 2013;112:302–313. doi: 10.1111/bcpt.12054. [DOI] [PubMed] [Google Scholar]
- Cerella C, Dicato M, Diederich M. Assembling the puzzle of anti-cancer mechanisms triggered by cardiac glycosides. Mitochondrion. 2013;13:225–234. doi: 10.1016/j.mito.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol. 2004;26:249–261. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- Dorai T, Aggarwal BB. Role of chemopreventive agents in cancer therapy. Cancer Lett. 2004;215:129–140. doi: 10.1016/j.canlet.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Eastmond DA, Tucker JD. Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody. Environ Mol Mutagen. 1989;13:34–43. doi: 10.1002/em.2850130104. [DOI] [PubMed] [Google Scholar]
- Felth J, Rickardson L, Rosén J, et al. Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J Nat Prod. 2009;72:1969–1974. doi: 10.1021/np900210m. [DOI] [PubMed] [Google Scholar]
- Fenech M. The in vitro micronucleus technique. Mutat Res. 2000;455:81–95. doi: 10.1016/S0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Fenech M. Cytokinesis-block micronucleus cytome assay. Nat Protoc. 2007;2:1084–1104. doi: 10.1038/nprot.2007.77. [DOI] [PubMed] [Google Scholar]
- Godard T, Deslandes E, Sichel F, et al. Detection of topoisomerase inhibitor-induced DNA strand breaks and apoptosis by the alkaline comet assay. Mutat Res. 2002;520:47–56. doi: 10.1016/S1383-5718(02)00174-2. [DOI] [PubMed] [Google Scholar]
- Gomes CC, Moreira LM, Santos VJSV, et al. Assessment of the genetic risks of a metallic alloy used in medical implants. Genet Mol Biol. 2011;121:116–121. doi: 10.1590/S1415-47572010005000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves A, de Lima A, da Silva Barbosa M, et al. Synthesis and biological evaluation of novel 3-alkylpyridine marine alkaloid analogs with promising anticancer activity. Mar Drugs. 2014;12:4361–4378. doi: 10.3390/md12084361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontijo VS, Espuri PF, Alves RB, et al. Leishmanicidal, antiproteolytic, and mutagenic evaluation of alkyltriazoles and alkylphosphocholines. Eur J Med Chem. 2015;101:24–33. doi: 10.1016/j.ejmech.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Gupta RS, Chopra A, Stetsko DK. Cellular basis for the species differences in sensitivity to cardiac glycosides (digitalis) J Cell Physiol. 1986;127:197–206. doi: 10.1002/jcp.1041270202. [DOI] [PubMed] [Google Scholar]
- Hajji N, Mateos S, Pastor N, et al. Induction of genotoxic and cytotoxic damage by aclarubicin, a dual topoisomerase inhibitor. Mutat Res. 2005;583:26–35. doi: 10.1016/j.mrgentox.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Hallböök H, Felth J, Eriksson A, et al. Ex vivo activity of cardiac glycosides in acute leukaemia. PLoS ONE. 2011;6:e15718. doi: 10.1371/journal.pone.0015718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YT, Chueh SC, Teng CM, Guh JH. Investigation of ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem Pharmacol. 2004;67:727–733. doi: 10.1016/j.bcp.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Hung Y, Liu HE. A hearty solution for acute myeloid leukemia. Acta Pharmacol Sin. 2012;33:1–2. doi: 10.1038/aps.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Liu S, Huang S, et al. Multiple effects of digoxin on subsets of cancer-associated genes through the alternative splicing pathway. Biochimie. 2014;106:131–139. doi: 10.1016/j.biochi.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Manoharan K, Banerjee MR. β-Carotene reduces sister chromatid exchanges induced by chemical carcinogens in mouse mammary cells in organ culture. Cell Biol Int Rep. 1985;9:783–789. doi: 10.1016/0309-1651(85)90096-7. [DOI] [PubMed] [Google Scholar]
- Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat Res Mol Mech Mutagen. 2000;455:29–60. doi: 10.1016/S0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- Nasu K, Nishida M, Ueda T, et al. Bufalin induces apoptosis and the G0/G1 cell cycle arrest of endometriotic stromal cells: a promising agent for the treatment of endometriosis. Mol Hum Reprod. 2005;11:817–823. doi: 10.1093/molehr/gah249. [DOI] [PubMed] [Google Scholar]
- Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- Olive PL, Banáth JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- Özdemir A, Şimay YD, İbişoğlu B, et al. Cardiac glycoside-induced cell death and Rho/Rho kinase pathway: implication of different regulation in cancer cell lines. Steroids. 2016;109:29–43. doi: 10.1016/j.steroids.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Rahimtoola S, Tak T. The use of digitalis in heart failure. Curr Probl Cardiol. 1996;21:781–853. doi: 10.1016/S0146-2806(96)80001-6. [DOI] [PubMed] [Google Scholar]
- Ramirez-Ortega M, Maldonado-Lagunas V, Melendez-Zajgla J, et al. Proliferation and apoptosis of HeLa cells induced by in vitro stimulation with digitalis. Eur J Pharmacol. 2006;534:71–76. doi: 10.1016/j.ejphar.2006.01.035. [DOI] [PubMed] [Google Scholar]
- Rascón-Valenzuela L, Velázquez C, Garibay-Escobar A, et al. Antiproliferative activity of cardenolide glycosides from Asclepias subulata. J Ethnopharmacol. 2015;171:280–286. doi: 10.1016/j.jep.2015.05.057. [DOI] [PubMed] [Google Scholar]
- Rocha SC, Pessoa MTC, Neves LDR, et al. 21-benzylidene digoxin: a proapoptotic cardenolide of cancer cells that up-regulates Na, K-ATPase and epithelial tight junctions. PLoS ONE. 2014;9:1–14. doi: 10.1371/journal.pone.0108776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos FV, Andreo M, Nasser ALM, et al. Absence of mutagenicity of plants used to treat gastrointestinal disorders. Arch Biol Sci. 2013;65:191–195. doi: 10.2298/ABS1301191S. [DOI] [Google Scholar]
- Schmitt AC, Ravazzolo AP, von Poser GL. Investigation of some Hypericum species native to Southern of Brazil for antiviral activity. J Ethnopharmacol. 2001;77:239–245. doi: 10.1016/S0378-8741(01)00314-2. [DOI] [PubMed] [Google Scholar]
- Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs. 2007;7:173–189. doi: 10.2165/00129784-200707030-00004. [DOI] [PubMed] [Google Scholar]
- Schoonen WGEJ, De Roos JADM, Westerink WMA, Débiton E. Cytotoxic effects of 110 reference compounds on HepG2 cells and for 60 compounds on HeLa, ECC-1 and CHO cells. II mechanistic assays on NAD(P)H, ATP and DNA contents. Toxicol In Vitro. 2005;19:491–503. doi: 10.1016/j.tiv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Sedigh-ardekani M, Saadat I, Saadat M. Evaluation of chromosome aberrations induced by digoxin in Chinese Hamster Ovary. EXCLI J. 2013;12:523–527. [PMC free article] [PubMed] [Google Scholar]
- Sloczyska K, Powroznik B, Pekala E, Waszkielewicz AM. Antimutagenic compounds and their possible mechanisms of action. J Appl Genet. 2014;55:273–285. doi: 10.1007/s13353-014-0198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder RD, Green JW. A review of the genotoxicity of marketed pharmaceuticals. Mutat Res. 2001;488:151–169. doi: 10.1016/S1383-5742(01)00055-2. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids) Fed Proc. 1976;35:1332–1338. [PubMed] [Google Scholar]
- Stenkvist B, Bengtsson E, Eriksson O, et al. Cardiac glycosides and breast cancer. Lancet. 1979;313:563. doi: 10.1016/S0140-6736(79)90996-6. [DOI] [PubMed] [Google Scholar]
- Stenkvist B, Bengtsson E, Dahlqvist B, et al. Cardiac glycosides and breast cancer, revisited. N Engl J Med. 1982;306:484. [PubMed] [Google Scholar]
- Thome RG, dos Santos HB, dos Santos FV, et al. Evaluation of healing wound and genotoxicity potentials from extracts hydroalcoholic of Plantago major and Siparuna guianensis. Exp Biol Med. 2012;237:1379–1386. doi: 10.1258/ebm.2012.012139. [DOI] [PubMed] [Google Scholar]
- Titenko-Holland N, Windham G, Kolachana P, et al. Genotoxicity of malathion in human lymphocytes assessed using the micronucleus assay in vitro and in vivo: a study of malathion-exposed workers. Mutat Res. 1997;388:85–95. doi: 10.1016/S1383-5718(96)00140-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Qiu Q, Shen J-J, et al. Cardiac glycosides induce autophagy in human non-small cell lung cancer cells through regulation of dual signaling pathways. Int J Biochem Cell Biol. 2012;44:1813–1824. doi: 10.1016/j.biocel.2012.06.028. [DOI] [PubMed] [Google Scholar]
- Waters MD, Brady AL, Stack HF, Brockman HE. Antimutagenicity profiles for some model compounds. Mutat Res Genet Toxicol. 1990;238:57–85. doi: 10.1016/0165-1110(90)90039-E. [DOI] [PubMed] [Google Scholar]
- Xue R, Han N, Xia M, et al. TXA9, a cardiac glycoside from Streptocaulon juventas, exerts a potent anti-tumor activity against human non-small cell lung cancer cells in vitro and in vivo. Steroids. 2015;94:51–59. doi: 10.1016/j.steroids.2014.12.015. [DOI] [PubMed] [Google Scholar]