

Abstract
Hepatocellular carcinoma (HCC) is a serious threat to human health. The carbohydrate recognition domain of Galectin-3 (Gal3C) has been reported to be an anti-tumour molecule. In this study, we aim to explore effects of Gal3C in HCC and its possible molecular mechanism with quantitative proteomics approach. We found that rGal3C stimulation could inhibit cell viability, migration and invasion of HepG2. After rGal3C stimulating, 190 proteins were differentially expressed. Eighty up-regulated proteins located mainly in extracellular exosome and involved in cell adhesion and metabolism, and 110 down-regulated proteins located in mitochondria and extracellular exosome, and related to processes of metabolism and oxidation-reduction. Of the differentially expressed proteins, CLU, NDRG1, CD166, S100A11 and Galectin-1 were carcinoma-related proteins affected by rGal3C. Potential receptors of rGal3C were explored by an UV cross-linking capture strategy. We showed that rGal3C could induce dephosphorylating of FAK/SRC. Blocking of the FAK/SRC pathway resulted in down-regulation of NDRG1. Immunofluorescence suggested that rGal3C could disrupt integrin clustering. Our study provides valuable insight into the anti-tumour mechanism of rGal3C in HCC on a proteomics level and is the first to reveal the possible mechanism involving integrin/FAK/SRC pathway and NDRG1. These results provide useful guidance of developing new therapies for HCC.
Introduction
Hepatocellular carcinoma (HCC) is the most common malignant liver cancer in the modern world1. The occurrence of HCC involves complex induction factors, such as Hepatitis B virus (HBV), Hepatitis C virus (HCV), alcohol and tobacco2. In general, most HCC cases develop from liver cirrhosis (LC)3.
Tumourigenesis of HCC is regulated by a series of pathways, such as Wnt/β-catenin, Notch, EGFR, RAS/MAPK/ERK, AKT/mTOR and FAK/SRC4–8. HCC progress is also accompanied by changes in protein expression; thus, some proteins can be used as potential biomarkers for diagnosis and treatment9. Currently, alpha-fetoprotein (AFP) is still the most common serum marker for HCC diagnosis, and its fucosylated fraction, AFP-L3, is an even more sensitive tumour marker10, 11. Other biomarkers used for HCC detection include GPC3, Osteopontin, and Golgi Protein 73 (GP73)12. Recently, N-myc downstream-regulated gene 1 protein (NDRG1)13 and Activated leukocyte cell adhesion molecule (ALCAM, CD166)14 have been identified as novel tumour markers for clinical diagnosis of HCC.
Galectin-3 is a mammalian lectin that is up-regulated in HCC and involves in tumour progression and poor prognosis in HCC. Therefore, it may serve as a prognostic marker for HCC15–19. Intracellular Galectin-3 is mostly a monomeric soluble protein regulating apoptosis20–24 and phosphorylation of AKT in cytoplasm25, 26, and Wnt/β-catenin pathway, transcription and pro-mRNA splicing in nucleus27–33. Extracellular monomer Galectin-3 could induce endothelial cell morphogenesis and angiogenesis in kinds of cancer34. Extracellular Galectin-3 is able to pentamerize. Pentamer Galectin-3 could crosslink glycosylated ligands to form a dynamic lattice on cell surface35. The lattice regulates the residing and endocytosis of plasma membrane glycoproteins and glycolipids. The lattice also regulates receptor kinase signaling and the functionality of membrane receptors, including the glucagon receptor, glucose and amino acid transporters, cadherins and integrin36. In addition, the lattice enhances tumour cell adhesion and migration in tumour progression37.
Cleavage of Galectin-3 by matrix metalloproteinases (MMPs) generates a single truncated Galectin-3 carbohydrate recognition domain (Gal3C), which binds to the glycoconjugate more tightly and could inhibit wound healing38, 39. In the bone remodelling process during skeletal metastasis of breast and prostate cancers, secreted intact Galectin-3 could induce osteoclastogenesis, whereas Gal3C could inhibit osteoclast differentiation, indicated that Gal3C may play an opposing role40. Gal3C inhibited tumour growth and metastasis in orthotopic nude mouse models of human breast cancer and in a murine model of human multiple myeloma41, 42.
Proteomics is a powerful tool to reveal the mechanism of diseases on the proteomics level. Stable Isotope Labelling with Amino acids in Cell culture (SILAC)43 provides an excellent quantitative proteomics approach for identifying dynamic alterations in protein expression and modification profiles under different stimuli44. Recently, quantitative proteomics approaches based on SILAC, liquid chromatography-mass spectrometry (LC-MS) and bioinformatics analysis have been widely applied in exploring mechanisms of tumourigenesis, metastasis and biomarker discovery of HCC45–48.
Although some research has been done to explore the anti-cancer or carcinogenic function of Gal3C in different types of cells, its potential anti-tumour mechanism in HCC is still unclear. Considering that Gal3C contains the same ligand binding site as Galectin-3, it may competitively bind the same ligands as Galectin-3 and disrupt Galectin-3-mediated processes in the extracellular space to provide an anti-Galectin-3 therapy. Here, for the first time, we report effects of Gal3C on HCC and its possible mechanism. Here, for the first time, we report effects of Gal3C on HCC and its possible mechanism. A recombinant carbohydrate recognition domain of Galectin-3 (rGal3C) was used to stimulate cells from the HepG2 HCC cell line. Changes in protein expression profiles were identified by quantitative proteomics based on SILAC and LC-MS/MS. Gene Ontology (GO), disease association and protein-protein interaction analysis found a series of carcinoma-related proteins affected by rGal3C. Immunoblotting and immunofluorescence results revealed that the possible anti-tumour mechanism of rGal3C in HCC involved the FAK/SRC pathway.
Results
rGal3C binds to cell surface of HepG2 and negatively affects viability, migration and invasion of HepG2
Expression of rGal3C was induced in BL21 (DE3, pET28a-AG/pBirA) with 0.1 mM IPTG at 30 °C for 8 hours. Then, the recombinant protein was purified by Ni-NTA agarose affinity chromatography and detected by SDS-PAGE (Fig. 1A and Supplementary Fig. S1). The molecular mass detected by matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) was 22.86 kDa (Fig. 1B), which is consistent with its theoretical molecular weight. Biotinylation was confirmed by streptavidin-biotin blotting (Fig. 1C). There was an obvious binding and residing of rGal3C on the lactose agarose, and binding could be dissociated by 100 mM galactose, while mannose can hardly elute rGal3C from the lactose agarose, indicating that rGal3C possessed galactose binding activity (Fig. 1D). Binding of rGal3C on the HepG2 cell surface was identified by immunofluorescence and Western blotting. Compared with control, rGal3C binding on HepG2 cell surfaces displayed obvious green fluorescence from FITC. Total extracellular galectin-3 and rGal3C binding on the surface of HepG2 cells displayed red fluorescence from Alexa 594, revealing cell morphology (Fig. 1E). Compared with control cells, rGal3C-treated HepG2 cells displayed an obvious rGal3C band, and galactose but not mannose washing could decrease this band (Fig. 1F). Galactose but not mannose washing could also decrease the galectin-3 band, possibly because extracellular galectin-3 was washed away. These results suggested that rGal3C could bind to the HepG2 cell surface due to its galactose binding activity.
Figure 1.
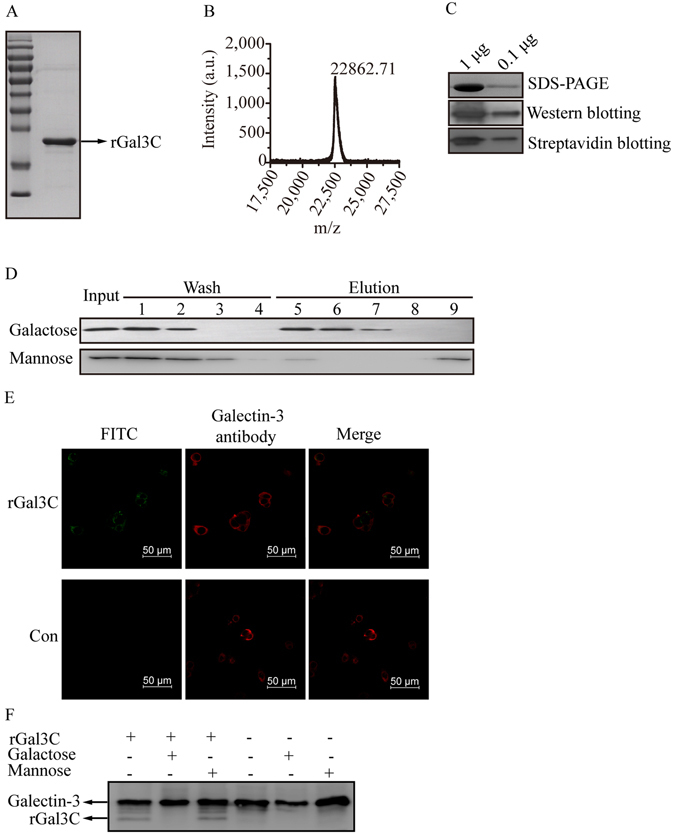
rGal3C is able to bind immobilized lactose and HepG2. (A) The 22.86 kDa band of rGal3C was detected. (B) Molecular weight of rGal3C detected by MALDI-TOF MS is 22862.71, which is consistent with its theoretical molecular weight. (C) Biotinylation of rGal3C was detected. (D) Binding of rGal3C to immobilize lactose was identified. Input fraction, washing fractions (band 1–4), eluting fractions (band 5–8) and NaOH eluting fraction (band 9) were detected. (E) Binding of rGal3C to cell surface of HepG2 was identified. Red fluorescence of Alexa 594 dye displayed total distribution of both Galectin-3 and rGal3C, while green fluorescence of FITC displayed only rGal3C binding to HepG2. (F) Galactose disrupted binding of rGal3C or Galectin-3 to HepG2. Cells washed with galactose displays a decreased Galectin-3 band, while mannose washing has no effect. HepG2 treated by rGal3C displays an rGal3C band which could be attenuated by galactose washing, but not mannose. Con, untreated HepG2 cells.
Effects of rGal3C on the viability, migration and invasion of HepG2 cells were determined. Compared with control cells, cells stimulated with rGal3C displayed a significant decrease in cell viability (Fig. 2A). With increasing treatment time and concentration, the inhibition effect became more obvious. We found that HepG2 treated with 50 or 100 μg/mL of rGal3C for 12 or 24 hours displayed similar attenuation of cell viability (54.4 ± 1.6% for 50 μg/mL, 12 hours; 53.2 ± 3.5% for 50 μg/mL, 24 hours; 52.6 ± 3.6% for 100 μg/mL, 12 hours; 52.2 ± 3.2% for 100 μg/mL, 24 hours; n = 3), thus we decided to use 50 μg/mL of rGal3C to treat HepG2 for 12 hours in next experiments.
Figure 2.
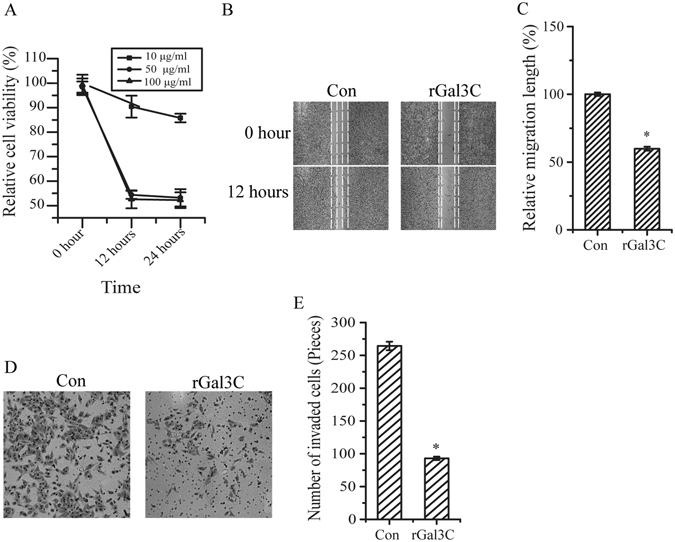
rGal3C negatively affects viability, migration and invasion of HepG2 cells. (A) The viability of HepG2 cells was inhibited after treated by 10, 50 and 100 μg/mL of rGal3C for 12 or 24 hours. (B) Cell migration was attenuated after stimulated by 50 μg/mL rGal3C for 12 hours. (C) Relative cell migration was attenuated to 59.8 ± 1.5% in HepG2 cells treated by 50 μg/mL rGal3C for 12 hours. Representative images were shown on the left, and the quantification of three replicates was shown. (D) The invasive capacity of HepG2 cells was inhibited after stimulated by by 50 μg/mL rGal3C for 12 hours. (E) The invaded cell munber was decreased (from 264 ± 6 to 93 ± 3) after stimulated by 50 μg/mL rGal3C for 12 hours. The quantification of three replicates was shown. Error bars indicate the mean ± SD. *p < 0.01. Con, untreated HepG2 cells.
Compared with rGal3C-treated cells, untreated cells displayed remarkable migration (Fig. 2B and C). The migration of HepG2 cells was attenuated (59.8 ± 1.5%, n = 3) under the continuous influence of rGal3C. HepG2 cells treated with rGal3C displayed decreased invasion, indicating that the cell invasion ability was attenuated (Fig. 2D and E). These results suggested that rGal3C could negatively affect HepG2 cell invasion and migration.
Protein identification and quantification using SILAC
The rGal3C-stimulated cells and control cells were subjected to quantitative proteomics analysis using the Stable Isotope Labelling with Amino acids in Cell culture (SILAC) technique to obtain an in-depth view of the proteomic regulation upon rGal3C stimulating. General workflow of the quantitative proteomics approach based on SILAC and LC-MS/MS is shown in Fig. 3A. HepG2 cells were cultured with DMEM containing 13C6-L-lysine and 13C6-L-arginine for at least 21 days to achieve a satisfactory labelling efficiency (Supplementary Fig. S2).
Figure 3.
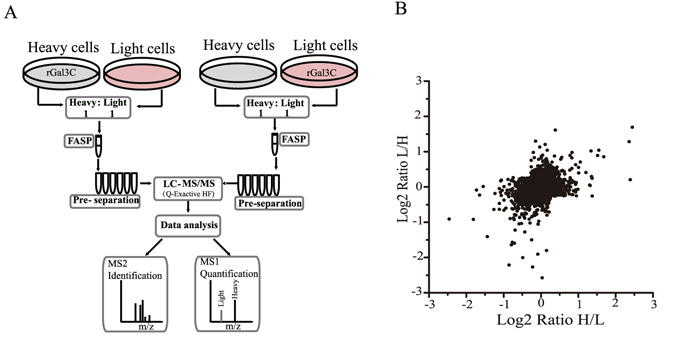
Quantitative proteomics analysis revealed the differentially expressed proteins after rGal3C treatment. (A) General workflow of the SILAC experiment. (B) Distribution of SILAC ratio of both experiments. Compared with untreated group, 80 proteins were up-regulated (log2 [ratio] > 0.58) and 110 were down-regulated (log2 [ratio] < −0.58).
In total, 4705 and 4435 proteins were identified in the labelled and reversed-labelled experiments, respectively, of which 3945 overlapping proteins were identified and 3611 proteins were quantified. Proteins with SILAC ratio greater than 1.5 or less than 0.67 in either experiment were considered differentially regulated proteins. Under this cutoff value, 80 up-regulated proteins and 110 down-regulated proteins were identified (Table 1 and Supplementary Tables S1 and S2). The SILAC ratio distribution of the quantified proteins is shown in Fig. 3B. Eleven up-regulated proteins and 24 down-regulated proteins in both experiments were considered highly reliable, and their ratio and unique peptide number are shown in Table 2.
Table 1.
The numbers of protein identified and quantified using SILAC.
| Experiment | Reverse experiment | Overlap proteins | Quantified proteins | Up-regutaed proteins (Ratio >1.5) | Down-regulated proteins (Ratio <0.67) | |
|---|---|---|---|---|---|---|
| Total identifications | 4705 | 4435 | 3945 | 3611 | 80 | 110 |
Table 2.
Information of up or down-regulated proteins in both experiments.
| Protein IDs | Gene names | Ratio | Unique peptides | |||
|---|---|---|---|---|---|---|
| H/L | L/H | H/L | L/H | |||
| Up-regulated proteins | Q14764 | MVP | 2.54 | 2.03 | 8 | 5 |
| P01011 | SERPINA3 | 3.21 | 1.81 | 6 | 6 | |
| P09382 | Galectin-1 | 2.87 | 1.85 | 4 | 4 | |
| Q13509 | TUBB3 | 2.01 | 1.46 | 4 | 4 | |
| Q99519 | NEU1 | 1.91 | 1.54 | 4 | 3 | |
| Q6PUV4 | CPLX2 | 2.93 | 2.05 | 4 | 2 | |
| Q9BQL6 | FERMT1 | 1.54 | 1.86 | 3 | 3 | |
| P37802 | TAGLN2 | 1.78 | 1.97 | 3 | 2 | |
| P52943 | CRIP2 | 1.47 | 1.79 | 3 | 2 | |
| P31949 | S100A11 | 5.16 | 2.43 | 2 | 2 | |
| O43301 | HSPA12A | 1.78 | 1.77 | 2 | 2 | |
| Down-regulated proteins | P04004 | VTN | 0.18 | 0.53 | 9 | 5 |
| P10909 | CLU | 0.29 | 0.53 | 11 | 6 | |
| Q92597 | NDRG1 | 0.37 | 0.38 | 9 | 9 | |
| P02671 | FGA | 0.42 | 0.64 | 9 | 8 | |
| O00584 | RNASET2 | 0.46 | 0.64 | 7 | 9 | |
| Q02338 | BDH1 | 0.50 | 0.63 | 8 | 7 | |
| P13674 | P4HA1 | 0.52 | 0.66 | 15 | 17 | |
| Q08AH3 | ACSM2A | 0.55 | 0.22 | 2 | 3 | |
| Q16877 | PFKFB4 | 0.57 | 0.54 | 5 | 4 | |
| P08833 | IGFBP1 | 0.58 | 0.32 | 2 | 4 | |
| P10646 | TFPI | 0.58 | 0.65 | 6 | 6 | |
| P05121 | SERPINE1 | 0.59 | 0.34 | 9 | 12 | |
| Q9C0E8 | KIAA1715 | 0.60 | 0.65 | 7 | 4 | |
| Q4G0N4 | NADKD1 | 0.60 | 0.54 | 6 | 5 | |
| P02787 | TF | 0.61 | 0.53 | 29 | 28 | |
| P07148 | FABP1 | 0.61 | 0.58 | 9 | 8 | |
| P08319 | ADH4 | 0.62 | 0.43 | 11 | 9 | |
| Q02252 | ALDH6A1 | 0.62 | 0.66 | 16 | 16 | |
| Q9Y646 | CPQ | 0.63 | 0.57 | 4 | 3 | |
| Q9UN36 | NDRG2 | 0.63 | 0.66 | 7 | 5 | |
| P09972 | ALDOC | 0.64 | 0.62 | 9 | 10 | |
| P28332 | ADH6 | 0.65 | 0.69 | 9 | 10 | |
| Q96A26 | FAM162A | 0.66 | 0.65 | 6 | 4 | |
| Q6UWM9 | UGT2A3 | 0.67 | 0.48 | 8 | 6 | |
Data mining of differentially expressed proteins
To obtain a biological view of 190 differentially expressed proteins, Gene Ontology (GO) enrichment analysis for biological process, cell component and molecular function was performed using DAVID Bioinformatics Resources 6.849, 50. Most of the up-regulated proteins located in extracellular exosome, vesicles, adherent junctions and anchoring junctions, and involved in cell-cell adhesion and metabolism. Most of the down-regulated proteins located in mitochondria and extracellular exosome, and involved in processes of metabolism and oxidation-reduction processes (Fig. 4A and B).
Figure 4.
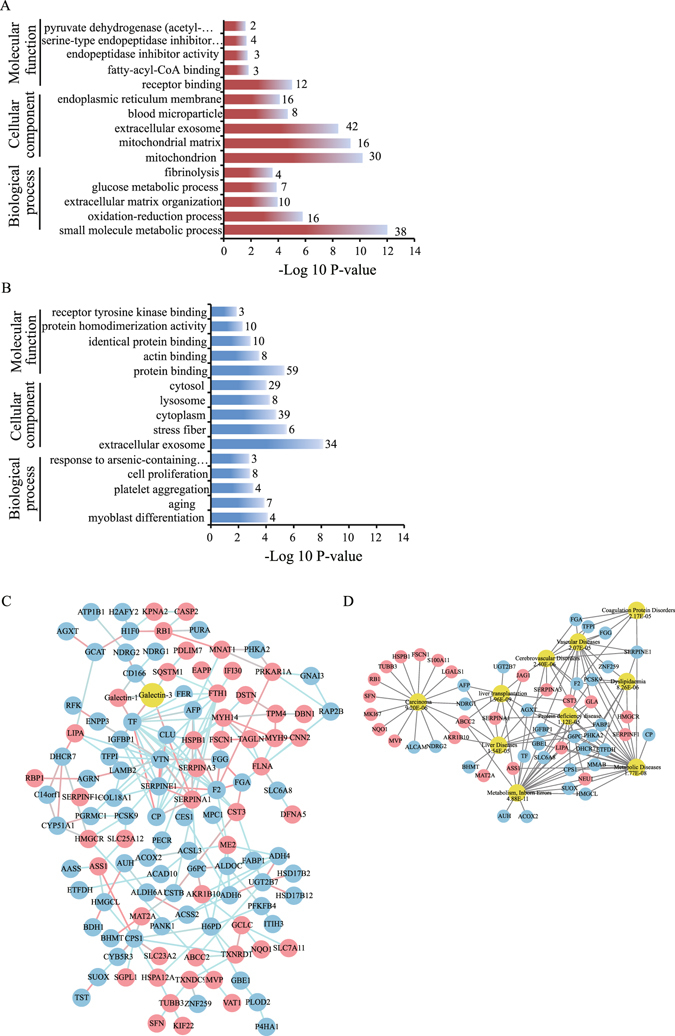
Gene Ontology, protein-protein interaction and disease association analysis of differentially expressed proteins upon rGal3C treatment. (A) Top five of biological process, cell component and molecular function of up-regulated proteins. (B) Top five of biological process, cell component and molecular function of down-regulated proteins. The −log10 p-value of the enrichment is shown on the x-axis, and the numbers represent the number of associated proteins associated with each term. (C) An interaction network of the proteins affected by rGal3C. The up-regulated proteins are represented by red nodes. The down-regulated proteins are represented by blue nodes and galectin-3 by yellow nodes. The red edge represents a strong close relationship between two nodes, and the blue edge represents an ordinary one. (D) Diseases associated with the differentially expressed proteins. The up-regulated proteins are represented by red nodes, and the down-regulated proteins are represented by blue nodes. Top ten of diseases associated with the differentially expressed proteins are represented by yellow ovals, and their P values are shown.
Considering that rGal3C is the carbohydrate recognition domain of Galectin-3, protein-protein interactions of differentially expressed proteins with Galectin-3 were analysed by String 1051 (Fig. 4C). The analysis showed that TF, CD166, Galectin-1, CLU and SQSTM1 were related to Galectin-3.
To reveal the relationship between differentially expressed proteins and diseases, disease-association analysis of differentially expressed proteins was processed by WebGestalt52, 53. According to the analysis results (Fig. 4D), differentially expressed proteins were mainly separated into two main groups, one was related to carcinoma (9.20E-06), liver diseases (1.54E-05) and liver transplantation (1.96E-09), while the other was related to metabolic diseases (1.77E-08), inborn errors (4.18E-11), protein deficiency diseases (1.12E-05), cerebrovascular diseases (2.40E-06), vascular diseases (2.40E-06), dyslipidaemia (8.26E-06) and coagulation protein disorders (2.17E-05). These results suggested that rGal3C mainly acted on HepG2 tumour-related processes and substance metabolism.
Of these disease-associated proteins and proteins related to Galectin-3, NDRG1 showed a close relationship to carcinoma and liver diseases, and CD166, S100A11 and Galectin-1 were also related to carcinoma. Based on quantitative proteomics results and their potential metastasis-related function, differential expression of CLU, NDRG1, CD166, Galectin-1 and S100A11 were further validated by a dose-response western blotting (Fig. 5A and B). Compared with non-stimulated HepG2, S100A11 and Galectin-1 showed significant up-regulation, while NDRG1, CD166 and CLU showed significant down-regulation, and alterations were correlated with the concentration of rGal3C. These results were consistent with the results of SILAC-based quantitative proteomics.
Figure 5.
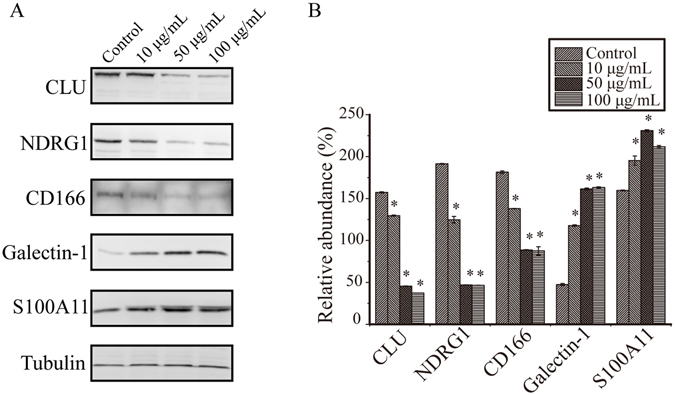
Dose-response western blotting verification of differentially expressed proteins. (A) The differential expression of CLU, NDRG1, ALCAM, Galectin-1 and S100A11 were displayed. (B) The western blot was quantitatively analyzed using Image J, and protein expression abundance was normalized to tubulin. Parallel experiments were progressed and representative results of three independent replicates were shown. Error bars indicate the mean ± SD. Control: untreated HepG2 cells; *p < 0.01.
Potential receptors of rGal3C on cell surface of HepG2
To capture the potential receptors of rGal3C binding on cell surface of HepG2, ultraviolet radiation dependent cross-link strategy was employed. Binding of photo-Leucine analogue labelled rGal3C to HepG2 cell surface was displayed by fluorescein isothiocyanate (FITC). There was a remarkable green fluorescent spreading around the HepG2 cell surface after photo-leucine analogue labelled rGal3C incubation and ultraviolet radiation (Supplementary Fig. S3A and B).
The released supernatant fraction enriched by streptavidin agarose was digested by FASP, and the peptide mixtures were identified by LC-MS/MS. Overall 177 proteins were identified in two independent repeated experiments, of which 30 proteins were predicted as transmembrane proteins by TMHMM Server v2.0 (Supplementary Fig. S3C and D and Supplementary Table S3). According to the label-free quantitative proteomics results (iBAQ value and unique peptide number) identified in two independent repeated experiments, we found that five integrins (ITA1, ITA2, ITA3, ITAV and ITB1) were enriched in 30 potential receptors of rGal3C on cell surface of HepG2, of which ITA3 was the most significant integrin, indicated that there may be interactions between rGal3C and ITA3 (Fig. 6A and B).
Figure 6.
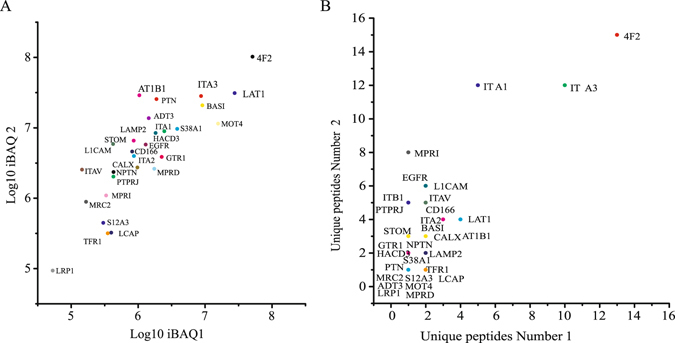
Label-free quantitative proteomics information of potential transmembrane receptors of rGal3C. (A) The distribution of iBAQ value in two independent repeated experiments. (B) The distribution of unique peptide number in two independent repeated experiments.
rGal3C induces decrease of NDRG1 through the integrin/FAK/SRC pathway
To explore the pathway by which rGal3C affects HepG2 cells, alteration of phosphorylation of Erk1/2, Akt and FAK/SRC were detected by western blotting (Fig. 7A and B). Increased phosphorylation of Erk1/2 and Akt were identified, while reduced phosphorylation of SRC at Tyr416 and FAK at Tyr397 and Tyr 925 were identified. These results suggested that Integrin/FAK/SRC pathway was inhibited after rGal3C stimulating.
Figure 7.
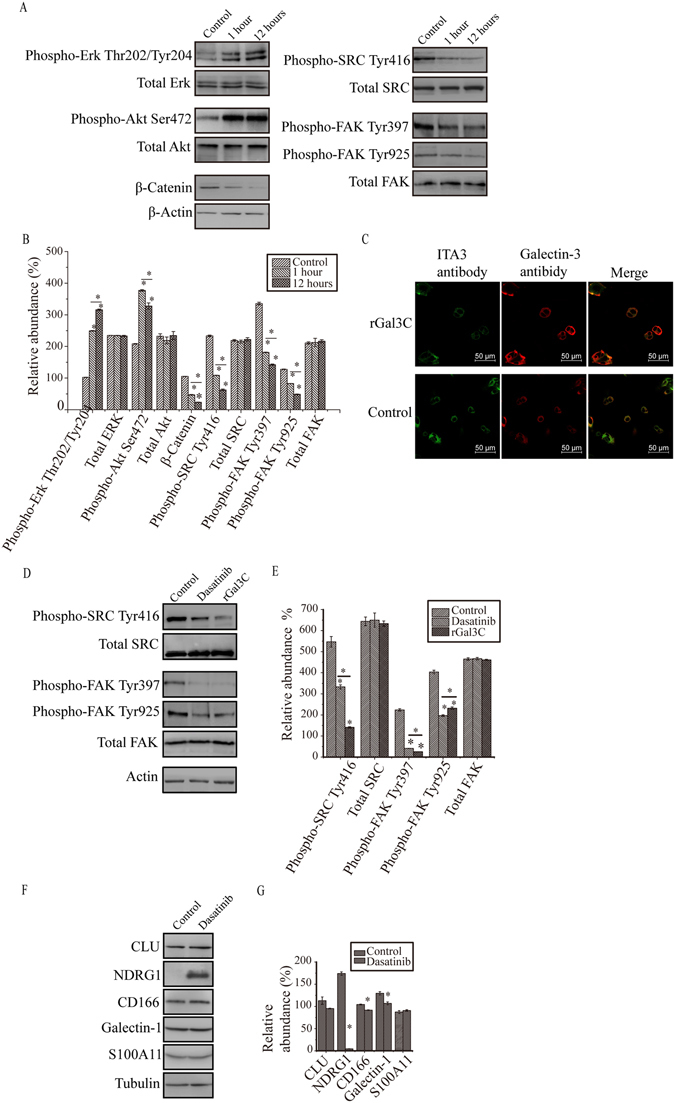
rGal3C regulates phosphorylation of FAK/SRC, integrin clustering and expression of proteins. (A) Phosphorylation of Erk1/2 at Thr202/Tyr204 and Akt at Ser472 were increased. Phosphorylation of FAK at Tyr397/Tyr925 and SRC at Tyr416 were suppressed, meanwhile expression of β-Catenin were decreased. Parallel experiments were progressed and representative results of three independent replicates were shown. (B) Relative abundance of β-Catenin and phosphorylated of Erk1/2, Akt, FAK/SRC and β-Catenin after rGal3C stimulating. Protein expression abundance was normalized to Actin. Results are representative of three independent experiments. (C) The total fluorescence of both galectin-3 and rGal3C of HepG2 cells treated by rGal3C was displayed by red fluorescence of Alexa 594 dye, while integrin clustering by green fluorescence of Alexa 488 dye. Normal HepG2 cells were used as control. (D) Both rGal3C and Dasatinib stimulating suppresses the phosphorylation of FAK at Tyr397/925 and SRC at Tyr416. Parallel experiments were progressed and representative results of three independent replicates were shown. (E) Relative abundance of phosphorylated FAK/SRC after rGal3C or Dasatinib stimulating. Protein expression abundance was normalized to Actin. Results are representative of three independent experiments. (F) Blocking of integrin/FAK/SRC signaling pathway by Dasatinib leads to the decrease of S100A11, Galectin-1, NDRG1, CD166 and CLU. Parallel experiments were progressed and representative results of three independent replicates were shown. (G) Relative abundance of CLU, NDRG1, CD166, Galectin-1 and S100A11 after Dasatinib stimulating. Protein expression abundance was normalized to tubulin. Results are representative of three independent experiments. Error bars indicate the mean ± SD. Con, untreated HepG2 cells; *p < 0.01.
Integrin clustering on the cell surface is important for activation of the FAK/SRC pathway. Our former result indicated the possible interaction between rGal3C and integrin (ITA1, ITA2, ITA3, ITAV and ITB1). Thus, integrin distribution on the surface of rGal3C-stimulated HepG2 and control cells was identified by immunofluorescence with confocal microscopy. Compared with control cells, cells stimulated with rGal3C displayed less fluorescent foci formed by integrin, indicated that integrin clustering might be partially disrupted after rGal3C stimulation (Fig. 7C).
We then investigated whether integrin/FAK/SRC pathway played a role in rGal3C regulated proteins. Dasatinib, a specific inhibitor of SRC in HepG2, was used to stimulate HepG2 cells for 12 hours at a concentration of 10 nM. Dasatinib treatment led to a remarkable decrease of phosphorylation in SRC at Tyr416 and FAK at Tyr397 and Tyr 925, as well as rGal3C did (Fig. 7D and E). NDRG1 displayed the most significant alteration after Dasatinib stimulation, suggested that NDRG1 was a main signature molecules regulated by rGal3C through the integrin/FAK/SRC signalling pathway (Fig. 7F and G).
Discussion
Liver cancer is one of the major health problems worldwide, especially in developing countries. Hepatocellular carcinoma (HCC) is the most common malignant liver cancer, and it has the highest cancer mortality rate, with increasing incidence in the modern world. During 2012, it is estimated that 782,500 new liver cancer patients and 745,500 deaths occurred worldwide, and 50% of the total number were in China1. Galectin-3, a mammalian lectin who could bind N-acetyllactosamine, is up-regulated in HCC and considered as a possible prognostic marker of HCC37. It could promote tumour progression based on its various functions in the cytoplasm, nucleus and extracellular microenvironment54. Thus, anti-Galectin-3 therapy may be an effective method for cancer treatment. Gal3C is the truncated carbohydrate recognition domain of Galectin-3, which contains the same ligand binding site as Galectin-3, but could not form pentamer, it has been reported that Gal3C is a potentially new molecule for treating breast cancer41, multiple myeloma42 and ovarian cancer55. Considering that Gal3C contains the same ligand binding site as Galectin-3, it may disrupt Galectin-3-mediated pathways on the cell surface through competitively bind the same ligands as Galectin-3.
Although some research has been done to explore the anti-tumour function of Gal3C in different types of cells, its potential anti-tumour mechanism in HCC is still unclear. In this study, we aim to explore the signature molecules and related pathways of Gal3C affecting HCC with quantitative proteomics approach.
Here, for the first time, we reported the signature molecules and related pathways of rGal3C affecting HCC. A recombinant carbohydrate recognition domain of Galectin-3 (rGal3C) was used to stimulate the HCC cell line, HepG2. Changes in protein expression profiles were identified by quantitative proteomics based on SILAC and LC-MS/MS. GO, disease association and protein-protein interaction analysis of differentially expressed proteins found a series of carcinoma-related proteins affected by rGal3C. Potential receptors of rGal3C were explored by an UV cross-linking capture strategy. Our results suggested the possible anti-tumour mechanism of rGal3C in HCC involved in Integrin/FAK/SRC pathway and NDRG1.
Firstly, we hope to prove that if Galectin-3 loses its N-terminal collagen domain and then could not form pentamer, it may loss its ability of promoting tumour procession, even inhibit cell growth and motility. We mainly focused on the effect of rGal3C on HepG2, thus HepG2 without rGal3C treatment was used as a suitable negative control in this study. rGal3C without N-terminal collagen domain was produced in E. coli, and its molecular weight detected by MALDI-TOF is 22.86 kDa (Fig. 1A and B and Supplementary Fig. S1). According to the reported structure of Gal3C56, 57, we consider that the N-terminus and C-terminus of the rGal3C would locate at the opposite side of its carbohydrate binding interface. According to our results, rGal3C retains its galactose binding characteristic (Fig. 1D–F).
Binding of rGal3C to HepG2 cells could inhibit cell viability, invasion and migration (Figs 1D–F and 2). HepG2 stimulated with rGal3C displayed a remarkable attenuation of cell viability, which positive correlated with the concentration of rGal3C and stimulating time (Fig. 2A). These results supported our former conjecture that Gal3C would inhibit cell growth and motility if it loses its oligomeric ability, suggested that rGal3C has the potential as an anti-tumour molecule in therapy of HCC.
Quantitative proteomics based on SILAC and LC-MS/MS offers a powerful tool to comprehensively discover the signature molecules of rGal3C-stimulated in HCC and explore the related pathway of anti-tumour mechanism of action (Fig. 3A). In this study, we identified 80 up-regulated proteins and 110 down-regulated proteins after rGal3C stimulating by quantitative proteomics approach (Table 1 and Supplementary Tables S1 and S2). GO analysis indicated that up-regulated proteins were located mainly in extracellular exosome/vesicle and involved in cell-cell adhesion and metabolism, and down-regulated proteins were located in mitochondria and extracellular exosome, and related to the processes of metabolism and oxidation-reduction (Fig. 4A and B). Protein-protein interaction analysis by String 10 indicated that the down-regulated proteins CD166, CLU and TF and the up-regulated proteins Galectin-1 and SQSTM1 were related rGal3C (Fig. 4C). Disease-association analysis results indicate that these differentially expressed proteins had a close relationship with carcinoma and metabolic disease (Fig. 4D).
Our quantitative proteomics results and subsequent bioinformatics analysis demonstrates a global view of rGal3C-stimulated HepG2 in protein expressing and biological process level. Extracellular rGal3C bound to HepG2 cell surface, then induced a series of changes in protein expression profile, and cell-cell adhesion, energy metabolism and exosome secretion were affected by rGal3C stimulating. These results suggest that rGal3C could regulate intercellular signature molecules and related pathways. CLU, NDRG1, CD166, S100A11 and Galectin-1 were associated with carcinoma and Galectin-3. Dose-response immunological experiment results displayed that their differential expression were correlated with the concentration of rGal3C, thus, they were possible signature molecules regulated by rGal3C (Fig. 5). These results were consistent with the results of SILAC-based quantitative proteomics.
NDRG1 is associated with carcinoma and liver transplantation, which strongly indicates that it might be closely related to HCC tumourigenesis and therapy. It has been reported that NDRG1 is a critical initiation factor for invasion of HepG2 cells13. In the present, the regulation of human NDRG1 gene expression has remained obscure, and limited studies have shown that the expression of NDRG1 is inhibited by Myc, and some chemical inducers such as NO, nickel (Ni2+) and cobalt (Co2+), as well as chemical iron chelators, can induce its expression58–61. Suppressed expression of NDRG1 significantly reduces cell proliferation and invasion in HepG2 cells and recovers senescence signaling62, 63. Overexpression of NDRG1 is often associated with tumour cell survival, significant portal vein invasion, intrahepatic metastasis, poor prognosis and shorter overall survival after liver transplantation13, 64, 65. These results strongly suggested that reducing intracellular NDRG1 levels is one possible therapeutic approach for HCC. In this study, we confirmed that rGal3C could effectively induce decrease of NDRG1 in HepG2, suggested that rGal3C could play its anti-tumour role through down-regulation of NDRG1 in HCC.
The related regulatory pathway of rGal3C towards NDRG1 in HCC is still unclear. Considering that rGal3C was located in extracellular space, it may process its regulatory role towards NDRG1 by binding receptors on HepG2 cell surface. It has been reported that extracellular Galectin-3 binds to a large number of membrane glycosylation ligands, affecting their distribution on the membrane and their signal transductionfunction54. Galectin-3 is able to pentamerize in the extracellular space and form lattice structures by crosslinking with its glycosylated receptors on the cell surface to regulate transmembrane signaling36, 66. Gal3C loses its oligomerization capacity, but gains higher ligand binding activity67, therefore rGal3C may competitive bind to the receptors of Galectin-3, and disrupt the lattice structure, resulting in negatively regulating the functions of extracellular Galectin-3. Thus, rGal3C may competitively inhibit Galectin-3 mediated transmembrane signaling through the same pathway.
In this study, 30 potential receptors of rGal3C on cell surface of HepG2 were identified by crosslinking and capturing rGal3C binding molecules using photo-leucine analogue labeled and biotinylated rGal3C (Supplementary Fig. S3 and Supplementary Table S3). We found that five integrin (ITA1, ITA2, ITA3, ITAV and ITB1) were enriched, of which ITA3 was one of the most significantly molecules according to their iBAQ value and unique peptide number (Fig. 6). These results suggested that integrin may be the main receptors of rGal3C on HepG2 cell surface. Integrin are glycoproteins on cell membrane which have been reported as receptors of Galectin-3, and Galectin-3-integrin interaction plays an important role in cell adhesion36. Galectin-3-integrin clustering could promote integrin activation as well as ligand-induced integrin activation68, 69. Integrin on membrane could regulate the activation of Erk, Akt and FAK/SRC pathway, thus the relationship between rGal3C stimulation and these pathways were identified.
Phosphorylation of Erk1/2, Akt and FAK/SRC were detected by western blotting. Decreased phosphorylation of FAK/SRC was identified in this study, while increased phosphorylation of Erk and Akt was also detected (Fig. 7A and B). These results are consistent with reported conclusions that Gal3C could induce Erk phosphorylation in HeLa cells70, and Galectin-3 is important for FAK/SRC signalling transduction71. The FAK/SRC pathway is always abnormally activated in HCC, and it contributes to invasion and metastasis, leading to poor survival5–8. These results suggested that decreased phosphorylation of FAK/SRC may be one main factor leading to decreased invasion and migration of HepG2 cells.
Auto-phosphorylation of FAK and activation of SRC is regulated by integrin clustering which dependents on Galectin-3 on the surface of cancer cells72–77. Auto-phosphorylation of FAK at Tyr397 induced by integrin clustering can create a high-affinity binding site for activated SRC, and activated SRC can trans-phosphorylate FAK at Tyr576/577, Tyr861 and Tyr925, thereby leading to the formation of a transient FAK/SRC signalling complex78. Therefore, a reasonable speculation is that rGal3C may induce decreased phosphorylation of FAK and SRC by disrupting Galectin-3-integrin clustering on the HepG2 cell surface. To confirm this inference, α3 integrin on the surface of HepG2 cells after rGal3C treatment were examined by immunofluorescence (Fig. 7C). Compared with control cells, rGal3C-stimulated HepG2 cells displayed less fluorescent foci, indicating that integrin clustering was partially disrupted. Thus, rGal3C can decrease integrin clustering and phosphorylation of FAK/SRC, thereby blocking FAK/SRC-mediated signalling.
We then determined that rGal3C-induced NDRG1 down-regulation was dependent on FAK/SRC phosphorylation by a signal blocking experiments using Dasatinib. Dasatinib is an ABL/SRC tyrosine kinase inhibitor that can specifically inhibit BCR-ABL and SRC family kinases79. Dasatinib inhibits phosphorylation of SRC, SFK/FAK and PI3K/PTEN/AKT but not MAPK/Erk and JAK/Stat pathways in HCC cells. In HepG2 cells, phosphorylation of SRC could be effectively inhibited by 10 μM Dasatinib80. Our results indicated that phosphorylation of both SRC at Tyr416 and FAK at Tyr397/925 were suppressed dramatically by either rGal3C or Dasatinib (Fig. 7D and E). Decreased phosphorylation of SRC by Dasatinib led to significant down-regulation of NDRG1 (Fig. 7F and G). These results suggest that NDRG1 is mainly SRC-regulated downstream signature molecule in HCC.
According to our results, a possible mechanism by which rGal3C incudes dephosphorylating of FAK/SRC and further down-regulates NDRG1 and invasion and migration of HCC was identified. rGal3C could disrupt Galectin-3-integrin clustering on the surface of HepG2 cells, thereby leading to decreased phosphorylation of FAK and SRC. Inhibition of integrin/FAK/SRC signalling causes down-regulation of NDRG1, thereby resulting in decreased migration and invasion of HepG2 cells (Fig. 8). This is the first report of the possible mechanism by which extracellular Gal3C negatively regulates HCC invasion and migration. This study is an initial research of the anti-tumor mechanism of rGal3C to HCC, and revealing the global functions of rGal3C needs to be progressed in different HCC cell lines.
Figure 8.
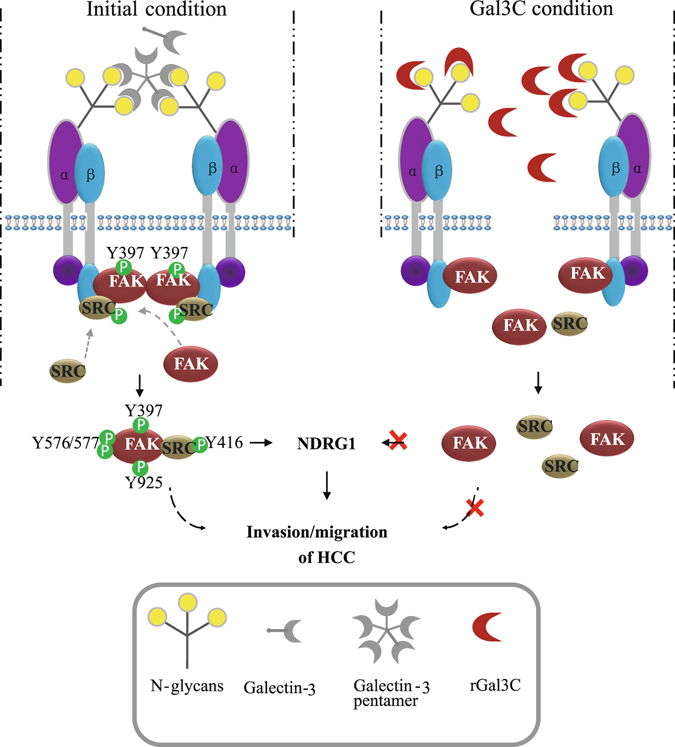
The diagram of rGal3C regulating NDRG1 through integrin/FAK/SRC pathway. rGal3C binds to the extracellular integrin, thereby resulting in the destruction of integrin clustering. Disintegrated integrin cannot induce phosphorylation of FAK/SRC to forming transient FAK-SRC signaling complex, and this may result in decreased expression of NDRG1 and attenuated migration and invasion of HCC.
Exosomes are membrane-derived nanovesicles of about 30~100 nm released by most types of cells81. Cancer-derived exosomes have displayed multifunction in invasion and metastasis of HCC, and considered as diagnosis target and therapeutic tools82. Our results suggested that rGal3C may affect the composition of exosome secreted by HepG2. Although the detailed mechanism was unclear, this phenomenon provides us with a possible research direction that extracellular Galectin-3 may involve in exosome secretion of HCC.
Differential expression of CLU, CD166, S100A11 and Galectin-1 were also identified in this study (Fig. 5). CLU is a conserved glycoprotein that is involved in tissue remodelling, adhesion, apoptosis, proliferation, senescence and cell aggregation83, 84. CD166 is a potential biomarker, which acts as a cell adhesion molecule and is involved in metastasis, proliferation and anti-apoptosis in different types of cancers14, 85, 86. S100A11 is a calcium-binding protein that plays a role in the inhibition of cell proliferation in different types of cells87–89. In the HCC cell line JHH-5, S100A11 can inhibit DNA replication in the nucleus90. Galectin-1 is an overexpressed mammalian lectin that is associated with tumour-invasive characteristics and cell adhesion and polarization, as well as in vivo tumour growth and EMT91–93. Both Galectin-1 and Galectin-3 are located in the extracellular space and have similar functions, such as lattice formation and regulation of tumour adhesion, invasion and migration94. In this study, down-regulation of NDRG1, CLU and CD166 and up-regulation of S100A11 may all lead to decreased migration and invasion of HepG2 cells, while up-regulation of Galectin-1 may occur in a compensatory manner in response to the blocking of Galectin-3-regulated functions and keep the cells alive. Detail roles of these potential signature molecules in HCC still need further study.
In summary, for the first time, our study provides valuable insight into the anti-tumour mechanism of rGal3C involving integrin/FAK/SRC pathway and NDRG1 in HCC on a proteomics level. Our results suggest that rGal3C is an effective but not comprehensive anti-tumour molecule to HepG2 cells. The possible anti-tumour mechanism is that rGal3C disrupts Galectin-3-integrin clustering on the cell surface, resulting in suppression of integrin/FAK/SRC pathway and subsequent down-regulation of NDRG1 expression. rGal3C also induces up-regulation of S100A11 and Galectin-1, and down-regulation of CD166 and CLU, which can partially contribute to inhibition of HepG2 cell migration and invasion. Our results also suggest the possible relationship between extracellular Galectin-3 and exosome secretion of HepG2. This study provides useful guidance for developing therapies based on rGal3C to treat HCC, and more work is needed to reveal a more detailed mechanism of action for rGal3C regulation of HCC.
Methods
Protein expression
Plasmid pET28a-AG (Supplementary Figure S1) for rGal3C expression was synthesized (The Beijing Genomics Institute, China). pBirA was a plasmid expressing BirA in E. coli under IPTG inducing which was a gift of Doctor Kurt Drickamer95. BL21(DE3, pET28a-AG/pBirA) was cultured in LB medium to OD600 = 0.5, and then 10 μM IPTG and 10 μg/mL biotin were added to the culture to induce the expression of rGal3C at 30 °C, 120 rpm for 8 hours. The Origami2(DE3, pET28a-AG/pBirA) was cultured in M9 medium to OD = 0.3, and then 10 μM IPTG (Sigma-Aldrich, USA) with 10 μg/ml biotin (Sigma-Aldrich, USA), 25 μg/ml photo-reactive-leucine analogue (Pierce, USA) and 25 μg/ml leucine (Sangon, China) were added to into the medium at 30 °C, 120 rpm for 8 hours in the dark.
The bacterial sediment was collected and then broken in binding buffer (20 mM Tris-HCl, 300 mM NaCl, pH 8.0) by ultrasonic wave at 4 °C. The cell supernatant was extracted by centrifuged at 10000 g for 30 minutes at 4 °C. rGal3C was captured by a Ni-NTA agarose column (MCLAB, USA), and washed with cold his-tag washing buffer (20 mM Tris-HCl, 300 mM NaCl, pH 6.0) containing 100 mM imidazole. Then, rGal3C was eluted with 1 mL washing buffer containing 250 mM imidazole. This step was repeated for ten times. The eluted fractions were detected using 12% SDS-PAGE, dialyzed for 24 hours in PBS with 10% glycerol in 4 °C and then stored at −80 °C.
Lactose binding assay
Ten μg of rGal3C was incubated with 100 μL of lactose-agarose (Sigma-Aldrich, USA) in 1 mL PBS in 1.5 mL column overnight at 4 °C. Lactose-agarose was washed with 1 mL of cold PBS for four times. Then, proteins bound to the lactose agarose were eluted with 1 mL PBS containing 100 mM galactose or 100 mM mannose for four times. The agarose was then washed with 1 mL 0.5N NaOH. Each fraction was collected. One fifth of each fraction was precipitated by incubation with 1 μg BSA and 800 μL acetone in 1.5 mL tubes at −20 °C. Tubes were centrifuged at 14000 g for 15 minutes at 4 °C, and the sediment was lyophilized and then detected by 12% SDS-PAGE and biotin-streptavidin blotting.
Cell culture and Stable isotope labeling with amino acids in cell culture (SILAC) and protein sample preparation
HepG2 cells were cultured in DMEM (Hyclone, USA) containing 10% fetal calf serum (Gibco, USA), 100 units/mL penicillin and streptomycin at 37 °C with 5% carbon dioxide and 100% humidity. Then, cells were transferred to a cell culture medium containing 100 mg/mL of 13C6-L-lysine (Invitrogen, USA) and 100 mg/mL of 13C6-L-lysine (Invitrogen, USA). The cells were grown in heavy stable isotope reagent for at least 21 days96.
Heavy isotope amine acid labeled HepG2 cells or light cells were cultured for 12 hours, and then washed with cold sterile PBS (Gibco, USA) for three times. The cells were then stimulated with rGal3C.
Cells were washed and collected, and then dissolved in cell lysis buffer [100 mM Tris, 4% sodium dodecyl sulfonate (SDS), 1% protease inhibitor cocktail (Sigma-Aldrich, USA), pH 8.0] using ultrasonic waves. Then, the mixture was centrifuged at 14000 g for 15 minutes at room temperature. Cell lysis supernatant was collected and total protein concentration was measured by Nanodrop 2000C (Thermo Fisher Scientific, USA).
Ultraviolet radiation dependent cross-link strategy for receptors capturing
HepG2 cells were cultured in 10 cm culture dishes (Corning, USA), and then washed PBS for three times. Then, 5 ml cold sterile PBS containing 250 μg photo-leucine analogue (Pierce, USA) labeled rGal3C was injected. Dishes were kept in dark for at least 4 hours at 4 °C, and then fixed with 30W, 365 nm UV for 15 minutes at a 5 cm distance. Fixed rGal3C were dtetcted by FTIC labelled streptavidin. Cells were collected and re-suspended by 1 mL PBS containing 0.5% triton X-100 and 1% protease inhibitor (Sigma-Aldrich, USA) and broken by ultrasonic wave at 4 °C. Cell supernatant was extracted, and captured fraction was enriched by 100 μL streptavidin agarose (Thermo Fisher Scientific, USA). Agarose was washed with 1M KCl and 0.1M Na2CO3, each for three times. Captured fraction released by heated at 95 °C for ten minutes in 100 μL 1% SDS solutionand then centrifuging at 10000 g for 15 minutes at room templature was loaded to FASP.
Filter-aided sample preparation (FASP)
The protein digestion was processed by FASP with slight adaptations97. For SILAC experiment, treated and untreated samples were mixed in equal amount. Samples were denatured at 95 °C with 10 mM DTT for 10 minutes. Then, 50 mM iodoacetamide was added for 30 minutes at room temperature in dark. UA (50 mM Tris, 8 M Urea, pH 8.0) was added to the mixture to dilute the SDS to a final concentration of 0.1%. The mixture was transferred into a 500 mL/30 kDa ultra-fraction tube (Merck-Millipore, Germany). The ultra-fraction tube was washed with 200 μL UA and succedent 200 μL ABC (50 mM ammonium bicarbonate), each for three times by centrifuging at 14000 g for 15 minutes at room temperature. Then, 100 μL ABC containing 0.1 μg/μL trypsin (Progema, USA) was injected into each ultra-fraction tube. Tubes were incubated at 37 °C overnight. Peptides were collected by centrifuging at 14000 g for 15 minutes. Tubes were washed twice with 100 μL of ABC for 15 minutes. The flowing fractions were collected together, and the concentration was measured using a Nanodrop 2000C. Peptides were acidized with 10 μL 0.1% trifluoroacetic acid (TFA, Sigma-Aldrich, USA) and freeze-dried, and then stored at −80 °C.
High pH reversed-phase prefractionation of the peptides
The peptides were pre-separated using high pH reversed-phase prefractionation. The pipet tip containing 5 mg of C18 reverse-phase material (Bonna-Agela Technologies, USA) was blocked by a C8 sieve plate. The tip was washed with 90 μL methanol, 90 μL acetonitrile, and then 90 μL alkaline water (pH = 10). Fifty μg peptides were re-dissolved in 160 μL of alkaline water (pH = 10) and injected into the tip. The tip was centrifuged at 1200 g for 5 minutes to remove the liquid. Then, 90 μL of alkaline water was injected into the tip, and then the tip was centrifuged at 1200 g for 5 minutes. Peptides were then eluted with increasing concentration (6%, 9%, 12%, 15%, 18%, 21%, 25%, 30%, 35%, 40%, 45% and 50%) of acetonitrile in alkaline water. Fractions were collected, and the last six fractions were mixed with the first six fractions. These six fractions were freeze-dried and stored at −80 °C.
Liquid chromatography tandem mass spectrometry analysis of peptide mixture
The LC-MS/MS detecting system consisted of a nanoflow HPLC chromatographic instrument (Easynano LC 1200, Thermo Fisher Scientific, USA) coupled to a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific, USA) with a Nano electrospray ion source (Thermo Fisher Scientific, USA) online. Briefly, 0.5 μg of peptide mixtures were transferred to the self-packed C18-reversed phase column (12 cm length, 150 μm inner diameter) using flowing phase A (99.9% water and 0.1% FA) and separated using a 78 min linear gradient of 6–40% flowing phase B (99.9% acetonitrile and 0.1% FA) at a flow rate of 600 nL/min. High-energy collisions induced dissociation mode and an Orbitrap mass analyzer were used in the analysis. The peptides were analyzed using data-dependent MS/MS acquisition with dynamic exclusion duration of 12 s. The resolution of MS1 was 120,000, and the maximum injection time was 80 ms. The resolution of MS2 was 15,000, and the maximum injection time was 45 ms, and the AGC target was 2E4. The scan range was 300–1,400 m/z for MS1 and 200–2,000 m/z for MS2. Twenty most intense precursor ions were selected for the MS/MS analysis.
Database searching and data mining
Raw files were processed to Maxquant 1.5.3 to quantitatively information analyze protein expression98. The enzyme digestion mode was set as Trypsin/P and two missed cleavage sites. The fragmentation spectra were searched against the Swiss prot Human database containing 20193 proteins. The multipilcity was set as two for the samples labeled with for 13C6-L-lysine and 13C6-L-lysine. The variable modifications were set as oxidation (M), acetylation (protein N-terminal) and Deamidation (NQ). The fixed modification was set as carbamidomethyl (C). Unique and razor peptides were used for the quantitative analysis. All of the peptides were used, regardless of their modifications. The PSM FDR was set as 1% and protein FDR was set as 1%. The remaining parameters were set to the default modes.
The gene ontology and pathway analyses were conducted using DAVID Bioinformatics Resources 6.8 online (https://david.ncifcrf.gov/) for gene ontology(GO), String 10 (http://string-db.org/) for protein interaction analysis, WebGestalt (http://www.webgestalt.org)53 for Disease Association analysis and TMHMM v2.0 (http://www.cbs.dtu.dk/services/TMHMM/) were used for bioinformatics analysis.
Cell viability, cell migration and invasion assay
For MTT assay, 104 cells in each well were cultured in 96-well plates (Corning, USA), and 200 μL of DMEM containing 10, 50 or 100 μg/mL rGal3C was injected. After stimulating, 0.5 mg/mL MTT (Sangon, China) was injected into each well for incubating 4 hours. Then, each well was gently washed three times with cold sterile PBS, and 200 μL of DMSO (Sigma-Aldrich, USA) was injected into each well to dissolve the treated cells. The plate was shocked at 200 rpm for 10 minutes, and the absorbance at 490 nm of each well was measured. Each well was repeated for three times.
For wound-healing assay, cells were maintained in the 12-well plates (Corning, USA) to form confluent cell monolayer. Then, cells were incubated with 1 mL DMEM containing rGal3C. The confluent cell monolayer was wounded by manually scraping the cells with a 10 μL pipette tip. Then, 1 mL DMEM (Hyclone, USA) containing 50 μg/mL of rGal3C was injected. Migration was assessed after 12 hours. Wells without rGal3C stimulating were the control group.
The basement membrane invasion assay was performed using Transwell (Corning, USA). Generally, 104 HepG2 cell was cultured in the insert coated with matrigel for 24 hours. Then, 600 μL DMEM containing 50 μg/mL of rGal3C and 10% FBS was added to the lower chamber, and 100 μL of DMEM containing 50 μg/mL of rGal3C was added into the insert. Cells were then incubated for 24 hours. The cells that invaded across and adhered on the bottom surface of the insert were fixed with 4% paraformaldehyde/PBS for 15 minutes at room temperature, then strained with 0.1% crystal violetand counted. There was no rGal3C in the control group.
Immunofluorescence analysis
HepG2 cells were maintained in the confocal culture dishes, and treated with 50 μg/mL of rGal3C for 12 hours. Dishes were gently washed three times with cold sterile PBS, and fixed in PBS with 4% paraformaldehyde for 15 minutes at room temperature. Fixed cells were gently washed three times with cold sterile PBS, then 400 μL cold sterile PBS containing 2 μg anti-Galectin-3 rabbit polyclone antibody (Cell Signaling Technology, USA) was injected. Dishes were incubated in dark and moist box overnight at 4 °C, then gently washed three times with cold sterile PBS for three times. One μg Alexa 594 dye labeled anti-mouse IgG (Cwbiotech, China) and 1 μg FITC labeled streptavidin (Cwbiotech, China) in 400 μL PBS was added into each dish. Dishes were incubated in dark and moist box for 1 hour at 37 °C.
For displaying the distribution of integrin clustering on cell surface, anti-ITA3 mouse polyclone antibody (Cell Signaling Technology, USA) and anti-Galectin-3 rabbit polyclone antibody (Cell Signaling Technology, USA) were added the same as described above. The following steps were mostly the same expect using Alexa 488 dye labeled anti-mouse IgG (Cwbiotech, China) and Alexa 561 labeled anti-rabbit IgG (Cwbiotech, China).
Dishes were gently washed with cold sterile PBS for three times, and then placed under a confocal microscope (Nikon, Japan). Images were taken under 60-fold objective lens.
Western blotting and biotin-streptavidin blotting
For the western blotting analysis, 30 μg of the total cell lysis was separated using 12% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Merck-Millipore, Germany) with a 0.22 μm aperture. After blocking with 5% bovine albumin dissolved in TBST (20 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween-20, pH 8.0), the membrane was washed three times with TBST. Then, the membrane was incubated with primary antibody at a 1:1000 dilution overnight. The membrane was washed three times with TBST for three times, and then incubated with HRP conjugated secondary antibody at a 1:5000 dilution for two hours in room temperature. The membrane was washed three times with TBST, and then washed three times with TBS. The target protein bands were detected using ImageQuant LAS 500 (GE Healthcare, USA). The primary antibodies and secondary antibodies used in this study were purchased from Cell Signaling Technology (Cell Signaling Technology, USA).
For the biotin-streptavidin blotting analysis, the protein samples separating and transferring steps were the same as western blotting, but the membrane was incubated with HRP labeled streptavidin at a 1:500 dilution for at least 1 hour at room temperature. The membrane was then washed three times with TBST, and then washed three times with TBS. The target protein bands were detected using ImageQuant LAS 500 (GE Healthcare, USA).
Statistical analysis
All tests were performed at least in triplicate, and all experiments were performed at least three times. The results are expressed as the mean ± standard deviation (SD). Differences between groups were analyzed using Student’s t-test or one-way analysis of variance (ANOVA). The analysis was conducted using Origin v9.0 software (OriginLab, USA), and differences were considered significant at *P ≤ 0.01.
Electronic supplementary material
Acknowledgements
We are grateful for financial support from the National Key Program for Basic Research of China (2016YFA0501300, 2014CBA02001, SQ2017YFSF090210), the National Natural Science Foundation of China (81530021).
Author Contributions
M.W. conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of manuscript; F.T. collection and/or assembly of data; W.Y. and X.Q. conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript. All authors approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05419-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wantao Ying, Email: proteomics@126.com.
Xiaohong Qian, Email: qianxh1@163.com.YING.
References
- 1.Torre LA, et al. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 4.Moeini A, Cornella H, Villanueva A. Emerging signaling pathways in hepatocellular carcinoma. Liver cancer. 2012;1:83–93. doi: 10.1159/000342405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao S, et al. The role of c-Src in the invasion and metastasis of hepatocellular carcinoma cells induced by association of cell surface GRP78 with activated alpha2M. BMC cancer. 2015;15:389. doi: 10.1186/s12885-015-1401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Toni EN, et al. P60-c-src suppresses apoptosis through inhibition of caspase 8 activation in hepatoma cells, but not in primary hepatocytes. Journal of hepatology. 2007;46:682–691. doi: 10.1016/j.jhep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Lau GM, et al. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Digestive diseases and sciences. 2009;54:1465–1474. doi: 10.1007/s10620-008-0519-0. [DOI] [PubMed] [Google Scholar]
- 8.Ito Y, et al. Activation of c-Src gene product in hepatocellular carcinoma is highly correlated with the indices of early stage phenotype. Journal of hepatology. 2001;35:68–73. doi: 10.1016/S0168-8278(01)00077-0. [DOI] [PubMed] [Google Scholar]
- 9.Ferrin G, Aguilar-Melero P, Rodriguez-Peralvarez M, Montero-Alvarez JL, de la Mata M. Biomarkers for hepatocellular carcinoma: diagnostic and therapeutic utility. Hepatic medicine: evidence and research. 2015;7:1–10. doi: 10.2147/HMER.S50161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato Y, et al. Early recognition of hepatocellular carcinoma based on altered profiles of alpha-fetoprotein. The New England journal of medicine. 1993;328:1802–1806. doi: 10.1056/NEJM199306243282502. [DOI] [PubMed] [Google Scholar]
- 11.Kuromatsu R, Tanaka M, Tanikawa K. Serum alpha-fetoprotein and lens culinaris agglutinin-reactive fraction of alpha-fetoprotein in patients with hepatocellular carcinoma. Liver. 1993;13:177–182. doi: 10.1111/j.1600-0676.1993.tb00627.x. [DOI] [PubMed] [Google Scholar]
- 12.Chaiteerakij R, Addissie BD, Roberts LR. Update on biomarkers of hepatocellular carcinoma. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2015;13:237–245. doi: 10.1016/j.cgh.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng J, et al. NDRG1 as a biomarker for metastasis, recurrence and of poor prognosis in hepatocellular carcinoma. Cancer letters. 2011;310:35–45. doi: 10.1016/j.canlet.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Ma L, et al. Serum CD166: a novel hepatocellular carcinoma tumor marker. Clinica chimica acta; international journal of clinical chemistry. 2015;441:156–162. doi: 10.1016/j.cca.2014.12.034. [DOI] [PubMed] [Google Scholar]
- 15.Serizawa N, et al. Galectin 3 regulates HCC cell invasion by RhoA and MLCK activation. Laboratory investigation; a journal of technical methods and pathology. 2015;95:1145–1156. doi: 10.1038/labinvest.2015.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang SS, et al. Galectin-3 is associated with a poor prognosis in primary hepatocellular carcinoma. Journal of translational medicine. 2014;12:273. doi: 10.1186/s12967-014-0273-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisa NH, Ebrahim MA, Ragab M, Eissa LA, El-Gayar AM. Galectin-3 and matrix metalloproteinase-9: Perspective in management of hepatocellular carcinoma. Journal of oncology pharmacy practice: official publication of the International Society of Oncology Pharmacy Practitioners. 2015;21:323–330. doi: 10.1177/1078155214532698. [DOI] [PubMed] [Google Scholar]
- 18.Zheng D, et al. Downregulation of galectin-3 causes a decrease in uPAR levels and inhibits the proliferation, migration and invasion of hepatocellular carcinoma cells. Oncology reports. 2014;32:411–418. doi: 10.3892/or.2014.3170. [DOI] [PubMed] [Google Scholar]
- 19.Song L, et al. Annexin A7 and its binding protein galectin-3 influence mouse hepatocellular carcinoma cell line in vitro. Biomedicine & pharmacotherapy=Biomedecine & pharmacotherapie. 2014;68:377–384. doi: 10.1016/j.biopha.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Yang R.Y, Hsu DK, Liu F-T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. 1996;93:6. doi: 10.1073/pnas.93.13.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukumori, T. et al. Endogenous Galectin-3 Determines the Routing of CD95 Apoptotic Signaling Pathways. Cancer research64, 4. [DOI] [PubMed]
- 22.Akahani, S., Nangia-Makker, P., Inohara, H., Kim, H. R. & Raz, A. Galectin-3: A Novel Antiapoptotic Molecule with A Functional BH1 (NWGR) Domain of Bcl-2 Family. Cancer Research57, 5 (1997). [PubMed]
- 23.Elad-Sfadia G, Haklai R, Balan E, Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. The Journal of biological chemistry. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 24.Shalom-Feuerstein R, Cooks T, Raz A, Kloog Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer research. 2005;65:7292–7300. doi: 10.1158/0008-5472.CAN-05-0775. [DOI] [PubMed] [Google Scholar]
- 25.Lee YJ, et al. Reconstitution of galectin-3 alters glutathione content and potentiates TRAIL-induced cytotoxicity by dephosphorylation of Akt. Experimental cell research. 2003;288:21–34. doi: 10.1016/S0014-4827(03)00211-8. [DOI] [PubMed] [Google Scholar]
- 26.Oka N, et al. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer research. 2005;65:7546–7553. doi: 10.1158/0008-5472.CAN-05-1197. [DOI] [PubMed] [Google Scholar]
- 27.Laing JG, Wang JL. Identification of carbohydrate binding protein 35 in heterogeneous nuclear ribonucleoprotein complex. Biochemistry. 1988;27:5329–5334. doi: 10.1021/bi00414a057. [DOI] [PubMed] [Google Scholar]
- 28.Dagher SF, W JL, Patfrerson RJ. Identification of galectin-3 as a factor in pre-mRNA splicing. Proc. Natl. Acad. Sci. 1995;92:5. doi: 10.1073/pnas.92.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi Kim H-R, L H-M, Biliran H, Raz A. Cell Cycle Arrest and Inhibition of Anoikis by Galectin-3 in Human Breast Epithelial Cells. Cancer research. 1999;59:7. [PubMed] [Google Scholar]
- 30.Paron I, et al. Nuclear localization of Galectin-3 in transformed thyroid cells: a role in transcriptional regulation. Biochemical and biophysical research communications. 2003;302:545–553. doi: 10.1016/S0006-291X(03)00151-7. [DOI] [PubMed] [Google Scholar]
- 31.Lin HM, Pestell RG, Raz A, Kim HR. Galectin-3 enhances cyclin D(1) promoter activity through SP1 and a cAMP-responsive element in human breast epithelial cells. Oncogene. 2002;21:8001–8010. doi: 10.1038/sj.onc.1205820. [DOI] [PubMed] [Google Scholar]
- 32.Tatsuo Shimura, Y. T., Souichi Tsutsumi, Victor Hogan, Akira Kikuchi, and Avraham Raz. Galectin-3, a Novel Binding Partner of β-Catenin. Cancer research64, 5 (2004). [DOI] [PubMed]
- 33.Tatsuo Shimura YT, et al. Implication of Galectin-3 in Wnt Signaling. Cancer research. 2005;65:3. doi: 10.1158/0008-5472.CAN-05-0104. [DOI] [PubMed] [Google Scholar]
- 34.Pratima Nangia-Makker YH, et al. Galectin-3 Induces Endothelial Cell Morphogenesis and Angiogenesis. American Journal of Pathology. 2000;156:11. doi: 10.1016/S0002-9440(10)64959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. The Journal of biological chemistry. 2007;282:1374–1383. doi: 10.1074/jbc.M604506200. [DOI] [PubMed] [Google Scholar]
- 36.Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. Journal of cell science. 2015;128:2213–2219. doi: 10.1242/jcs.151159. [DOI] [PubMed] [Google Scholar]
- 37.Fortuna-Costa A, Gomes AM, Kozlowski EO, Stelling MP, Pavao MS. Extracellular galectin-3 in tumor progression and metastasis. Frontiers in oncology. 2014;4:138. doi: 10.3389/fonc.2014.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puthenedam M, et al. Matrilysin-1 (MMP7) cleaves galectin-3 and inhibits wound healing in intestinal epithelial cells. Inflammatory bowel diseases. 2011;17:260–267. doi: 10.1002/ibd.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ochieng J, Green B, Evans S, James O, Warfield P. Modulation of the biological functions of galectin-3 by matrix metalloproteinases. Biochimica et biophysica acta. 1998;1379:97–106. doi: 10.1016/S0304-4165(97)00086-X. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima K, et al. Galectin-3 Cleavage Alters Bone Remodeling: Different Outcomes in Breast and Prostate Cancer Skeletal Metastasis. Cancer research. 2016;76:1391–1402. doi: 10.1158/0008-5472.CAN-15-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.John CM, Leffler H, Kahl-Knutsson B, Svensson I, Jarvis GA. Truncated galectin-3 inhibits tumor growth and metastasis in orthotopic nude mouse model of human breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:2374–2383. [PubMed] [Google Scholar]
- 42.Mirandola L, et al. Galectin-3C inhibits tumor growth and increases the anticancer activity of bortezomib in a murine model of human multiple myeloma. PloS one. 2011;6:e21811. doi: 10.1371/journal.pone.0021811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nature protocols. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 44.Chen X, Wei S, Ji Y, Guo X, Yang F. Quantitative proteomics using SILAC: Principles, applications, and developments. Proteomics. 2015;15:3175–3192. doi: 10.1002/pmic.201500108. [DOI] [PubMed] [Google Scholar]
- 45.Chen N, et al. Quantitative proteome analysis of HCC cell lines with different metastatic potentials by SILAC. Proteomics. 2008;8:5108–5118. doi: 10.1002/pmic.200800280. [DOI] [PubMed] [Google Scholar]
- 46.Chakraborty S, et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nature communications. 2015;6:6184. doi: 10.1038/ncomms7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yeh CC, et al. Integrated Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) and Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) Quantitative Proteomic Analysis Identifies Galectin-1 as a Potential Biomarker for Predicting Sorafenib Resistance in Liver Cancer. Molecular & cellular proteomics: MCP. 2015;14:1527–1545. doi: 10.1074/mcp.M114.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren F, et al. Quantitative proteomics identification of phosphoglycerate mutase 1 as a novel therapeutic target in hepatocellular carcinoma. Molecular cancer. 2010;9:81. doi: 10.1186/1476-4598-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 51.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic acids research. 2005;33:W741–748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic acids research. 2013;41:W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochimica et biophysica acta. 2006;1760:616–635. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 55.Mirandola L, et al. Anti-galectin-3 therapy: a new chance for multiple myeloma and ovarian cancer? International reviews of immunology. 2014;33:417–427. doi: 10.3109/08830185.2014.911855. [DOI] [PubMed] [Google Scholar]
- 56.Collins PM, Hidari KI, Blanchard H. Slow diffusion of lactose out of galectin-3 crystals monitored by X-ray crystallography: possible implications for ligand-exchange protocols. Acta crystallographica. Section D, Biological crystallography. 2007;63:415–419. doi: 10.1107/S090744490605270X. [DOI] [PubMed] [Google Scholar]
- 57.Saraboji K, et al. The carbohydrate-binding site in galectin-3 is preorganized to recognize a sugarlike framework of oxygens: ultra-high-resolution structures and water dynamics. Biochemistry. 2012;51:296–306. doi: 10.1021/bi201459p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kovacevic Z, Richardson DR. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis. 2006;27:2355–2366. doi: 10.1093/carcin/bgl146. [DOI] [PubMed] [Google Scholar]
- 59.Melotte V, et al. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4153–4166. doi: 10.1096/fj.09-151464. [DOI] [PubMed] [Google Scholar]
- 60.Ellen TP, Ke Q, Zhang P, Costa M. NDRG1, a growth and cancer related gene: regulation of gene expression and function in normal and disease states. Carcinogenesis. 2008;29:2–8. doi: 10.1093/carcin/bgm200. [DOI] [PubMed] [Google Scholar]
- 61.Hickok JR, Sahni S, Mikhed Y, Bonini MG, Thomas DD. Nitric oxide suppresses tumor cell migration through N-Myc downstream-regulated gene-1 (NDRG1) expression: role of chelatable iron. The Journal of biological chemistry. 2011;286:41413–41424. doi: 10.1074/jbc.M111.287052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan X, Chua MS, Sun H, So S. N-Myc down-regulated gene 1 mediates proliferation, invasion, and apoptosis of hepatocellular carcinoma cells. Cancer letters. 2008;262:133–142. doi: 10.1016/j.canlet.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 63.Lu WJ, Chua MS, So SK. Suppressing N-Myc downstream regulated gene 1 reactivates senescence signaling and inhibits tumor growth in hepatocellular carcinoma. Carcinogenesis. 2014;35:915–922. doi: 10.1093/carcin/bgt401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akiba J, et al. N-myc downstream regulated gene 1 (NDRG1)/Cap43 enhances portal vein invasion and intrahepatic metastasis in human hepatocellular carcinoma. Oncology reports. 2008;20:1329–1335. [PubMed] [Google Scholar]
- 65.Lu WJ, Chua MS, Wei W, So SK. NDRG1 promotes growth of hepatocellular carcinoma cells by directly interacting with GSK-3beta and Nur77 to prevent beta-catenin degradation. Oncotarget. 2015;6:29847–29859. doi: 10.18632/oncotarget.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Earl LA, Bi S, Baum LG. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology. 2011;21:6–12. doi: 10.1093/glycob/cwq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ochieng J, et al. Galectin-3 is a novel substrate for human matrix metalloproteinases-2 and -9. Biochemistry. 1994;33:14109–14114. doi: 10.1021/bi00251a020. [DOI] [PubMed] [Google Scholar]
- 68.Fukushi J, Makagiansar IT, Stallcup WB. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Molecular biology of the cell. 2004;15:3580–3590. doi: 10.1091/mbc.E04-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lagana A, et al. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Molecular and cellular biology. 2006;26:3181–3193. doi: 10.1128/MCB.26.8.3181-3193.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao X, Balan V, Tai G, Raz A. Galectin-3 induces cell migration via a calcium-sensitive MAPK/ERK1/2 pathway. Oncotarget. 2014;5:2077–2084. doi: 10.18632/oncotarget.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Park GB, et al. Silencing of galectin-3 represses osteosarcoma cell migration and invasion through inhibition of FAK/Src/Lyn activation and beta-catenin expression and increases susceptibility to chemotherapeutic agents. International journal of oncology. 2015;46:185–194. doi: 10.3892/ijo.2014.2721. [DOI] [PubMed] [Google Scholar]
- 72.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Current opinion in cell biology. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 73.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 74.Ochieng J, Leite-Browning ML, Warfield P. Regulation of cellular adhesion to extracellular matrix proteins by galectin-3. Biochemical and biophysical research communications. 1998;246:788–791. doi: 10.1006/bbrc.1998.8708. [DOI] [PubMed] [Google Scholar]
- 75.Lei CX, Zhang W, Zhou JP, Liu YK. Interactions between galectin-3 and integrinbeta3 in regulating endometrial cell proliferation and adhesion. Human reproduction (Oxford, England) 2009;24:2879–2889. doi: 10.1093/humrep/dep250. [DOI] [PubMed] [Google Scholar]
- 76.Friedrichs J, Manninen A, Muller DJ, Helenius J. Galectin-3 regulates integrin alpha2beta1-mediated adhesion to collagen-I and -IV. The Journal of biological chemistry. 2008;283:32264–32272. doi: 10.1074/jbc.M803634200. [DOI] [PubMed] [Google Scholar]
- 77.Saravanan C, Liu FT, Gipson IK, Panjwani N. Galectin-3 promotes lamellipodia formation in epithelial cells by interacting with complex N-glycans on alpha3beta1 integrin. Journal of cell science. 2009;122:3684–3693. doi: 10.1242/jcs.045674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochimica et biophysica acta. 1692;77-102:2004. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 79.Lindauer M, Hochhaus A. Dasatinib. Recent results in cancer research. Fortschritte der Krebsforschung. Progres dans les recherches sur le cancer. 2014;201:27–65. doi: 10.1007/978-3-642-54490-3_2. [DOI] [PubMed] [Google Scholar]
- 80.Chang AY, Wang M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC cancer. 2013;13:267. doi: 10.1186/1471-2407-13-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mohammed, H. R. et al. Exosomes: From Garbage Bins to Promising Therapeutic Targets. International journal of molecular sciences18, doi:10.3390/ijms18030538 (2017). [DOI] [PMC free article] [PubMed]
- 82.Zhitong Wu, Q. Z., Ke Cao & Yifan Sun. Exosomes: small vesicles with big roles in hepatocellular carcinoma. 7, 11 (2016). [DOI] [PMC free article] [PubMed]
- 83.Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003;22:5093–5107. doi: 10.1038/sj.onc.1206557. [DOI] [PubMed] [Google Scholar]
- 84.Trougakos IP, Gonos ES. Clusterin/apolipoprotein J in human aging and cancer. The international journal of biochemistry & cell biology. 2002;34:1430–1448. doi: 10.1016/S1357-2725(02)00041-9. [DOI] [PubMed] [Google Scholar]
- 85.Levin, T. G. et al. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology139, 2072–2082 e2075, doi:10.1053/j.gastro.2010.08.053 (2010). [DOI] [PMC free article] [PubMed]
- 86.Ma L, Pan Q, Sun F, Yu Y, Wang J. Cluster of differentiation 166 (CD166) regulates cluster of differentiation (CD44) via NF-kappaB in liver cancer cell line Bel-7402. Biochemical and biophysical research communications. 2014;451:334–338. doi: 10.1016/j.bbrc.2014.07.128. [DOI] [PubMed] [Google Scholar]
- 87.Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. European journal of surgical oncology: the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2008;34:357–364. doi: 10.1016/j.ejso.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 88.Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nature reviews. Cancer. 2015;15:96–109. doi: 10.1038/nrc3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sakaguchi M, et al. Relationship between contact inhibition and intranuclear S100C of normal human fibroblasts. The Journal of cell biology. 2000;149:1193–1206. doi: 10.1083/jcb.149.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyazaki M, et al. Involvement of interferon regulatory factor 1 and S100C/A11 in growth inhibition by transforming growth factor beta 1 in human hepatocellular carcinoma cells. Cancer research. 2004;64:4155–4161. doi: 10.1158/0008-5472.CAN-03-2750. [DOI] [PubMed] [Google Scholar]
- 91.Wu H, et al. Overexpression of galectin-1 is associated with poor prognosis in human hepatocellular carcinoma following resection. Journal of gastroenterology and hepatology. 2012;27:1312–1319. doi: 10.1111/j.1440-1746.2012.07130.x. [DOI] [PubMed] [Google Scholar]
- 92.Espelt MV, et al. Novel roles of galectin-1 in hepatocellular carcinoma cell adhesion, polarization, and in vivo tumor growth. Hepatology (Baltimore, Md.) 2011;53:2097–2106. doi: 10.1002/hep.24294. [DOI] [PubMed] [Google Scholar]
- 93.Bacigalupo ML, et al. Galectin-1 triggers epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Journal of cellular physiology. 2015;230:1298–1309. doi: 10.1002/jcp.24865. [DOI] [PubMed] [Google Scholar]
- 94.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nature reviews. Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 95.Powlesland AS, et al. A novel mechanism for LSECtin binding to Ebola virus surface glycoprotein through truncated glycans. The Journal of biological chemistry. 2008;283:593–602. doi: 10.1074/jbc.M706292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harsha HC, Molina H, Pandey A. Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nature protocols. 2008;3:505–516. doi: 10.1038/nprot.2008.2. [DOI] [PubMed] [Google Scholar]
- 97.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 98.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.