

Abstract
Cell migration drives cell invasion and metastatic progression in prostate cancer and is a major cause of mortality and morbidity. However the mechanisms driving cell migration in prostate cancer patients are not fully understood. We previously identified the cancer-associated cell migration protein Tetraspanin 1 (TSPAN1) as a clinically relevant androgen regulated target in prostate cancer. Here we find that TSPAN1 is acutely induced by androgens, and is significantly upregulated in prostate cancer relative to both normal prostate tissue and benign prostate hyperplasia (BPH). We also show for the first time, that TSPAN1 expression in prostate cancer cells controls the expression of key proteins involved in cell migration. Stable upregulation of TSPAN1 in both DU145 and PC3 cells significantly increased cell migration and induced the expression of the mesenchymal markers SLUG and ARF6. Our data suggest TSPAN1 is an androgen-driven contributor to cell survival and motility in prostate cancer.
Introduction
Cancer, in its most aggressive form, is not only a disease of uncontrolled cell growth, but also a disease of inappropriate cell migration. Activating invasion and metastasis is a hallmark of cancer progression1, 2 and is the leading cause of mortality among cancer patients3. Metastasis involves cancer cells detaching from the primary tumour, and travelling as circulating tumour cells through the bloodstream or lymphatic system to other parts of the body. Prostate cancer is the most common male cancer in Europe, with around 50,000 new cases in the UK each year4. At initial diagnosis 37–43% of men have late stage disease and 17–34% of prostate cancer patients have metastasis (http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/prostate-cancer/incidence#ref-8). The development of prostate cancer is initially driven by androgen steroid hormones via the androgen receptor (AR) transcription factor. The first line treatment for prostate cancer that is no longer organ confined is androgen deprivation therapy (ADT). However, after 2–3 years many patients develop castrate resistant prostate cancer (CRPC) for which treatment options are limited and prognosis is poor5, meaning there is an urgent need to develop new treatments for advanced prostate cancer. Prognostic heterogeneity is an important feature of prostate cancer; while some prostate cancers can progress very rapidly, others can remain indolent for many years, hence there also a major unmet clinical need to identify new biomarkers to help distinguish indolent from aggressive disease6.
The mechanisms underlying the development and progression of prostate cancer are poorly understood. We recently used RNA-Sequencing to comprehensively profile how the prostate cancer transcriptome responds to androgens7. Our approach directly correlated gene expression data from LNCaP cells before and after androgen exposure, with data from a small cohort of 7 prostate cancer patients before and after ADT. We identified a set of nearly 700 genes which were reciprocally regulated between the two datasets and so were strong candidates to be clinically relevant androgen-regulated genes in prostate cancer. This set of 700 genes included the gene for the cancer-associated cell migration protein Tetraspanin 1 (TSPAN1) which had not previously been shown to be regulated by androgens in prostate cancer.
Tetraspanins, also known as the transmembrane 4 superfamily, are small transmembrane glycoproteins which were first described in studies of tumour associated proteins8–13. As a member of the tetraspanin family, TSPAN1 has been reported to regulate cancer progression in many human cancers. TSPAN1 is upregulated in human hepatocellular carcinoma14, gastric carcinoma15, colorectal adenocarcinoma16, ovarian carcinomas17 and cervical cancer18, 19. Tetraspanins reportedly play a role in a range of biological processes including cell proliferation9, cell adhesion20, cell migration and motility21, 22 and signal transduction23, 24. Here, we show that expression of TSPAN1 is controlled by androgens in prostate cancer cells, is upregulated in prostate cancer tissue and is important for prostate cancer cell survival and migration. Our findings are in agreement with numerous studies showing that TSPAN1 is upregulated in several other cancer types15, 17, 25–28, but are in contrast to a recent publication suggesting that decreased TSPAN1 is linked to prostate cancer progression29.
Results
TSPAN1 is an early target of the AR in vitro and in vivo
Previous RNA-Seq analysis of LNCaP cells identified the cancer-associated TSPAN1 gene as being under control of androgens after 24 hours treatment with 10 nM of the synthetic androgen analogue R1881 (methyltrienolone)7. Using a time course and real-time PCR we found that androgen mediated induction of the TSPAN1 gene could be detected in LNCaP cells 9 hours after androgen exposure suggesting it is directly regulated by the AR. The early expression profile of TSPAN1 following androgen exposure had similar dynamics to the known directly AR-regulated gene KLK3 (PSA) (Fig. 1A). Androgen-mediated induction of TSPAN1 expression in LNCaP cells was also induced by treatment with a range of R1881 concentrations for 24 hours, consistent with TSPAN1 induction also occurring under physiological androgen concentrations within the prostate (Fig. 1B), and was blocked by treatment with the AR antagonist CasodexR (bicalutamide) (Fig. 1C). To test whether androgen-mediated induction of TSPAN1 expression was a result of AR activity, we treated LNCaP cells with 10 mM R1881 (androgens) in the presence and absence of cycloheximide to inhibit de novo protein synthesis. Androgen mediated up-regulation of TSPAN1 mRNA expression was observed in the presence or absence of the protein synthesis inhibitor cycloheximide indicating that TSPAN1 induction might be directly mediated by the AR (Fig. 1D). Consistent with this, analysis of previously published AR ChIP-Seq data30 revealed an AR binding site which is overlapping with the start of the TSPAN1 gene for both LNCaP and VCaP cells (Supplementary Figure 1).
Figure 1.
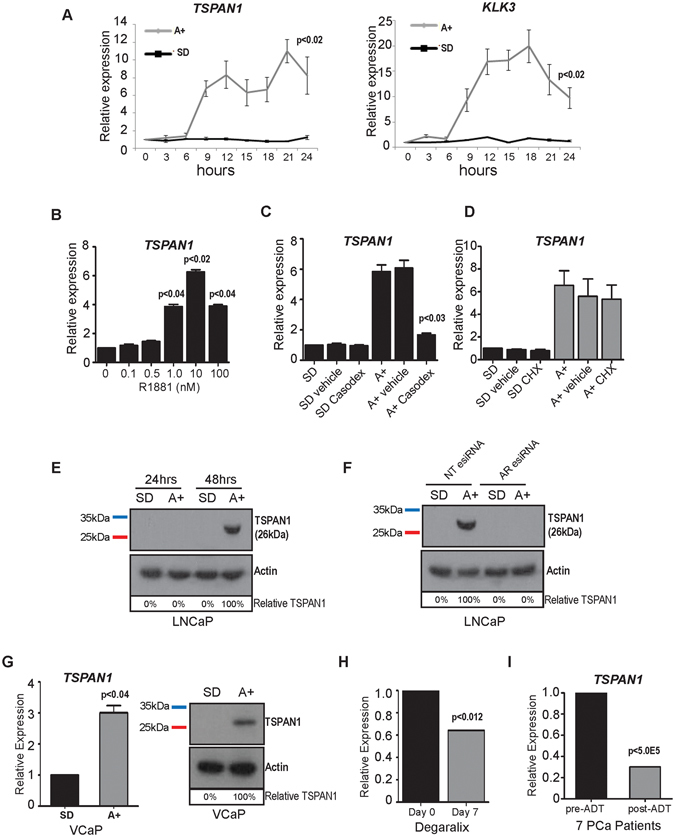
TSPAN1 is regulated by androgens in in vitro and in vivo. (A) Analysis of TSPAN1 and KLK3 (PSA) mRNA in LNCaP cells treated with androgens (A+) over a 24 hour time course. (B) Upregulation of TSPAN1 is also evident in LNCaP cells treated with 1 to 100 nM of R1881. (C) Induction of TSPAN1 mRNA by the AR is inhibited in the presence of 10 μM of the anti-androgen Casodex (bicalutamide) (lane 6). (D) The increase in TSPAN1 mRNA expression in response to androgens is still seen in the presence of 1 μg/ml cycloheximide (CHX). Relative TSPAN1 expression was detected by real-time PCR and normalised to three reference genes. (E) Expression of TSPAN1 protein is induced in LNCaP cells treated with 10 nM R1881 for 48 hours as detected by western blot. Actin was used as a loading control. (F) Depletion of AR protein in LNCaP cells by esiRNA shows that when the AR is depleted there is no induction of the TSPAN1 protein in response to androgens. (G) Real-time PCR and western blot analysis of TSPAN1 mRNA and protein in VCaP cells grown in steroid deplete (SD) or 10 nM R1881 (androgens, A+) treated conditions for 48 hours. (H) Analysis of TSPAN1 mRNA expression before and after 7 days of serum/tissue androgen depletion in prostate cancer patients using degarelix31. (I) Analysis of TSPAN1 expression in previously published RNA-Seq data32 shows that TSPAN1 mRNA is significantly down-regulated in 7 PCa patients following androgen deprivation treatment (ADT). The Y axis shows the relative expression level of TSPAN1 (calculated by comparing the FPKM expression values of genes after ADT to the value before treatment). Please note: cropped western blots are displayed in the figure, but full length blots are included in the Supplementary Information file.
Expression of TSPAN1 protein was also induced by treatment of LNCaP cells with 10 nM R1881 (Fig. 1E). Confirming this effect on protein levels was mediated by the AR, we found that androgen-mediated induction of the TSPAN1 protein is prevented when cells are depleted of the AR using esiRNA (Fig. 1F). Interestingly, although induction of TSPAN1 mRNA was detected within 9 hours of androgen exposure, induction at the protein level was not detected until 48 hours. Specificity of our TSPAN1 antibody was confirmed via siRNA mediated protein depletion and detection of over-expressed protein by western blotting (Supplementary Figure 2A,B). Supporting our data obtained in LNCaP cells, TSPAN1 expression was also androgen responsive in VCaP cells at both the RNA and protein level (Fig. 1G). Analysis of previously published RNA-Seq data from before and after 7 days of androgen depletion in prostate cancer patients using Degarelix31 shows a significant reduction in TSPAN1 mRNA indicating a tissue based response (Fig. 1H). Similarly, analysis of previously published RNA-Seq data from 7 prostate cancer patients32 showed that TSPAN1 gene expression is strongly down-regulated following ADT (Fig. 1I), suggesting that TSPAN1 is also regulated (either directly or indirectly) by androgens in vivo within patients.
TSPAN1 is upregulated in prostate cancer tissue
We carried out meta-analysis of 1012 prostate tissue samples using data from 14 previously published studies33–46 to monitor how expression of TSPAN1 changes in clinical prostate cancer. We found that 11/14 datasets showed significant up-regulation of TSPAN1 mRNA expression in prostate carcinoma versus normal prostate tissue (Supplementary Table 1). TSPAN1 expression showed an average mean fold change of 2.214 (p = 1.31E-6) in 122 primary prostate carcinoma samples studied by Grasso et al.33, and a mean fold change of 1.844 (p = 1.72E-7) in 185 samples studied by Taylor et al.34. TSPAN1 expression analysis by real-time PCR showed that TSPAN1 mRNA was significantly up-regulated in prostate carcinoma samples relative to BPH samples (p = 0.049), and in primary prostate tumour tissue relative to matched normal tissue from the same patient (p = 0.0004) (Fig. 2A,B).
Figure 2.
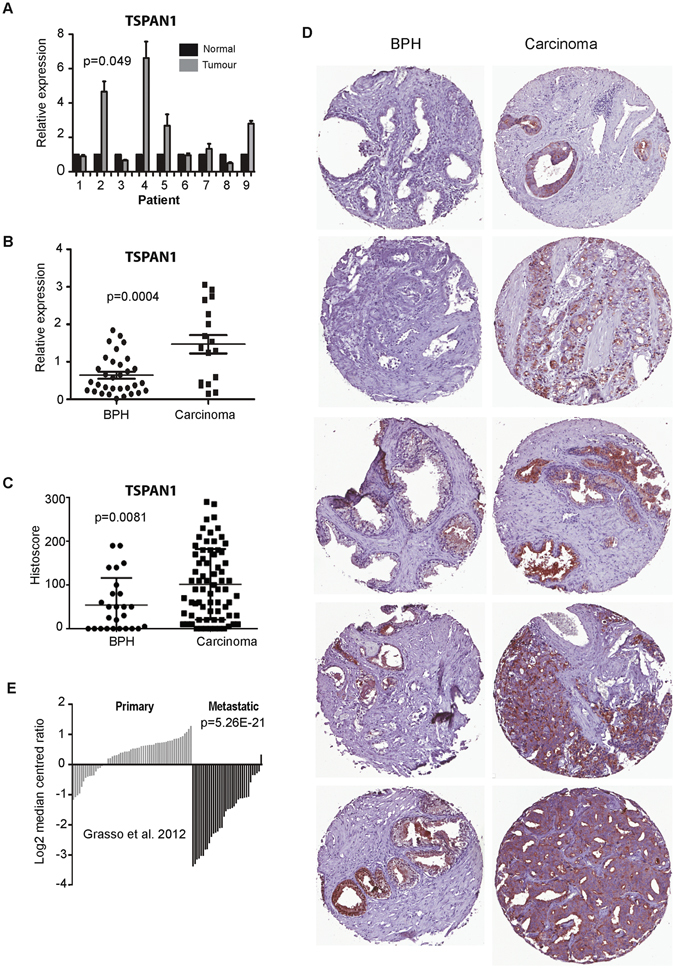
TSPAN1 is upregulated in prostate cancer tissue. (A) Real-time PCR analysis of TSPAN1 mRNA from normal and matched prostate cancer tissue from 9 patients obtained from radical prostatectomy. (B) We also analysed 32 benign samples from patients with benign prostate hyperplasia (BPH) and 17 malignant samples from transurothelial resection of the prostate (TURP) samples. (C) Analysis of TSPAN1 protein levels in patients with BPH or prostate cancer by Tissue Microarray (TMA). (D) Representative cores from each TMA. (E) TSPAN1 gene expression levels in prostate tissue from the primary or metastatic site in data published by Grasso et al.33.
To develop a rigorous test for TSPAN1 protein we extensively validated our TSPAN1 antibody in multiple cell lines using western blotting, siRNA mediated protein depletion, and detection of over-expressed protein (Supplementary Figure 2A,B). To examine TSPAN1 protein expression in clinical prostate cancer tissue we further showed that staining by this antibody was blocked by pre-incubation with a blocking peptide (Supplementary Figure 2C) and by detection of siRNA mediated protein depletion by immunohistochemistry in Formalin Fixed Paraffin Embedded (FFPE) cell pellets (Supplementary Figure 2D). TSPAN1 is predicted to be a transmembrane protein (http://www.uniprot.org/uniprot/O60635#subcellular_location), and the localisation of TSPAN1 within prostate tissue was mainly cytoplasmic and membrane bound. Staining a tissue microarray (TMA) containing 26 samples from patients with BPH and 72 samples from patients with prostate cancer confirmed that TSPAN1 protein is also expressed at significantly higher levels in prostate cancer patients compared to benign controls (p = 0.0081) (Fig. 2C) (further information on the clinical samples used in the TMA is given in Supplementary Table 2). Representative cores from each TMA are shown in Fig. 2D.
A previous study suggested decreased TSPAN1 could be used to predict biochemical recurrence after radical prostatectomy29, however, our data indicates no significant correlation between both overall survival and relapse free survival for patients with either low or high TSPAN1 (Supplementary Figure 3). To determine whether TSPAN1 is associated with cancer metastasis, we studied TSPAN1 gene expression in prostate cancer tissue obtained from either the primary or metastatic site using a previously-published clinical dataset33. We observed a changing pattern of TSPAN1 expression between primary and metastatic tumours with a striking decrease in TSPAN1 expression in prostate cancer tissue from the metastatic site (p = 5.26E-21) (Fig. 2E).
TSPAN1 increases prostate cancer cell migration and can upregulate expression of Slug and ARF6
The above data indicated that prostate cancer cells over-express TSPAN1 compared to normal or benign prostate tissue. To investigate why upregulation of TSPAN1 might be important in prostate cancer cells, we created DU145 and PC3 cell lines that over-express TSPAN1 protein (Fig. 3A,C). DU145 and PC3 cells were chosen for these analyses as they have low endogenous TSPAN1 levels (Supplementary Figure 4). Although there were no significant differences in cell proliferation or adhesion with upregulated TSPAN1 (Supplementary Figure 5), the rate of cell migration was significantly increased in both DU145 and PC3 cell backgrounds (Fig. 3B,D).
Figure 3.
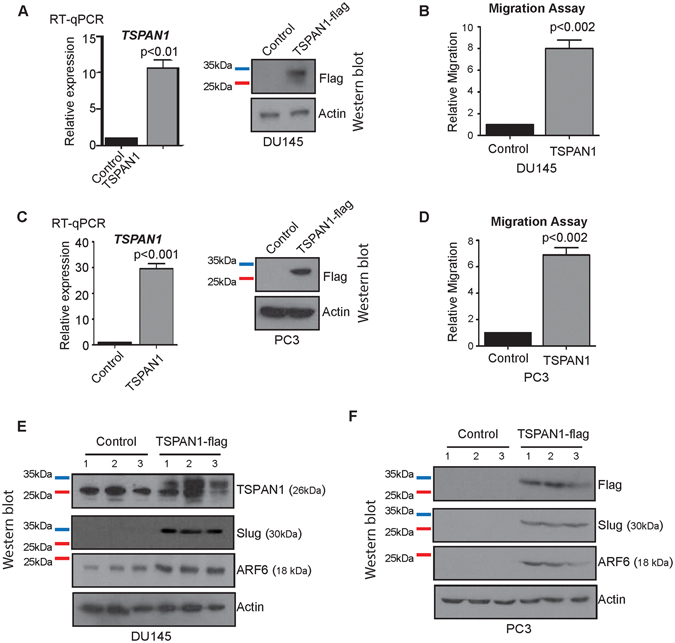
TSPAN1 increases prostate cancer cell migration and induces the expression of Slug and ARF6. (A,C) Validation of TSPAN1 overexpression in DU145 and PC3 cells by real-time PCR and western blot. (B,D) Migration assays for DU145 and PC3 cells overexpressing TSPAN1. (E,F) Western blot analysis of Slug and ARF6 protein expression in DU145 and PC3 cells overexpressing TSPAN1. Please note: cropped western blots are displayed in the figure, but full length blots are included in the Supplementary Information file.
To probe the regulatory mechanism governing TSPAN1-dependent prostate cancer cell motility, we analysed expression of 77 genes with established roles in the epithelial to mesenchymal transition (EMT) and/or cell motility and migration. In DU145 cells 10 out of 77 genes showed significant changes in gene expression when TSPAN1 was overexpressed (Supplementary Table 3), including genes associated with EMT. Increased expression of the EMT-related transcriptional repressor Slug and the small GTP-binding protein ARF6 was also seen at the protein level (Fig. 3E), and in PC3 cells with upregulated TSPAN1 (Fig. 3F). Slug expression is increased in advanced-stage primary prostate cancer where it has been shown to play a role in the EMT transition47. Slug is also highly expressed in prostate cancers associated with a neuroendocrine phenotype48. Consistent with this, using data generated by Beltran et al.49 we found TSPAN1 and Slug are co-amplified in 28% and 37% of neuroendocrine prostate cancer tumours respectively (Supplementary Table 4).
TSPAN1 is essential for prostate cancer cell viability
Previous studies on other cancer cell types have indicated that TSPAN1 can be essential for cell survival and proliferation28, 50, 51. To test whether changes in the expression of TSPAN1 can influence prostate cancer cell viability, we depleted LNCaP and CWR22RV1 cells of TSPAN1 protein using two independent siRNAs. Loss of TSPAN1 resulted in a significant reduction in cell number and significantly reduced cell viability for both cell lines 96 hours after siRNA treatment (Fig. 4A,B). To examine whether TSPAN1 is upstream of key cell signalling pathways in prostate cancer, we tested whether depletion of TSPAN1 influenced PIK3 signalling (detected using phospho-AKT) or RAS/ERK1/2 signalling (detected using phospho-ERK1/2). We observed a significant reduction in the levels of phospho-ERK1/2 levels in both LNCaP and CWR22RV1 cells in LNCaP cells with depleted TSPAN1, but there was no difference in the levels of phospho-AKT (Fig. 4C,D).
Figure 4.
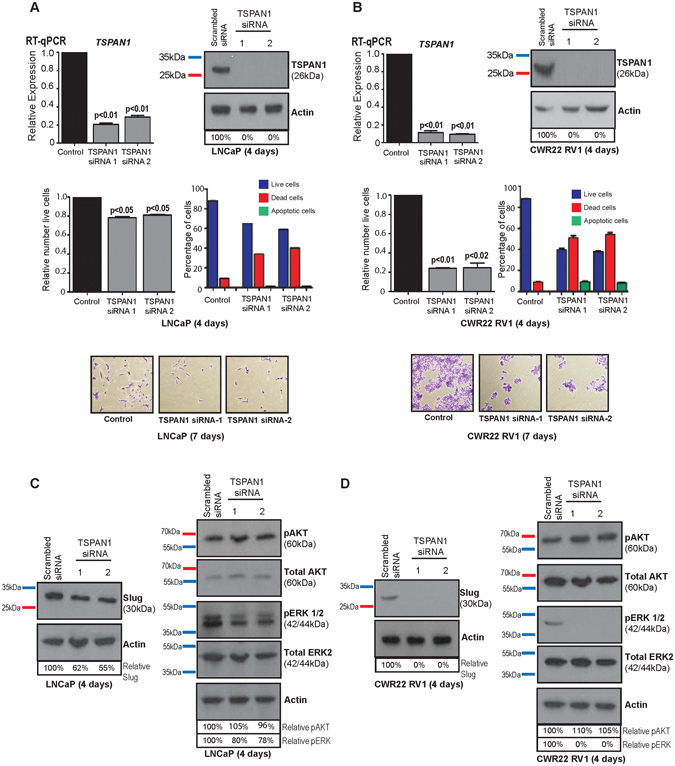
TSPAN1 is essential for prostate cancer cell viability. Depletion of TSPAN1 in (A) LNCaP and (B) CWR22 RV1 cells by transient transfection with siRNA. Protein depletion was carried out using two different siRNAs in cells grown in full media. 96 hours after transfection knockdown of TSPAN1 was confirmed by real-time PCR and western blot and the relative number of live cells was calculated. Representative crystal violet stained images are shown below for each cell line after 7 days (due to a dramatic decrease in cell viability we were unable to harvest enough live cells to confirm TSPAN1 knockdown after 96 hours). (C) Western blot analysis of various proteins in LNCaP and (D) CWR22 RV1 cells following siRNA mediated protein depletion of TSPAN1 for 96 hours. Please note: cropped western blots are displayed in the figure, but full length blots are included in the Supplementary Information file.
Discussion
The progression to advanced prostate cancer is thought to involve persistence of AR signalling, and prostate cancer treatment is dominated by strategies to control AR activity52. The AR controls prostate cancer development through the regulation of transcription, meaning that identifying both transcriptional targets of the AR and the factors involved will provide opportunities for both cancer detection and therapeutic intervention. Here, we show that the Tetraspanin 1 gene (TSPAN1) is regulated by androgens and is significantly upregulated in primary prostate cancer tissue. We find that while normal expression of TSPAN1 is important for prostate cancer cell viability, induced upregulated expression drives increased cell migration. The data presented in this study are in contrast to a recent publication suggesting that decreased TSPAN1 is linked to prostate cancer progression and that TSPAN1 plays a negative role in prostate cancer cell proliferation and migration (which used a different antibody to detect TSPAN129), but are in agreement with studies in numerous other cancer types where upregulated TSPAN1 is linked to cancer progression, cell survival, proliferation, migration and invasion15, 17, 25–28, 50, 51, 53.
Tetraspanins are reported to play a role in a range of biological processes including cell proliferation9, cell adhesion20, cell migration and motility11, 21, 22 and signal transduction23, 24. Previous studies have indicated that tetraspanins can regulate the function of proteins involved in all aspects of cell migration11. For example, tetraspanins can directly interact with integrins and modulate their downstream signalling in response to migratory signals54, 55. Tetraspanin CD9 has been shown to regulate expression of MMP9 via the JNK pathway56, 57, and tetraspanin CD82 can inhibit cancer cell retraction and motility via deregulation of the Rac1/RhoA signaling network58, 59. Tetraspanins also play a role in E-cadherin based cell–cell junctions60–62, and can directly interact with and influence the activity of matrix metalloproteinases63, 64.
TSPAN1 has been shown to promote invasiveness of cervical cancer cells27, increase the proliferation and invasion of colon cancer cells in vitro 50, promote survival and invasion of skin carcinoma cells51, and to play a role in survival, proliferation and carcinogenesis of pancreatic cancer28. In addition, regulation of TSPAN1 by micro-RNAs has been shown to promote cell proliferation and invasion in colorectal cancer65, and cell migration in non-small cell lung cancer26. Taken together, these studies suggest a positive role for TSPAN1 in cancer progression, but the mechanisms underlying this are currently poorly understood. Here, we find for the first time, that TSPAN1 expression controls the expression of key proteins with roles in cell migration. We show that upregulation of TSPAN1 in prostate cancer cells results in increased expression of the zinc finger transcription factor Slug (SNAI2) which has previously been shown to promote migration and invasion, and a transition to a more mesenchymal state in prostate cancer cells47, 66. Slug expression is increased in advanced primary prostate tumours47, and clusters in cancer cells at the invasive front in neuroendocrine areas of high-grade prostate cancer48. TSPAN1 also modulates the expression of small GTPase ADP-ribosylation factor 6 (ARF6) which is linked to cancer cell motility67, and is upregulated in prostate cancer tissue68. To the best of our knowledge this is the first time these genes have been shown to be regulated by TSPAN1 in any cancer type. Inhibition of TSPAN1 in prostate cancer has been previously linked with increased phosphorylation of AKT29, however we found no evidence of this in our study. A study in cervical cancer excluded a role for TSPAN1 in focal adhesion kinase (FAK), phosphoinositide-3-kinase (PI3K) and in EGFR-dependent signalling to the RAS/ERK pathway, arguing against a role for TSPAN1 as a mediator of cell surface receptor signalling27.
By identifying a mechanism through which this protein can modulate cell migration this current study thus sheds new light on the role of TSPAN1 in cancer. EMT and MET are processes by which cells transit between epithelial and mesenchymal states and play an integral role in cancer metastasis69. Activation of EMT is important for cancer cell dissemination, whereas the reversal of EMT (or MET) is necessary for efficient metastatic colonisation70, 71. Taken together our data suggest dynamic TSPAN1 expression levels might contribute to transitions between a more migratory mesenchymal like cell phenotype and establishing stable metastases. We find TSPAN1 expression in prostate cancer is biphasic, with upregulation at the primary site and downregulation in metastatic lesions. Similarly, a study in breast cancer has shown that although TSPAN1 is upregulated in most primary tumours it is more likely to be downregulated in metastatic lesions72.
TSPAN1 is part of a larger gene family. It has been speculated that distinct combinations of tetraspanins could act in concert to control various aspects of cell behaviour in cancer cells27. Tetraspanin CD151 is elevated in patients with prostate cancer73 and has been shown to contribute to spontaneous metastasis in mouse models74. TSPAN8 is also upregulated in prostate cancer tissue and associated with cell invasion75. Conversely, tetraspanin CD9 is downregulated during prostate cancer progression76 and is a suppressor of metastasis in prostate cancer mouse models77. Prostate cancer is an extremely heterogeneous disease and it is likely that the role of tetraspanins is multifaceted in the development and progression of prostate cancer. Interestingly, TSPAN1 is listed in the top 30 most common and abundant glycoproteins detected in prostate cancer secretions by mass spectrometry and has been studied as part of a predictive signature for prostate cancer aggressiveness78, 79. The overlapping TMA data for TSPAN1 between benign and prostate cancer groups presented in this study do not suggest that detection of TSPAN1 alone could be used clinically, but indicate it may still be worthwhile to examine TSPAN1 levels as part of a ‘tetraspanin signature’ in future studies.
In conclusion, our study shows that expression of TSPAN1 is controlled by androgens in prostate cancer cells and is upregulated in prostate cancer tissue. The AR has been shown to be essential for prostate cancer cell viability, proliferation and invasion80–82, however the mechanisms controlling this are poorly understood. Our data supports TSPAN1 as an androgen-driven contributor to prostate cancer cell survival and motility, which can control the expression of the mesenchymal proteins Slug and ARF6. Future studies will help elucidate the role TSPAN1 plays in these processes in relation to other AR regulated genes and whether TSPAN1 can be targeted therapeutically.
Methods
Cell Culture
Cell culture and the treatment of cells with androgens were as described previously7, 83–86. LNCaP cells (CRL-1740, ATCC) were maintained in phenol red free RPMI-1640 with L-Glutamine (PAA Laboratories, R15–802) supplemented with 10% Fetal Bovine Serum (FBS) (PAA Laboratories, A15-101). For androgen treatment of LNCaP cells, medium was supplemented with 10% dextran charcoal stripped FBS (PAA Laboratories, A15-119) to produce a steroid-deplete medium. Following culture for 72 hours, 10 nM synthetic androgen analogue methyltrienolone (R1881) (Perkin–Elmer, NLP005005MG) was added (Androgen+) or absent (Steroid deplete) for the times indicated. Stable cell lines were generated by transfecting cells using Lipofectamine 2000 (11668-027, Invitrogen), followed by selection with 300 µg/ml Geneticin (Invitrogen, 10131019) (reduced to 150 µg/ml following the death of untransfected cells) for at least four weeks.
siRNA
Knockdown of TSPAN1 was carried out using predesigned siRNA sequences purchased from IDT (hs.Ri.TSPAN1.13.1 and hs.Ri.TSPAN1.13.2). AR esiRNA was obtained from Sigma-Aldrich (EHU025951). Transfections were carried out using LipofectamineR RNAi Max (Thermo Fisher Scientific 13778075) as per the manufacturer’s instructions.
RT-qPCR
Cells were harvested and total RNA extracted using TRI-reagent (Invitrogen, 15596-026), according to the manufacturer’s instructions. RNA was treated with DNase 1 (Ambion) and cDNA was generated by reverse transcription of 200ng of total RNA using the Superscript VILO cDNA synthesis kit (Invitrogen, 11754-050). Quantitative PCR (qPCR) was performed in triplicate on cDNA using SYBR® Green PCR Master Mix (Invitrogen, 4309155) using the QuantStudio™ 7 Flex Real-Time PCR System (Life Technologies). Gene expression changes were analysed using relative quantification (comparative CT method) to a given control sample. Samples were normalised using the average of three reference genes: GAPDH, β –tubulin and actin. All primer sequences are listed in Supplementary Table 5.
Antibodies
The following antibodies were used in the study: anti-TSPAN1 antibody produced in rabbit (Sigma-Aldrich HPA011909), anti-AR mouse antibody (BD Bioscience, 554226), anti-p21 Waf1/Cip1 rabbit antibody (Cell Signaling 2947), anti-slug rabbit antibody (Cell Signaling 9585), anti- phospho-p44/42 MAPK mouse monoclonal (Erk1/2 Thr202/Tyr204) (Cell signaling 9106), anti-ERK2 mouse monoclonal (1647 Santa Cruz), anti-AKT rabbit antibody (Santa Cruz, sc-8312), anti-phospho-AKT1 (pSer473) rabbit antibody (Sigma, SAB4300042), anti-ARF6 rabbit antibody (Cell Signaling 5740), anti-FLAG mouse monoclonal (F3165, Sigma), anti-actin rabbit polyclonal (A2668, Sigma), normal rabbit IgG (711-035-152 Jackson labs) and normal mouse IgG (715-036-150 Jackson labs). Quantifications of western blots were carried out using ImageJ. Pixel densities for the bands of interest were calculated relative to background levels and the normalising control (actin). The concentrations of antibodies used are given in Supplementary Table 6.
DNA Constructs
For stable transfection of DU145 and PC3 cells TSPAN1 was cloned into pX3flag CMV 10 (E7658, Sigma) using EcoR1 and Xba1. The plasmid used underwent DNA sequencing to confirm correct insertion of the TSPAN1 gene.
Cell Viability Assay
Cell viability analysis was carried out using the TaliR Cell Viability Kit (Life Technologies A10786) and the TaliR Image-based Cytometer. Relative cell numbers following siRNA treatment were determined using the TaliR Image-based Cytometer (Life Technologies).
Migration Assays
Cell migration assays were carried out using the collagen coated OrisTM Pro Cell Migration Assay (Platypus Technologies PROCMACC1) as per the manufacturer’s instructions. For both DU145 and PC3 cells 30,000 cells per well were seeded at the start of the experiment. The results presented show the relative migration for each cell line over 41 hours (DU145 cells) or 30 hours (PC3 cells).
Adhesion Assays
For adhesion assays cells were labelled with 5 µM Calcein-AM (BD Biosciences). 50,000 cells were then allowed to adhere to collagen coated plates for two hours, washed 4 times with PBS (to remove any non-adherent cells), and the absorbance measured at 485/538 nm using a plate reader. The percentage adhesion was then calculated by comparing a washed plate (representative of the number of adherent cells) to an identical unwashed plate (representative of the total number of cells seeded).
Immunohistochemistry
A tissue microarray (TMA) containing 0.6 mm cores of benign prostatic hyperplasia (BPH) (n = 26), PC (n = 72), and control tissues including breast, kidney, placenta, ovary, and liver was used87. A specialised urological pathologist selected representative tissue pieces to be included in the TMA based on percentage of tumours cells assessed from the H&E stained sections. For each patient two separate representative areas were selected and included on the TMA to account for tumour heterogeneity. Antigen retrieval was performed by pressure cooking the TMA for 90 seconds in 10 mM citrate pH 6.0 followed by staining the tissues with anti-TSPAN1 antibody (Sigma-Aldrich HPA011909) at a 1:1500 dilution. Nuclei were counterstained with haematoxylin. The TMA was scored by Urszula L. McClurg using the 0-300 Histoscore score method88. Only epithelial cells were scored. Sections were scored based on their staining intensity with 0 being assigned to cells with absent staining, 1 to weak staining, 2 to moderate staining and 3 to strong staining. Within each staining intensity percentage of epithelial cells (0–100%) with this staining intensity was assigned. This resulted in a Histoscore calculated from the following equation H = 0x (% of cells scored at 0) + 1x (% of cells scored at 1) + 2x (% of cells scored at 2) + 3x (% of cells scored at 3).
Clinical Samples
Our study made use of RNA from 32 benign samples from patients with benign prostatic hyperplasia (BPH) and 17 malignant samples from transurethral resection of the prostate (TURP) samples. Malignant status and Gleason score were obtained for these patients by histological analysis. We also analysed normal and matched prostate tissue from 9 patients obtained by radical prostatectomy. The samples were obtained with ethical approval through the Exeter NIHR Clinical Research Facility tissue bank (Ref: STB20). Our TMA study made use of 26 BPH and 72 prostate cancer tissue samples. These samples were obtained with full ethical approval from the Northumberland, Tyne and Wear NHS Strategic Health Authority Local Research Ethics Committee (Ref: 2003/11). Written informed consent for the use of surgically obtained tissue was provided by all patients. All methods were performed in accordance with the relevant guidelines and regulations.
Electronic supplementary material
Acknowledgements
This work was funded by Prostate Cancer UK (PG12-34), The J. G. W Patterson Foundation, Newcastle University Special Trustees, the BBSRC (grants BB/1006923/1 and BB/K018957/1) and the Breast Cancer Now (grant number 2014NovPR355). The authors would like to thank Exeter NIHR Clinical Research Facility, and Mr Ben Lee for technical assistance.
Author Contributions
J.M. was involved in the conception of the study, carried out the majority of the experiments, participated in experimental design and interpretation of results, and drafted the manuscript. K.E.L. and I.E. assisted with some of the molecular experiments. U.L.M. carried out the immunohistochemistry analysis of prostate tissue and interpretation of results. B.K., P.M., J.Mc. and M.C. collected the prostate tissue which was used for RNA analysis. C.N.R., H.Y.L., P.R., L.W.H. and I.G.M. were involved in the analysis and interpretation of data, and were involved in critically revising the manuscript. D.J.E. conceived of the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05489-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 4.Center MM, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012;61:1079–1092. doi: 10.1016/j.eururo.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 5.Livermore KE, Munkley J, Elliott DJ. Androgen receptor and prostate cancer. AIMS Molecular Science. 2016;3:280–299. doi: 10.3934/molsci.2016.2.280. [DOI] [Google Scholar]
- 6.Munkley J, Mills IG, Elliott DJ. The role of glycans in the development and progression of prostate cancer. Nat Rev Urol. 2016;13:324–333. doi: 10.1038/nrurol.2016.65. [DOI] [PubMed] [Google Scholar]
- 7.Munkley J, et al. Glycosylation is an Androgen-Regulated Process Essential for Prostate Cancer Cell Viability. EBioMedicine. 2016;8:103–116. doi: 10.1016/j.ebiom.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotta H, et al. Molecular cloning and characterization of an antigen associated with early stages of melanoma tumor progression. Cancer Res. 1988;48:2955–2962. [PubMed] [Google Scholar]
- 9.Oren R, Takahashi S, Doss C, Levy R, Levy S. TAPA-1, the target of an antiproliferative antibody, defines a new family of transmembrane proteins. Mol Cell Biol. 1990;10:4007–4015. doi: 10.1128/MCB.10.8.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang S, et al. The phylogenetic analysis of tetraspanins projects the evolution of cell-cell interactions from unicellular to multicellular organisms. Genomics. 2005;86:674–684. doi: 10.1016/j.ygeno.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Jiang X, Zhang J, Huang Y. Tetraspanins in cell migration. Cell adhesion & migration. 2015;9:406–415. doi: 10.1080/19336918.2015.1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 13.Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. J Cell Sci. 2014;127:3641–3648. doi: 10.1242/jcs.154906. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, et al. Association of NET-1 gene expression with human hepatocellular carcinoma. International journal of surgical pathology. 2007;15:346–353. doi: 10.1177/1066896907306083. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, et al. Clinicopathological significance of overexpression of TSPAN1, Ki67 and CD34 in gastric carcinoma. Tumori. 2008;94:531–538. doi: 10.1177/030089160809400415. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, et al. TSPAN1 protein expression: a significant prognostic indicator for patients with colorectal adenocarcinoma. World J Gastroenterol. 2009;15:2270–2276. doi: 10.3748/wjg.15.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scholz CJ, et al. Tspan-1 is a tetraspanin preferentially expressed by mucinous and endometrioid subtypes of human ovarian carcinomas. Cancer letters. 2009;275:198–203. doi: 10.1016/j.canlet.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 18.Nees M, van Wijngaarden E, Bakos E, Schneider A, Durst M. Identification of novel molecular markers which correlate with HPV-induced tumor progression. Oncogene. 1998;16:2447–2458. doi: 10.1038/sj.onc.1201785. [DOI] [PubMed] [Google Scholar]
- 19.Wollscheid V, et al. Identification of a new proliferation-associated protein NET-1/C4.8 characteristic for a subset of high-grade cervical intraepithelial neoplasia and cervical carcinomas. Int J Cancer. 2002;99:771–775. doi: 10.1002/ijc.10442. [DOI] [PubMed] [Google Scholar]
- 20.Masellis-Smith A, Jensen GS, Seehafer JG, Slupsky JR, Shaw AR. Anti-CD9 monoclonal antibodies induce homotypic adhesion of pre-B cell lines by a novel mechanism. J Immunol. 1990;144:1607–1613. [PubMed] [Google Scholar]
- 21.Rubinstein E, Le Naour F, Billard M, Prenant M, Boucheix C. CD9 antigen is an accessory subunit of the VLA integrin complexes. Eur J Immunol. 1994;24:3005–3013. doi: 10.1002/eji.1830241213. [DOI] [PubMed] [Google Scholar]
- 22.Miyake M, Koyama M, Seno M, Ikeyama S. Identification of the motility-related protein (MRP-1), recognized by monoclonal antibody M31-15, which inhibits cell motility. The Journal of experimental medicine. 1991;174:1347–1354. doi: 10.1084/jem.174.6.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olweus J, Lund-Johansen F, Horejsi V. CD53, a protein with four membrane-spanning domains, mediates signal transduction in human monocytes and B cells. J Immunol. 1993;151:707–716. [PubMed] [Google Scholar]
- 24.Schick MR, Nguyen VQ, Levy S. Anti-TAPA-1 antibodies induce protein tyrosine phosphorylation that is prevented by increasing intracellular thiol levels. J Immunol. 1993;151:1918–1925. [PubMed] [Google Scholar]
- 25.Chen L, et al. Clinicopathological significance of expression of Tspan-1, Jab1 and p27 in human hepatocellular carcinoma. Journal of Korean medical science. 2010;25:1438–1442. doi: 10.3346/jkms.2010.25.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, et al. MiR-200a enhances the migrations of A549 and SK-MES-1 cells by regulating the expression of TSPAN1. Journal of biosciences. 2013;38:523–532. doi: 10.1007/s12038-013-9351-6. [DOI] [PubMed] [Google Scholar]
- 27.Holters S, et al. Tetraspanin 1 promotes invasiveness of cervical cancer cells. Int J Oncol. 2013;43:503–512. doi: 10.3892/ijo.2013.1980. [DOI] [PubMed] [Google Scholar]
- 28.Hou FQ, Lei XF, Yao JL, Wang YJ, Zhang W. Tetraspanin 1 is involved in survival, proliferation and carcinogenesis of pancreatic cancer. Oncol Rep. 2015 doi: 10.3892/or.2015.4272. [DOI] [PubMed] [Google Scholar]
- 29.Xu F, et al. Decreased TSPAN1 promotes prostate cancer progression and is a marker for early biochemical recurrence after radical prostatectomy. Oncotarget. 2016 doi: 10.18632/oncotarget.11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Massie CE, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw GL, et al. The Early Effects of Rapid Androgen Deprivation on Human Prostate Cancer. Eur Urol. 2016;70:214–218. doi: 10.1016/j.eururo.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajan P, et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur Urol. 2014;66:32–39. doi: 10.1016/j.eururo.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh, D. et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell1, 203–209, doi:S1535610802000302 (2002). [DOI] [PubMed]
- 36.Welsh JB, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 37.Yu YP, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 38.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–3882. [PubMed] [Google Scholar]
- 39.LaTulippe E, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 40.Varambally, S. et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell8, 393–406, doi:S1535-6108(05)00305-3 (2005). [DOI] [PubMed]
- 41.Wallace TA, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 42.Arredouani MS, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15:5794–5802. doi: 10.1158/1078-0432.CCR-09-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lapointe J, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu P, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;66:4011–4019. doi: 10.1158/0008-5472.CAN-05-3055. [DOI] [PubMed] [Google Scholar]
- 45.Luo JH, et al. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33:25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 46.Holzbeierlein J, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu YN, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor beta-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol. 2012;32:941–953. doi: 10.1128/MCB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esposito S, et al. SNAI2/Slug gene is silenced in prostate cancer and regulates neuroendocrine differentiation, metastasis-suppressor and pluripotency gene expression. Oncotarget. 2015;6:17121–17134. doi: 10.18632/oncotarget.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beltran H, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen L, Yuan D, Zhao R, Li H, Zhu J. Suppression of TSPAN1 by RNA interference inhibits proliferation and invasion of colon cancer cells in vitro. Tumori. 2010;96:744–750. doi: 10.1177/030089161009600517. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, et al. Knockdown of TSPAN1 by RNA silencing and antisense technique inhibits proliferation and infiltration of human skin squamous carcinoma cells. Tumori. 2010;96:289–295. doi: 10.1177/030089161009600217. [DOI] [PubMed] [Google Scholar]
- 52.Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat Rev Cancer. 2014;14:187–198. doi: 10.1038/nrc3678. [DOI] [PubMed] [Google Scholar]
- 53.Lu Z, et al. TSPAN1 functions as an oncogene in gastric cancer and is downregulated by miR-573. FEBS Lett. 2015;589:1988–1994. doi: 10.1016/j.febslet.2015.05.044. [DOI] [PubMed] [Google Scholar]
- 54.Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci. 2001;114:4143–4151. doi: 10.1242/jcs.114.23.4143. [DOI] [PubMed] [Google Scholar]
- 55.Bassani S, Cingolani LA. Tetraspanins: Interactions and interplay with integrins. The international journal of biochemistry & cell biology. 2012;44:703–708. doi: 10.1016/j.biocel.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, et al. CD9 is critical for cutaneous wound healing through JNK signaling. The Journal of investigative dermatology. 2012;132:226–236. doi: 10.1038/jid.2011.268. [DOI] [PubMed] [Google Scholar]
- 57.Jiang XP, et al. Downregulation of CD9 in keratinocyte contributes to cell migration via upregulation of matrix metalloproteinase-9. PLoS One. 2013;8:e77806. doi: 10.1371/journal.pone.0077806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takahashi M, Sugiura T, Abe M, Ishii K, Shirasuna K. Regulation of c-Met signaling by the tetraspanin KAI-1/CD82 affects cancer cell migration. Int J Cancer. 2007;121:1919–1929. doi: 10.1002/ijc.22887. [DOI] [PubMed] [Google Scholar]
- 59.Liu WM, et al. Tetraspanin CD82 inhibits protrusion and retraction in cell movement by attenuating the plasma membrane-dependent actin organization. PLoS One. 2012;7:e51797. doi: 10.1371/journal.pone.0051797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stipp CS. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert reviews in molecular medicine. 2010;12:e3. doi: 10.1017/S1462399409001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greco C, et al. E-cadherin/p120-catenin and tetraspanin Co-029 cooperate for cell motility control in human colon carcinoma. Cancer Res. 2010;70:7674–7683. doi: 10.1158/0008-5472.CAN-09-4482. [DOI] [PubMed] [Google Scholar]
- 62.Johnson JL, Winterwood N, DeMali KA, Stipp CS. Tetraspanin CD151 regulates RhoA activation and the dynamic stability of carcinoma cell-cell contacts. J Cell Sci. 2009;122:2263–2273. doi: 10.1242/jcs.045997. [DOI] [PubMed] [Google Scholar]
- 63.Takino T, et al. Tetraspanin CD63 promotes targeting and lysosomal proteolysis of membrane-type 1 matrix metalloproteinase. Biochem Biophys Res Commun. 2003;304:160–166. doi: 10.1016/S0006-291X(03)00544-8. [DOI] [PubMed] [Google Scholar]
- 64.Lafleur MA, Xu D, Hemler ME. Tetraspanin proteins regulate membrane type-1 matrix metalloproteinase-dependent pericellular proteolysis. Molecular biology of the cell. 2009;20:2030–2040. doi: 10.1091/mbc.E08-11-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, et al. MicroRNA-638 inhibits cell proliferation, invasion and regulates cell cycle by targeting tetraspanin 1 in human colorectal carcinoma. Oncotarget. 2014;5:12083–12096. doi: 10.18632/oncotarget.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uygur B, Wu WS. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol Cancer. 2011;10:139. doi: 10.1186/1476-4598-10-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hongu T, et al. Pathological functions of the small GTPase Arf6 in cancer progression: Tumor angiogenesis and metastasis. Small GTPases. 2016;7:47–53. doi: 10.1080/21541248.2016.1154640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morgan C, et al. Increased expression of ARF GTPases in prostate cancer tissue. SpringerPlus. 2015;4:342. doi: 10.1186/s40064-015-1136-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heerboth S, et al. EMT and tumor metastasis. Clinical and translational medicine. 2015;4:6. doi: 10.1186/s40169-015-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ocana OH, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 71.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Desouki MM, et al. Identification of metastasis-associated breast cancer genes using a high-resolution whole genome profiling approach. J Cancer Res Clin Oncol. 2011;137:795–809. doi: 10.1007/s00432-010-0937-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ang J, Lijovic M, Ashman LK, Kan K, Frauman AG. CD151 protein expression predicts the clinical outcome of low-grade primary prostate cancer better than histologic grading: a new prognostic indicator? Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2004;13:1717–1721. [PubMed] [Google Scholar]
- 74.Copeland BT, Bowman MJ, Ashman LK. Genetic ablation of the tetraspanin CD151 reduces spontaneous metastatic spread of prostate cancer in the TRAMP model. Molecular cancer research: MCR. 2013;11:95–105. doi: 10.1158/1541-7786.MCR-12-0468. [DOI] [PubMed] [Google Scholar]
- 75.Bhansali M, Zhou J, Shemshedini L. TM4SF3 and AR: A Nuclear Complex that Stabilizes Both Proteins. Molecular endocrinology. 2016;30:13–25. doi: 10.1210/me.2015-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang JC, et al. Down-regulation of CD9 expression during prostate carcinoma progression is associated with CD9 mRNA modifications. Clin Cancer Res. 2007;13:2354–2361. doi: 10.1158/1078-0432.CCR-06-1692. [DOI] [PubMed] [Google Scholar]
- 77.Copeland BT, Bowman MJ, Boucheix C, Ashman LK. Knockout of the tetraspanin Cd9 in the TRAMP model of de novo prostate cancer increases spontaneous metastases in an organ-specific manner. Int J Cancer. 2013;133:1803–1812. doi: 10.1002/ijc.28204. [DOI] [PubMed] [Google Scholar]
- 78.Drake RR, Jones EE, Powers TW, Nyalwidhe JO. Chapter Ten – Altered Glycosylation in Prostate Cancer. Advances in Cancer Research. 2015;126:345–382. doi: 10.1016/bs.acr.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y, et al. Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol Cell Proteomics. 2014;13:1753–1768. doi: 10.1074/mcp.M114.038273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Snoek R, et al. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res. 2009;15:39–47. doi: 10.1158/1078-0432.CCR-08-1726. [DOI] [PubMed] [Google Scholar]
- 81.Hara T, Miyazaki H, Lee A, Tran CP, Reiter RE. Androgen receptor and invasion in prostate cancer. Cancer Res. 2008;68:1128–1135. doi: 10.1158/0008-5472.CAN-07-1929. [DOI] [PubMed] [Google Scholar]
- 82.Haag P, Bektic J, Bartsch G, Klocker H, Eder IE. Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen independent prostate cancer cells. The Journal of steroid biochemistry and molecular biology. 2005;96:251–258. doi: 10.1016/j.jsbmb.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 83.Munkley, J. et al. Androgen-regulation of the protein tyrosine phosphatase PTPRR activates ERK1/2 signalling in prostate cancer cells. BMC Cancer15, 9, doi:s12885-015-1012-8 (2015). [DOI] [PMC free article] [PubMed]
- 84.Munkley J, et al. The PI3K regulatory subunit gene PIK3R1 is under direct control of androgens and repressed in prostate cancer cells. Oncoscience. 2015;2:755–764. doi: 10.18632/oncoscience.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Munkley, J. et al. A novel androgen-regulated isoform of the TSC2 tumour suppressor gene increases cell proliferation. Oncotarget5, 131-139, doi:1405 (2014). [DOI] [PMC free article] [PubMed]
- 86.Munkley J, et al. The androgen receptor controls expression of the cancer-associated sTn antigen and cell adhesion through induction of ST6GalNAc1 in prostate cancer. Oncotarget. 2015;6:34358–34374. doi: 10.18632/oncotarget.6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burska UL, et al. Deubiquitinating enzyme Usp12 is a novel co-activator of the androgen receptor. J Biol Chem. 2013;288:32641–32650. doi: 10.1074/jbc.M113.485912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kirkegaard T, et al. Observer variation in immunohistochemical analysis of protein expression, time for a change? Histopathology. 2006;48:787–794. doi: 10.1111/j.1365-2559.2006.02412.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.