

Abstract
Perfluorooctane sulfonate (PFOS) is an environmental contaminant that has been manufactured to be used as surfactants and repellents in industry. Due to long half-life for clearance and degradation, PFOS is accumulative in human body and has potential threat to human health. Previous studies have shown the development and function of immune cells can be affected by PFOS. Although PFOS has a high chance of being absorbed through the oral route, whether and how PFOS affects immune cells in the gut is unknown. Using mouse model of Citrobacter rodentium infection, we investigated the role of PFOS on intestinal immunity. We found at early phase of the infection, PFOS inhibited the expansion of the pathogen by promoting IL-22 production from the group 3 innate lymphoid cell (ILC3) in an aryl hydrocarbon receptor dependent manner. Nevertheless, persistent PFOS treatment in mice finally led to a failure to clear the pathogen completely. At late phase of infection, enhanced bacterial counts in PFOS treated mice were accompanied by increased inflammatory cytokines, reduced mucin production and dysbiosis, featured by decreased level of Lactobacillus casei, Lactobacillus johnsonii and increased E. coli. Our study reveals a deleterious consequence in intestinal bacterial infection caused by PFOS accumulation.
Introduction
Perfluorooctane sulfonate (PFOS) is widely used as surface treatment chemical, polymerization acid and surfactant in industry, due to its chemical stability, high surface activity and water and oil repellence property. The 3 M Company, main manufacturer of PFOS, phased out the product in 2002 because of toxic effects of PFOS to the human endocrine, reproductive and immune systems1–4. In 2009, the 4th meeting of the Conference of the Parties to the Stockholm Convention listed PFOS to the Annex B to limit the use of PFOS5. Though the application of PFOS has been abandoned by its main manufacturer, emission of PFOS still persists in many countries due to a lack of cost-efficient alternatives6, 7. Besides, the half-life for clearance of PFOS in the serum is as long as 4.8 years on average8. Thus, PFOS is bio-accumulative in water and ground and remains to be a potential threat to human health worldwide.
PFOS has been reported to affect the immune cells in the central and peripheral lymphoid organs by various mechanisms9–16. Animal studies have shown high dose of PFOS treatment causes atrophy of the thymus and spleen10, 15. And in vivo PFOS treatment inhibits Th1 responses while Th2 responses are promoted9, 13, 14. Being a paper-packaging material and a contaminant in the water, PFOS can frequently be absorbed through the oral route and accumulate in the intestine, thus modulate intestinal immunity under physiological and pathological conditions. However, it is not known whether and how PFOS affects the intestinal immune cells, especially during pathological conditions such as intestinal bacterial infections.
Mouse Citrobacter rodentium infection has been widely used as a model for studying human intestinal infections, such as enteropathogenic E. coli and enterohemorrhagic E. coli infection17–19. Innate and adaptive immune cells are activated by antigens derived from C. rodentium and exhibit immune defensive function to clear the pathogen. Th17 cells, one subset of T helper cells, are characterized by the expression of master transcription factor RAR-related orphan receptor gamma t (RORγt) and are important for protective immunity against C. rodentium 20–22. Besides Th17 cells, group 3 innate lymphoid cells (ILC3s) are believed to be crucial for controlling the expansion of C. rodentium at early phase of infection before Th17 cell responses are primed21, 23, 24. Both Th17 cells and ILC3s secrete IL-17 and IL-22, which are key cytokines required for clearing C. rodentium by stimulating epithelial cells to secrete anti-microbial peptides or through recruitment of neutrophils25–27. Th17 cells and ILC3s share a lot of features including cytokine production and profiles of transcription factor expression28, 29. Besides RORγt, aryl hydrocarbon receptor (Ahr) is another well-established transcription factor expressed by both Th17 cells and ILC3s, and is known to be a key factor regulating the function of Th17 cells and ILC3s24, 30–35. Notably, dioxins from the environmental contaminants act as agonistic or antagonistic ligands for Ahr36. Interestingly, some of the perfluoroalkyl acids have been reported to be able to activate Ahr37, raising the possibility that PFOS may regulate Th17 cells and ILC3s through activating Ahr in the intestine.
In this study, we determined the effect of PFOS on mouse C. rodentium infection. We found PFOS prevented the growth of C. rodentium at early stage of infection by promoting IL-22 production from ILC3 in an Ahr-dependent manner. However, PFOS exposure caused persistent inflammation in the intestine accompanied by decreased mucin production from goblet cells and dysbiosis, which finally led to a failure to clear C. rodentium at late phase of infection. Our finding reveals that PFOS exposure leads to a detrimental consequence in intestinal bacterial infection.
Results
Perfluorooctane sulfonate (PFOS) exhibits differential roles at different stages of intestinal bacterial infection
To determine the effect of PFOS on intestinal infection, we infected mice with Citrobacter rodentium while treating mice with PFOS by oral gavage before and during the infection. We gavaged mice daily with PFOS at 2 mg/kg or vehicle control for 7 days before infecting mice with C. rodentium. Mice were continuously treated with PFOS during the whole course of observation. Though both PFOS treated and control group showed no obvious weight loss during C. rodentium infection, PFOS treated mice had less gain of weight after infection with C. rodentium compared to control, indicating potential sickness of PFOS treated mice (Fig. 1A). Under the steady state without C. rodentium, the difference in the change of body weight was not observed in PFOS treated mice compared to control, implying the pathogenic role of PFOS mainly exists in intestinal infection (Figure S1). On day 5 after C. Rodentium infection, we observed a significantly lower pathogen burden in PFOS treated mice compared to control group (Fig. 1B). This data suggests PFOS has a protective effect at early phase of C. rodentium infection. However, C. rodentium load in PFOS treated mice reached a comparable level to control group at day 8 after infection, which is considered to be the peak phase of this model (Fig. 1B)38. And on day 12 after infection, although both control and PFOS treated mice showed a sign for clearance of C. rodentium, PFOS gavaged mice manifested a much less extent of pathogen clearance compared to control group (Fig. 1B). The enhanced C. rodentium burden in PFOS treated mice compared to control group lasted till as late as day 18 post infection, suggesting a pathogenic role of PFOS at late phase of C. rodentium infection (Fig. 1B). The increased level of C. rodentium in PFOS treated mice was also observed in the liver and the spleen compared to control, although the absolute amount of bacteria burden was not high enough to cause lethality of any individual mouse (Fig. 1C and D). The above data suggest PFOS treatment limits the expansion of C. rodentium at early phase of the infection. However, it causes a failure to clear the pathogen efficiently at late stage of infection.
Figure 1.
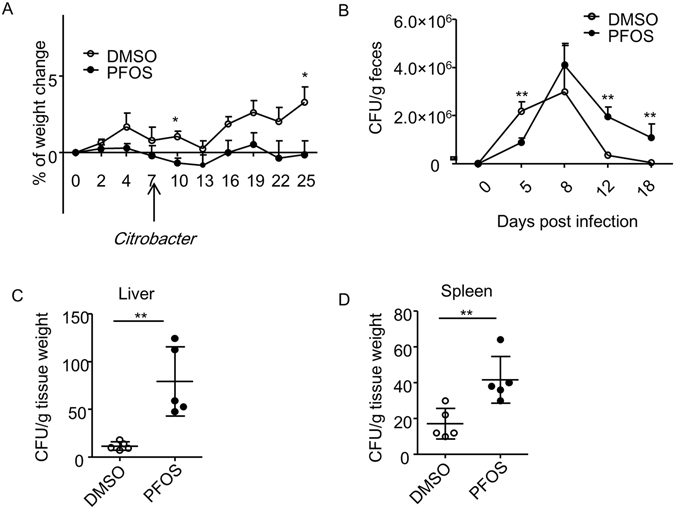
PFOS exhibits differential effects during different stages of mouse C. rodentium infection. (A) Wild-type mice were treated daily by oral gavage with DMSO or PFOS (2 mg/kg) in water for 7 days. Mice were then infected with 1010 Colony-forming unit (CFU) of C. rodentium. PFOS treatment continued on a daily basis through the whole time course of observation. Percentages of weight change of the two groups of mice before and after C. rodentium infection at indicated time points were shown. Data were pooled from 4 mice from each group. Error bars represent SEM. Data are representative of at least 3 independent experiments. (B) CFU counts of C. rodentium in the fecal pellets at indicated time points after infection were shown. Data were pooled from at least 4–8 mice from each group for all time points from two experiments. Error bars represent SEM. (C) CFU counts of C. rodentium from spleen and liver homogenates cultures at 21 days after infection were shown. Horizontal lines show the mean. Error bars represent SEM. The data are representative of three independent experiments.
PFOS promotes anti-microbial defense at early phase of C. rodentium infection by enhancing IL-22 production from ILC3 cells
ILC3s has been shown to play a key role in controlling C. rodentium infection, specifically at early phase of the infection, while Th17 cells exhibit immune defensive effect at late stage of infection21. Both ILC3s and Th17 cells produce IL-17 and IL-22, two major cytokines that are required for controlling intestinal pathogen infection25–27. Therefore, we analyzed the percentage of ILC3s and Th17 cells, as well as functional cytokine production from ILC3s and Th17 cells of large intestinal lamina proprial lymphocytes (LPLs) by flow cytometry. The percentage of ILC3s gated on CD3− non-T cell population was similar between control and PFOS treated group (Fig. 2A and B). But the IL-22 production by ILC3 significantly enhanced in PFOS treated mice compared to control (Fig. 2A and C). Consistent with previous findings24, 39, ILC3 produced limited amount of IL-17, which was not affected by PFOS treatment (Fig. 2A and C). Percentage of IFN-γ production from CD3− non-T cells in PFOS treated mice was comparable to control (Fig. 2D). Percentage of Th17 cells among all the CD4+ T cells was not changed in PFOS treated group compared to control mice (Fig. 2E and F). No obvious difference was observed in IL-17 or IL-22 production from Th17 cells in PFOS treated mice compared to control group (Fig. 2E and G). The production of IFN-γ by CD4+T cells in PFOS treated mice was on average but not significantly higher than control group, indicating a possibly pro-inflammatory role of PFOS in the intestine (Fig. 2E and H). We further evaluated the role on PFOS on ILC3 under the steady state without infection. Mice were treated with PFOS continuously for 11 days to match the same dose of PFOS at early phase of infection. No difference in the percentage of ILC3 was found in PFOS treated mice compared to control (Figure S2A). Interestingly, the production of IL-22 but not IL-17 from ILC3 was similarly enhanced by PFOS treatment without infection (Figure S2B and S2C). This suggests the promotion of IL-22 production by ILC3 was independent of infection. We further measured the expression of anti-microbial peptides, which are downstream of IL-22 and play essential role in clearance of C. rodentium during infection25. We consistently observed enhanced level of mRNA expression of RegIIIβ and RegIIIγ in colon tissues of PFOS treated mice (Fig. 2I). From above data, we reasoned the protective effect of PFOS at early stage of C. rodentium infection was mainly mediated by enhanced IL-22 production from ILC3.
Figure 2.
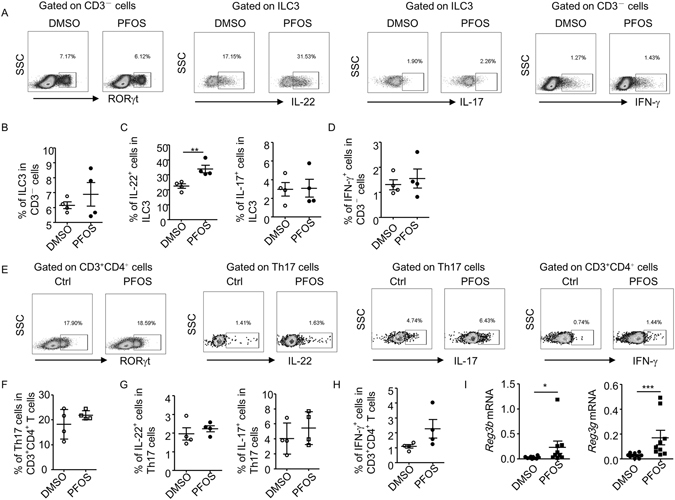
PFOS enhances IL-22 production from ILC3 at early phase of C. rodentium infection. On day 5 post C. rodentium infection, large intestinal lamina proprial lymphocytes were isolated from each group and analyzed by flow cytometry. Cells were stimulated with PMA and ionomycin for 4 hr before analysis. (A and E) The expression of CD3, CD4, RORγt, IL-17, IL-22 and IFN-γ was analyzed by flow cytometry. (B) Percentages of ILC3 (CD3−RORγt+ cells) gated on CD3− cells were shown. (C) Percentages of IL-22 and IL-17 expression gated on ILC3 were shown. (D) Percentages of IFN-γ gated on CD3− cells were shown. (F) Percentages of Th17 cells (CD3+CD4+RORγt+ cells) gated on CD3+CD4+ cells were shown. (G) Percentages of IL-22 and IL-17 production gated on Th17 cells were shown. (H) Percentages of IFN-γ gated on CD3+CD4+ cells were shown. Horizontal lines show the mean. Error bars represent SEM. Data are representative of two independent experiments. (I) mRNA expression of RegIIIβ and RegIIIγ in colon tissues was analyzed using realtime RT-PCR. Statistical analyses were performed using Mann-Whitney unpaired U test. Horizontal lines show the mean. Error bars represent SEM. Data are representative of two independent experiments.
PFOS induces cytokine production by ILC3s and Th17 cells in vitro
IL-1β and IL-23 produced from antigen presenting cells are considered to be key drivers for IL-22 production from ILC3 during C. rodentium infection25, 40, 41. To determine whether PFOS promoted the function of ILC3s and Th17 cells by inducing IL-1β or IL-23, we isolated LPL from C. rodentium infected mice on day 5 post infection and analyzed the mRNA expression of IL-1β and IL-23 by real-time RT-PCR. We found no significant difference in IL-1β or IL-23 mRNA expression in the LPL of PFOS treated mice compared to control group (Figure S3A), suggesting these two cytokines were less likely to be involved in promoting IL-22 production from ILC3 by PFOS. We then isolated LPL cells from wild-type mice under the steady state and treated the cells with different doses of PFOS in vitro. We found PFOS potently promoted the production of IL-22 by ILC3 cells at the concentration of 50 uM and 100 uM after culturing for 20 hr, without cytotoxic effect (Fig. 3A,B and Figure S4). Consistently, IL-17 production by ILC3 was not affected by PFOS treatment in vitro (Fig. 3A and B). Interestingly, both IL-17 and IL-22 production from Th17 cells were enhanced by PFOS treatment at 100 uM (Fig. 3C and D). Moreover, no difference in IL-23 and IL-1β mRNA was affected by PFOS treatment in vitro (Figure S3B). Thus, PFOS was more likely to promote the function of ILC3 and Th17 cell through a cell-intrinsic mode.
Figure 3.
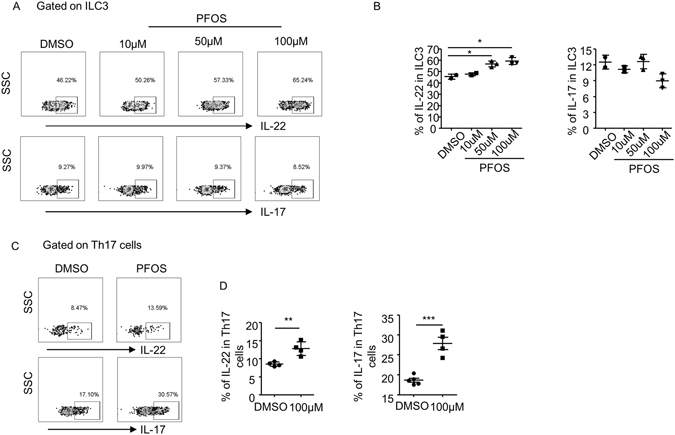
PFOS promotes cytokine production from intestinal ILC3 and Th17 cells in vitro. Large intestinal LPLs were isolated from wild-type mice and treated with indicated concentrations of PFOS or DMSO for 20 hr. Cells were stimulated with PMA and ionomycin for the last 4 h before harvested for flow cytometry analysis. (A and C) The expression of CD3, CD4, RORγt, IL-17 and IL-22 from indicated samples was analyzed by flow cytometry. (B) Percentages of IL-22 and IL-17 expression gated on ILC3 (CD3−RORγt+ cells) were shown. (D) Percentages of IL-22 and IL-17 expression gated on Th17 (CD3+CD4+RORγt+) cells were shown. Horizontal lines show the mean. Error bars represent SEM. Data are representative of two independent experiments.
PFOS promotes the function of ILC3s and Th17 cells in an Ahr-dependent manner
Previous study suggests perfluoroalkyl acids act as an agonist for aryl hydrocarbon receptor (Ahr)37, a nuclear transcriptional factor which is highly expressed by various immune cells and crucial for the development and function of ILC3s and Th17 cells24, 30–35. Indeed, the mRNA expression of Cyp1a1, a direct target gene of Ahr, was significantly elevated in the LPL by PFOS treatment (Fig. 4A). To further determine whether PFOS promoted the function of Th17 cells and ILC3s through Ahr in a cell-intrinsic mechanism, we crossed Ahr f/f mouse to RORc-cre mouse to delete Ahr specifically in ILC3 and T cells (Ahr f/f RORc-cre). We then treated LPL isolated from either Ahr f/f RORc-cre or Ahr f/f mice with PFOS in vitro. While PFOS potently enhanced the IL-22 production by ILC3 from LPL of Ahr f/f mice, this effect was ablated in Ahr f/f RORc-cre mice (Fig. 4B and C). IL-22 production from ILC3 on a per-cell-based level was also enhanced by PFOS in ILC3 from Ahr f/f but not Ahr f/f RORc-cre mice indicated by the mean fluorescence intensity (MFI) of IL-22 gated on ILC3s (Figure S5). Similarly, IL-22 and IL-17 production from Th17 cells was enhanced in Ahr f/f mice but not in Ahr f/f RORc-cre mice by PFOS treatment (Fig. 4D and E). IL-17 production from ILC3 was not obviously changed by PFOS treatment in both Ahr f/f RORc-cre mice and Ahr f/f mice (Fig. 4B and C). Thus, we conclude PFOS promotes the function of ILC3 and Th17 in vitro through Ahr.
Figure 4.
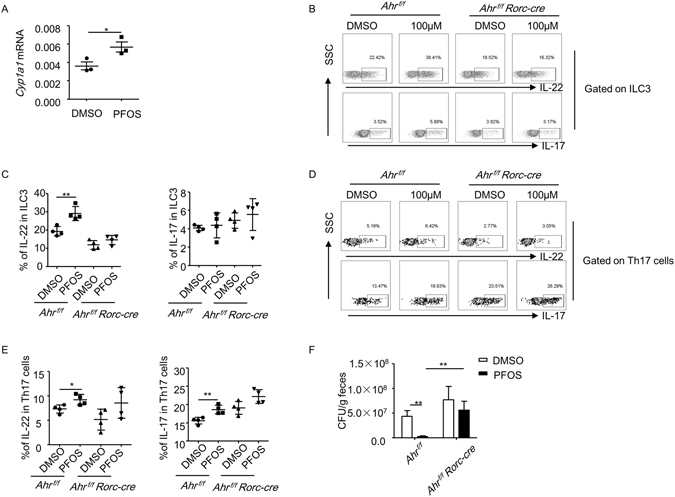
PFOS inhibites the growth of C. rodentium by promoting IL-22 production from ILC3 through an Ahr-dependent manner. (A) Large intestinal LPLs were isolated from wild-type mice and treated with PFOS at 100 uM or DMSO for 20 hr. mRNA expression of Cyp1a1 in cultured LPLs was analyzed by real-time RT-PCR. (B to E) Large intestinal LPLs were isolated from Ahr f/f RORc-cre or Ahr f/f mice and treated with PFOS at 100 uM or DMSO for 20 hr. Cells were stimulated with PMA and ionomycin for the last 4 hr before harvested for flow cytometry analysis. (B and D) The expression of CD3, CD4, RORγt, IL-17 and IL-22 from indicated samples was analyzed by flow cytometry. (C) Percentages of IL-22 and IL-17 expression gated on ILC3 (CD3−RORγt+ cells) were shown. (E) Percentages of IL-22 and IL-17 expression gated on Th17 (CD3+CD4+RORγt+) cells were shown. (F) Ahr f/f RORc-cre or Ahr f/f mice were treated with PFOS at 2 mg/kg for 7 days before mice were infected with 1010 CFU of C. rodentium. CFU counts of C. rodentium in the fecal pellets from mice of indicated genotypes at day 5 post-infection were shown. Horizontal lines show the mean. Error bars represent SEM. Data are representative of two independent experiments.
We then investigated whether PFOS protected mice from C. rodentium infection through activating Ahr in vivo. We found while PFOS efficiently inhibited the expansion of C. rodentium in Ahr f/f mice on day 5 post infection, C. rodentium count was significantly higher in Ahr f/f RORc-cre mice and PFOS failed to inhibit the growth of the pathogen (Fig. 4F). This data suggests Ahr expression on ILC3s and T cells is required for the protective effect of PFOS at early phase of C. rodentium infection. Since Th17 cell immunity was not affected by PFOS treatment during early phase of C. rodentium infection, PFOS mainly activated Ahr in ILC3 to promote the production IL-22 by ILC3, which is essential for controlling the expansion of C. rodentium.
Pro-inflammatory effect of PFOS at late stage of C. rodentium infection
Although PFOS controlled the growth of C. rodentium at early phase of infection, the pathogen greatly expanded at peak phase (day 8 after infection) and PFOS treated mice failed to clear C. rodentium efficiently after day 12 post infection (Fig. 1B). We examined whether the defective defense against C. rodentium in PFOS treated mice was caused by loss of function of ILC3s and Th17 cells at late stage of the infection. On day 12 post infection, we found the percentage of ILC3s gated on CD3− non-T cell population increased in PFOS treated group compared to control mice (Fig. 5A and B). Moreover, the production of IL-17 and IL-22 by ILC3 was significantly higher than control mice (Fig. 5A and C). Notably, IFN-γ production from CD3− non-T cells also increased in PFOS treated mice compared to control, indicating a pro-inflammatory role of PFOS during C. rodentium infection (Fig. 5A and D). Th17 cell response on day 12 of infection was much higher than early phase (day 5) (Figs 2E and 5E). No difference in the percentage of Th17 cells gated on CD4+T cells was changed by PFOS treatment (Fig. 5E and F). However, the level of IL-17 and IL-22 produced by Th17 significantly enhanced in PFOS treated mice compared to control group (Fig. 5E and G). IFN-γ production by CD4+ T cells was on average but not significantly higher in PFOS treated mice than control at late stage of infection (Fig. 5E and H). Notably, PFOS consistently enhanced IFN-γ production from both CD3− non-T cells and CD4+ T cells in vitro, further indicating the pro-inflammatory role of PFOS in intestinal immunity (Figure S6A–S6D). The above data indicate IL-17 and IL-22 from both the innate and adaptive sources are boosted by PFOS treatment, likely due to a further accumulation of the compound locally. Nevertheless, the expression of RegIIIβ and RegIIIγ, which are downstream of IL-22, was not higher in PFOS treated mice compared to control (Fig. 5I). And PFOS treated mice failed to clear the pathogen efficiently despite an enhanced defensive immune responses during late phase of C. rodentium infection.
Figure 5.
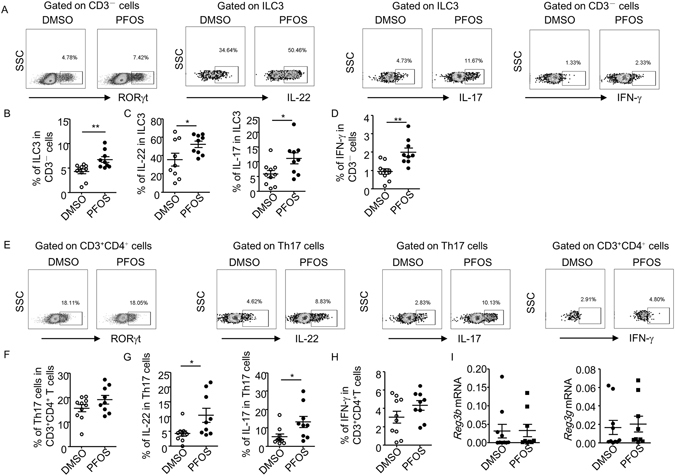
PFOS promotes both innate and adaptive production of IL-17 and IL-22 at late phase of C. rodentium infection. On day 12 post C. rodentium infection, large intestinal lamina proprial lymphocytes were isolated from each group and analyzed by flow cytometry. Cells were stimulated with PMA and ionomycin for 4 hr before analysis. (A and E) The expression of CD3, CD4, RORγt, IL-17, IL-22 and IFN-γ was analyzed by flow cytometry. (B) Percentages of ILC3 (CD3−RORγt+ cells) gated on CD3− cells were shown. (C) Percentages of IL-22 and IL-17 expression gated on ILC3 were shown. (D) Percentages of IFN-γ gated on CD3− cells were shown. (F) Percentages of Th17 cells (CD3+CD4+RORγt+ cells) gated on CD3+CD4+ cells were shown. (G) Percentages of IL-22 and IL-17 production gated on Th17 cells were shown. (H) Percentages of IFN-γ gated on CD3+CD4+ cells were shown. Horizontal lines show the mean. Error bars represent SEM. Data were pooled from three independent experiments. (I) mRNA expression of RegIIIβ and RegIIIγ in colon tissues was analyzed using realtime RT-PCR. Statistical analyses were performed using Mann-Whitney unpaired U test. Horizontal lines show the mean. Error bars represent SEM. Data are representative of two independent experiments.
We also evaluated the role of long-term PFOS treatment on immune subsets and cytokine production without C. rodentium infection. We treated mice for 17 consecutive days to match the dose of PFOS at late phase of infection. Compared to the infection status where an extensive pro-inflammatory cytokine production from both ILC3 and Th17 cells was induced in PFOS treated mice (Fig. 5), only the induction of IL-22 from ILC3 and Th17 cells was consistently found in PFOS treated group under the steady state (Figure S7A, S7C, S7E and S7G). There was no difference in percentages of ILC3 or Th17 cells in PFOS treated mice compared to control (Figure S7A, S7B, S7E and S7F). The levels of IL-17 and IFN-γ production from both innate cells and T cells were comparable between two groups (Figure S7). The above data indicate the induction of IL-22 by PFOS in vivo is independent of infection. However, the pro-inflammatory effect of PFOS to enhance IL-17 and IFN-γ production only occurs during infectious status.
Persistent PFOS exposure results in reduced mucin production and dysbiosis
The paradoxical phenotype of enhanced ILC3 and Th17 responses and increased bacterial burden in PFOS treated mice made us to think other mechanisms should be accounted for the failure to clear C. rodentium. Previous reports have suggested the colonization of C. rodentium is associated with distribution of commensal flora in the gut42–45. And dysbiosis may cause increase of susceptibility to C. rodentium infection46. Therefore, we checked the expression of different bacterial commensals in the feces of PFOS treated mice and control mice at both early and late phase during C. rodentium infection using real-time PCR. We first compared the expression of major bacterial phylum in PFOS treated mice and control mice during infection. No difference was found in the expression of Actinobacteria, Bacteroidetes and Firmicutes in PFOS treated mice and control mice at both early and late phase of C. rodentium infection (Fig. 6A and B). However, the expression of Proteobacteria was significantly higher in PFOS treated mice at the late but not early stage of C. rodentium infection (Fig. 6B). We further analyzed the expression of commensals at the genus or family level which belong to the Bateroidetes, Firmicutes and Proteobacteria phylum. No difference was found in all the analyzed commensals at the early stage of infection between PFOS treated group and control (Fig. 6C). Nevertheless, Enterobacteriaceae was found to be significantly higher in PFOS treated mice than control mice at the late stage of infection (Fig. 6D). The increase of Enterobacteriaceae and Proteobacteria was likely due to expansion of C. rodentium in PFOS treated mice since C. rodentium belongs to the Proteobacteria phylum and Enterobacteriaceae family47. We thus analyzed the expression of commensals at the species level (Fig. 6E and F). Interestingly, E. coli was found to increase in PFOS treated group compared to control group at late but not early phase of infection, suggesting the presence of dysbiosis specifically at late phase of infection (Fig. 6F). We further analyzed 4 types of bacterial species in the Lactobacillus genus, which has been shown to play regulatory roles during intestinal inflammation42–45. We found Lactobacillus johnsonni and Lactobacillus casei decreased in PFOS treated mice at late but not early phase of infection, while Lactobacillus acidophilus and Lactobacillus reuteri was not affected by PFOS at both early and late phase of the infection (Fig. 6E and F). Given the reported protective effect of Lactobacillus johnsonni and Lactobacillus casei in C. rodentium infection and epithelial cell functions48, 49, dysbiosis caused by PFOS treatment at late stage of C. rodentium infection could contribute to the failure of the host to clear C. rodentium efficiently.
Figure 6.
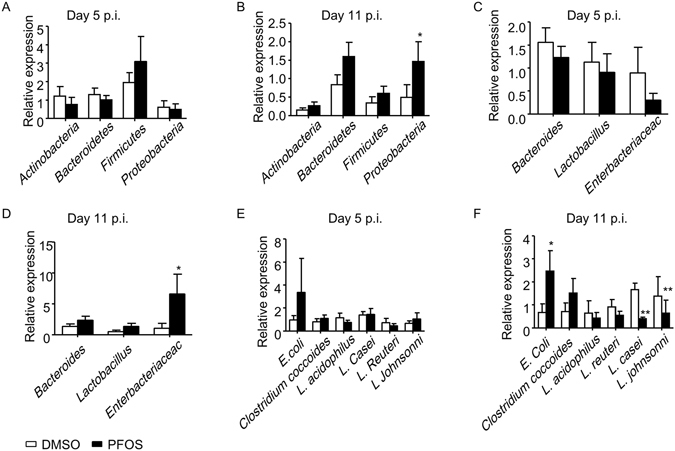
PFOS treatment results in dysbiosis at late phase of C. rodentium infection. Wild-type mice were treated with PFOS and infected with C. rodentium as described in Fig. 1. (A,C and E) The expression of indicated commensals in the feces of PFOS treated mice and control at day 5 post C. rodentium infection (p.i.) was analyzed by real-time PCR. (B,D and F) The expression of indicated commensals in the feces of PFOS treated mice and control at day 11 post C. rodentium infection (p.i.) was analyzed by real-time PCR. Statistical analyses were performed using Mann-Whitney unpaired U test. Error bars represent SEM. Data were pooled from three independent experiments.
Mucins produced by goblet cells in the intestinal epithelium are crucial for maintaining a healthy community of microbiota in the gut50, 51. Pro-inflammatory cytokines including IL-17 and IFN-γ have been indicated to cause tissue damage or result in loss of goblet cells during intestinal inflammation52, 53. We suspect the enhanced inflammation induced by PFOS at late stage of C. Rodenium infection may cause a defective mucin production by goblet cells. We then analyzed the mRNA expression of mucins in colon tissues by real-time RT-PCR. We observed significant reduction of mRNA expression of mucin 1 and mucin 2 at late stage but not early stage of C. rodentium infection in PFOS treated mice compared to control (Fig. 7A and B), while expression of mucin 3 was comparable between two groups (Fig. 7A and B). Interestingly, we found the mRNA expression of RELM-β, a resistin-like molecule specifically expressed by goblet cells, decreased in PFOS treated mice at late but not early phase of C. rodentium infection (Fig. 7C and D). The combinatorial downregulation of mucins and RELM-β may result in dysbiosis featured by increased E. coli and decreased Lactobacillus species51.
Figure 7.
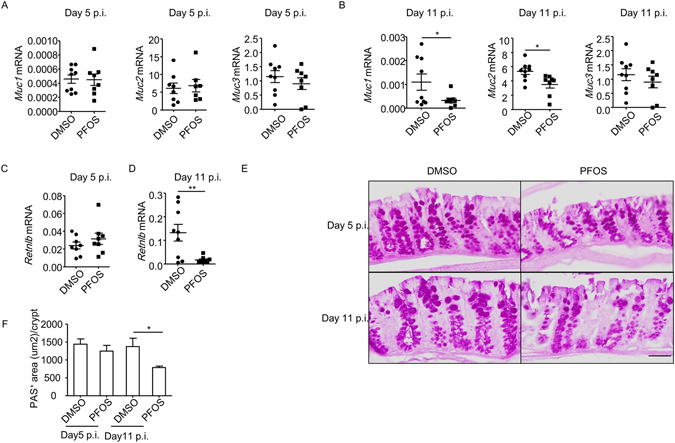
PFOS ablates function of goblet cells at late phase of C. rodentium infection. Wild-type mice were treated with PFOS and infected with C. rodentium as described in Fig. 1. (A) mRNA expression of Muc1, Muc2 and Muc3 at day 5 post C. rodentium infection in colon tissues was analyzed by realtime RT-PCR. (B) mRNA expression of Muc1, Muc2 and Muc3 at day 11 post C. rodentium infection in colon tissues was analyzed by realtime RT-PCR. (C) mRNA expression of Retnlb at day 5 post C. rodentium infection in colon tissues was analyzed by realtime RT-PCR. (D) mRNA expression of Retnlb at day 11 post C. rodentium infection in colon tissues was analyzed by realtime RT-PCR. (A–D) Horizontal lines show the mean. Error bars represent SEM. Data were pooled from two independent experiments. (E) Paraffin-embedded colon sections were stained with periodic acid-schiff (PAS). Magnification is 20 × (Scale bar, 50 um). (F) PAS+ areas per crypt were quantified by ImageJ Software. Data were pooled from three mice from indicated group. Error bars represent SEM. Data are representative of two independent experiments.
We also measured the level of mucins and RELM- β under the steady state without infection. Short-term PFOS treatment didn’t affect expression of mucins (Figure S8A). Intriguingly, REML-β was found to be enhanced by PFOS treatment (Figure S8C). Furthermore, a reduction of mucin 2 was found in mice with long-term PFOS treatment compared to control (Figure S8B). However, no difference was observed in mucin 1, 3 or RELM- β expression with long-term PFOS treatment (Figure S8B and S8D). The above data suggest long-term treatment of PFOS suppresses the expression of mucin 2 independent of infection. Nevertheless, infection resulted in a more profound dysfunction of epithelial cells, indicated by decreased level of mucin 1 and RELM- β at the late phase (Fig. 7B and D). This was likely caused by a more extensive inflammation triggered by PFOS during infection, which finally led to dysbiosis.
We further examined the mucin production from intestinal epithelial cells using periodic acid-schiff (PAS) staining during infection. Consistently, PFOS treated mice manifested reduced PAS+ area per crypt, indicating reduced mucin or number of goblet cells, compared to control group at late but not early stage of C. rodentium infection (Fig. 7E and F). From above data, we conclude that PFOS causes decreased mucin production from intestinal epithelial cells, which may lead to dysbiosis and exacerbation of C. rodentium infection at late phase.
Discussion
Perfluorooctane sulfonate (PFOS) has been manufactured to be used as surfactants and repellents in the industry for its desirable properties of high surface activity and water and oil repellence. Due to its long half-life for serum clearance and resistance to environmental degradation, PFOS is bio-accumulative and remains to be a threat to human health3, 4, 8, 54, 55. Being widely used in food-packaging material and non-stick pans, PFOS absorption through the oral route is considerably common. Thus, the accumulation of PFOS in the intestine may have deleterious effect on intestinal diseases, such as intestinal infection, intestinal autoimmune diseases and tumor. In this study, we determined the effect of PFOS on intestinal immunity and intestinal infection using a mouse model of Citrobacter rodentium infection, which recapitulates human enteropathogenic E. coli and enterohemorrhagic E. coli infection17–19. We have found PFOS affects the outcome of C. rodentium infection through modulating intestinal immunity and microbiota. At early stage of infection, PFOS prevents the expansion of C. rodentium by promoting the IL-22 production from the group 3 innate lymphoid cells (ILC3s) through activating aryl hydrocarbon receptor (Ahr), which is a key transcription factor known to regulate the development and function of ILC3s. However, consistent exposure to PFOS finally leads to a failure to clear the C. rodentium at late stage, mainly due to dysbiosis accompanied by persistent inflammation and reduced mucin production by goblet cells. Our study brought out the caution that the pro-inflammatory effect of PFOS in the intestine may result in dysbiosis and failure of clearing intestinal pathogen.
In our study, we have found PFOS promoted the cytokine production from both ILC3s and Th17 cells, which are two major immune cell subsets that are required for defense against C. rodentium 21, 23–27. At the primary expansion phase of C. rodentium infection (day 5), PFOS boosted the production of IL-22 from ILC3 without having any effects on Th17 cells. At late stage of infection, PFOS exhibited more pro-inflammatory effect indicated by increased IL-17, IL-22 and IFN-γ by innate cells and enhanced Th17 cell immunity. The delayed effect of PFOS on Th17 cells in vivo maybe because Th17 cells are less sensitive to PFOS mediated effect than ILC3s. Thus, a longer time of exposure and accumulation of PFOS is required to boost Th17 cell responses. Indeed, in vitro stimulation of ILC3 to secrete IL-22 required a much lower concentration of PFOS than Th17 cells, indicating ILC3 is more sensitive than Th17 to PFOS treatment.
Given the immune defensive effect of IL-17, IL-22 and IFN-γ, the increased level of the above inflammatory cytokines appeared contradictory to the failure to clear C. rodentium at late phase of infection in PFOS treated mice. However, the persistent inflammation caused by accumulative PFOS during the infection may be overt, thus result in tissue damage and loss of goblet cells, which may finally lead to dysbiosis and failure to clear C. rodentium. Notably, despite enhanced IL-22 level at both early and late stage of infection in PFOS treated mice, the expression of anti-microbial peptides was higher only at early phase of infection. Since anti-microbial peptides are well-known as targets of IL-2225, the failed induction of anti-microbial peptides in response to higher IL-22 in PFOS treated mice at late phase of infection may due to a damage of epithelial cells caused by overt inflammation. Previous studies have revealed the pathogenic role of IL-17 and IFN-γ in tissue damage and loss of goblet cells52, 53. We found IFN-γ production from CD3−non-T cells significantly increased at late phase of C. rodentium infection. A trend of increased IFN-γ production by CD4+ T cells was also observed at both early and late phase of C. rodentium infection. And IL-17 production from both ILC3 and Th17 cells was upregulated during late phase of C. rodentium infection. In addition, we found the innate and T cell production of IFN-γ was consistently enhanced by PFOS in vitro, suggesting a promotive effect of PFOS on type 1 immunity. The molecular mechanism of how PFOS promoted type 1 immunity in the intestine remains to be determined.
The failure to clear C. rodentium in PFOS treated mice at late phase of infection was accompanied by dysbosis featured by enhanced level of E. coli and decreased level of Latobacillus johnsonni and Lactobacillus casei. Though the causal link between E. coli expansion and C. rodentium growth is unclear, the protective effect of Latobacillus johnsonni and Lactobacillus casei in C. rodentium infection and epithelial barrier function has been shown in previous studies48, 49. Since mucins produced by goblet cells have been shown to be essential for the maintenance of microbial homeostasis and defense against C. rodentium in the gut50, 51, we reasoned goblet cell loss and dysbiosis at late phase of infection contributes to the failure of clearing C. rodentium by PFOS treated mice. Indeed, histology analysis revealed decreased mucin production by goblet cells in PFOS treated mice at late but not early phase of C. rodentium infection, which was commensurate with no observed dysbiosis in PFOS treated group at early phase of infection. And mRNA production of mucin 1 and mucin 2 was significantly lower in PFOS treated mice. In addition, RELM-β, another protein specifically produced by goblet cells and required for prevention of dysbiosis in synergy with mucin 2, also decreased in PFOS treated mice at late stage of infection51. Loss of goblet cells in PFOS treated mice may be caused by the persistence of pro-inflammatory responses, including increased IFN-γ, which may also be the reason for reduced RELM-β52, 56.
The effect of PFOS on ILC3 is organ-specific because ILC3s and Th17 cells are specifically abundant in the intestine but rare in other organs under the steady state in mice57–59. PFOS has been reported to affect the immune cells in the central and peripheral lymphoid organs by various mechanisms9–12, 14–16. High dose of PFOS exposure has been shown to cause atrophy of the thymus and spleen, as well as the percentages of T cell subsets in the spleen10, 15. In this study, we used a previously reported low dose of PFOS to avoid direct toxicity to the thymus and spleen10. Except for the role of PFOS in promoting Th17 cell responses in the gut, similar effect is likely to occur in different autoimmune disorders where Th17 cells are pathogenic, such as multiple sclerosis and rheumatoid arthritis60, 61. Thus, it brings out an alert that PFOS accumulation may be detrimental for autoimmune diseases. Epidemiologic and experimental studies are called for further evaluation for the correlation of PFOS accumulation and autoimmune diseases.
Methods
Mice
Wild-type mice were purchased from Shanghai SLAC Laboratory Animal Co. Ahr f/f and RORc-cre mice were purchased from Jackson Laboratory. Ahr f/f RORc-cre mice were generated by crossing Ahr-floxed mice62 with RORc-cre mice63. All mice used in this study are on C57BL/6 background and maintained in specific pathogen-free conditions. All mice used in this study were littermate controlled, gender-matched and were 6–8 weeks old. All animal experiments were performed in compliance with the guide for the care and use of laboratory animals and were approved by the institutional biomedical research ethics committee of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Chemicals
PFOS (heptadecafluorooctanesulfonic acid potassium salt, CAS 2795–39–3, purity >98%) was purchased from Sigma-Aldrich, dissolved in DMSO (100 mM), and stored at −20 °C as stock solution for in vivo and in vitro experiments. For in vivo treatments, PFOS (2 mg/kg) or same volume of DMSO was dissolved in water containing 0.5% Tween 20 and mice were gavaged 200 ul per day.
C. rodentium Infection and Colony-Forming Unit (CFU) Counts
C. rodentium strain DBS100 (ATCC 51459; American Type Culture Collection) was cultured overnight and bacterial concentration was calculated by measuring optical density at a wavelength of 600 nm (OD600) with spectrometer. Mice were treated by oral gavage with 1010 CFU C. rodentium in 200 ul PBS. Body weight was measured. Fecal pellets, liver and spleen were collected, weighed and then homogenized in sterile PBS. Serially diluted homogenates were plated on MacConkey agar plates. C. rodentium colonies were identified based on morphology after 24 hr of incubation at 37 °C.
Isolation of Large Intestinal Lamina Proprial Lymphocytes (LPLs)
The isolation of large intestinal lamina proprial lymphocytes was done as previously described24. Briefly, large intestines were dissected. Fat tissues were removed. Intestines were cut open longitudinally and washed in PBS. Intestines were then cut into 3 pieces, washed and shaken in PBS containing 1 mM DTT for 10 min at RT. Intestines were incubated with shaking in PBS containing 30 mM EDTA and 10 mM HEPES at 37 °C for 10 min for two cycles. The tissues were then digested in RPMI1640 medium (Invitrogen) containing DNase I (Sigma) (150 ug/ml) and collagenase VIII (Sigma) (150 U/ml) at 37 °C in 5% CO2 incubator for 1.5 hr. The digested tissues were homogenized by vigorous shaking and passed through 100 um cell strainer. Mononuclear cells were then harvested from the interphase of an 80% and 40% Percoll gradient after a spin at 2500 rpm for 20 min at room temperature.
Flow Cytometry and Antibodies
Anti-mouse CD16/32 antibody was used to block the non-specific binding to Fc receptors before all surface stainings. All antibodies used for flow cytometry were purchased from eBioscience except for α-IL-22, which was purified from a hybridoma cell line (ATCC, PTA-7319) and labeled with biotin with a EZ-Link Micro Sulfo-NHS-LC-Biotinylation Kit (Pierce). For nuclear stainings, cells were fixed and permeabilized using a Mouse Regulatory T Cell Staining Kit (eBioscience). For cytokine stainings, cells were stimulated by PMA (Sigma) (50 ng/ml) and ionomycin (Sigma) (500 ng/ml) for 2 hr, and then treated with Brefeldin A (Sigma) (2 µg/ml) for another 2 hr before cells were harvested for analysis. Dead cells were stained with Live and Dead violet viability kit (Invitrogen) and were gated out in analysis. Flow cytometry data were collected using the Gallios flow cytometer (Beckman) and analyzed by FlowJo software (Tree Star Inc.)
Cell culture
Purified large intestinal lamina proprial lymphocytes were cultured with DMSO or PFOS for 20 hr in IMDM medium (Hyclone), supplemented with 10% FBS (Gibco), 2 mM L-Glutamine (Gibco) and 100 U/ml Penicillin (Gibco), 100 ug/ml Streptomycin (Gibco) at 37 °C with 5% CO2. The concentration of DMSO in both control and PFOS group was 0.1%.
Detection of mRNA by Real-time RT-PCR
RNA from control or PFOS treated large intestinal LPLs was isolated with Trizol reagent (Invitrogen). cDNA was synthesized using GoScript™ Reverse Transcription kit (Promega). Real-time PCR was performed using SYBR Green (Bio-rad). Reactions were run with the Mx 3000 P Q-PCR System (Angilent). The results were displayed as relative expression values normalized to β-actin. Primers used in this study were shown in Table S1.
Bacterial DNA extraction and Real-time PCR
Fecal pellets were collected and total bacterial DNA was extracted using the Stool DNA Kit (Omega Biotek). Quantitative PCR for the 16 S rRNA gene was performed with SYBR Green (Bio-Rad) and normalized to total bacterial DNA. Reactions were run with the Mx 3000 P Q-PCR System (Angilent). Primers used in this study were shown in Table S2.
Histological Analysis
The colonic swiss-roll was fixed with 4% formaldehyde and embedded in paraffin. Tissues were cut into 7 μm sections for periodic acid-schiff (PAS) staining to evaluate mucin production from epithelial cells. Sections were examined by Olympus light microscope (vs120). ImageJ software (National Institutes of Health, Bethesda, MD USA) was used to quantify area of PAS positive cells. PAS+ area in four fields of around 2.25 mm2 from each section was quantified and divided by number of crypts to obtain area of PAS+ cells per crypt.
Statistical methods
Unless otherwise noted, statistical analyses were performed with the unpaired Student’s t test on individual biological samples. Linear regression analysis was performed with GraphPad Prism. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Electronic supplementary material
Acknowledgements
We thank the entire J.Q. laboratory for their help and suggestions. This study was supported by grants 2015CB943400 and 2014CB943300 from the Ministry of Science and Technology of China, grant XDB19000000 from the “Strategic priority research program of the Chinese Academy of Sciences”, grants 91542102 and 31570887 from the National Natural Science Foundation of China, grant 14140902400 from the Experimental Animal of Shanghai Science and Technology Committee and China’s Youth 1000-Talent Program to J.Q. The work was also supported by the National Institutes of Health (DK105562 L.Z.), and by a Cancer Research Institute Investigator Award (L.Z.). Liang Zhou is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts, and an Investigator in the Pathogenesis of Infectious Disease, supported by Burroughs Wellcome Fund.
Author Contributions
Q.J. and S.C. designed the research. S.C. conducted the experiments and analyzed the data. F.Z. helped with experiments. Q.J. and S.C. wrote the manuscript. Z.L. revised the manuscript and supported the project.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04091-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Su TC, et al. Serum perfluorinated chemicals, glucose homeostasis and the risk of diabetes in working-aged Taiwanese adults. Environment international. 2016;88:15–22. doi: 10.1016/j.envint.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 2.Dallaire R, Dewailly E, Pereg D, Dery S, Ayotte P. Thyroid function and plasma concentrations of polyhalogenated compounds in Inuit adults. Environmental health perspectives. 2009;117:1380–1386. doi: 10.1289/ehp.0900633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen MS, et al. Phthalates and perfluorooctanesulfonic acid in human amniotic fluid: temporal trends and timing of amniocentesis in pregnancy. Environmental health perspectives. 2012;120:897–903. doi: 10.1289/ehp.1104522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grandjean P, et al. Serum vaccine antibody concentrations in children exposed to perfluorinated compounds. Jama. 2012;307:391–397. doi: 10.1001/jama.2011.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Secretariat of the Stockholm Convention: The new POPs under the Stockholm Convention, 2011).
- 6.Xie S, et al. Industrial source identification and emission estimation of perfluorooctane sulfonate in China. Environment international. 2013;52:1–8. doi: 10.1016/j.envint.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Eriksen KT, et al. Association between plasma PFOA and PFOS levels and total cholesterol in a middle-aged Danish population. PloS one. 2013;8:e56969. doi: 10.1371/journal.pone.0056969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen GW, et al. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environmental health perspectives. 2007;115:1298–1305. doi: 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng L, Dong GH, Jin YH, He QC. Immunotoxic changes associated with a 7-day oral exposure to perfluorooctanesulfonate (PFOS) in adult male C57BL/6 mice. Archives of toxicology. 2009;83:679–689. doi: 10.1007/s00204-008-0361-3. [DOI] [PubMed] [Google Scholar]
- 10.Qazi MR, et al. The atrophy and changes in the cellular compositions of the thymus and spleen observed in mice subjected to short-term exposure to perfluorooctanesulfonate are high-dose phenomena mediated in part by peroxisome proliferator-activated receptor-alpha (PPARalpha) Toxicology. 2009;260:68–76. doi: 10.1016/j.tox.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Qazi MR, et al. High-dose, short-term exposure of mice to perfluorooctanesulfonate (PFOS) or perfluorooctanoate (PFOA) affects the number of circulating neutrophils differently, but enhances the inflammatory responses of macrophages to lipopolysaccharide (LPS) in a similar fashion. Toxicology. 2009;262:207–214. doi: 10.1016/j.tox.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 12.DeWitt JC, Peden-Adams MM, Keller JM, Germolec DR. Immunotoxicity of perfluorinated compounds: recent developments. Toxicologic pathology. 2012;40:300–311. doi: 10.1177/0192623311428473. [DOI] [PubMed] [Google Scholar]
- 13.Zheng L, et al. Type 1 and Type 2 cytokines imbalance in adult male C57BL/6 mice following a 7-day oral exposure to perfluorooctanesulfonate (PFOS) Journal of immunotoxicology. 2011;8:30–38. doi: 10.3109/1547691X.2010.537287. [DOI] [PubMed] [Google Scholar]
- 14.Dong GH, et al. Sub-chronic effect of perfluorooctanesulfonate (PFOS) on the balance of type 1 and type 2 cytokine in adult C57BL6 mice. Archives of toxicology. 2011;85:1235–1244. doi: 10.1007/s00204-011-0661-x. [DOI] [PubMed] [Google Scholar]
- 15.DeWitt JC, et al. Immunotoxicity of perfluorooctanoic acid and perfluorooctane sulfonate and the role of peroxisome proliferator-activated receptor alpha. Critical reviews in toxicology. 2009;39:76–94. doi: 10.1080/10408440802209804. [DOI] [PubMed] [Google Scholar]
- 16.Peden-Adams MM, et al. Suppression of humoral immunity in mice following exposure to perfluorooctane sulfonate. Toxicological sciences: an official journal of the Society of Toxicology. 2008;104:144–154. doi: 10.1093/toxsci/kfn059. [DOI] [PubMed] [Google Scholar]
- 17.Collins JW, et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nature reviews. Microbiology. 2014;12:612–623. doi: 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 18.Borenshtein D, McBee ME, Schauer DB. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr Opin Gastroenterol. 2008;24:32–37. doi: 10.1097/MOG.0b013e3282f2b0fb. [DOI] [PubMed] [Google Scholar]
- 19.Koroleva EP, et al. Citrobacter rodentium-induced colitis: A robust model to study mucosal immune responses in the gut. Journal of immunological methods. 2015;421:61–72. doi: 10.1016/j.jim.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol. 2007;19:377–382. doi: 10.1016/j.smim.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu, J. et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity36, 92–104, doi:10.1016/j.immuni.2011.11.011 S1074-7613(11)00505-X [pii] (2012). [DOI] [PMC free article] [PubMed]
- 25.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 26.Ishigame H, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Takatori H, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. The Journal of experimental medicine. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van de Pavert SA, Vivier E. Differentiation and function of group 3 innate lymphoid cells, from embryo to adult. International immunology. 2016;28:35–42. doi: 10.1093/intimm/dxv052. [DOI] [PubMed] [Google Scholar]
- 30.Qiu J, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiss, E. A. et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science334, 1561–1565, doi:10.1126/science.1214914 science.1214914 [pii] (2011). [DOI] [PubMed]
- 32.Lee, J. S. et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature immunology13, 144–151, doi:10.1038/ni.2187 ni.2187 [pii] (2012). [DOI] [PMC free article] [PubMed]
- 33.Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. The Journal of experimental medicine. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veldhoen M, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long M, Ghisari M, Bonefeld-Jorgensen EC. Effects of perfluoroalkyl acids on the function of the thyroid hormone and the aryl hydrocarbon receptor. Environmental science and pollution research international. 2013;20:8045–8056. doi: 10.1007/s11356-013-1628-7. [DOI] [PubMed] [Google Scholar]
- 38.Kamada N, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawa, S. et al. RORgammat(+) innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nature immunology12, 320–326, doi:ni.2002 [pii]10.1038/ni.2002 (2011). [DOI] [PubMed]
- 40.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 41.Longman RS, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. The Journal of experimental medicine. 2014;211:1571–1583. doi: 10.1084/jem.20140678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackos AR, Eubank TD, Parry NM, Bailey MT. Probiotic Lactobacillus reuteri attenuates the stressor-enhanced severity of Citrobacter rodentium infection. Infection and immunity. 2013;81:3253–3263. doi: 10.1128/IAI.00278-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson-Henry KC, et al. Amelioration of the effects of Citrobacter rodentium infection in mice by pretreatment with probiotics. The Journal of infectious diseases. 2005;191:2106–2117. doi: 10.1086/430318. [DOI] [PubMed] [Google Scholar]
- 44.Mackos AR, et al. Social stress-enhanced severity of Citrobacter rodentium-induced colitis is CCL2-dependent and attenuated by probiotic Lactobacillus reuteri. Mucosal immunology. 2016;9:515–526. doi: 10.1038/mi.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vong L, et al. Selective enrichment of commensal gut bacteria protects against Citrobacter rodentium-induced colitis. American journal of physiology. Gastrointestinal and liver physiology. 2015;309:G181–192. doi: 10.1152/ajpgi.00053.2015. [DOI] [PubMed] [Google Scholar]
- 46.Gil-Cruz C, et al. Fibroblastic reticular cells regulate intestinal inflammation via IL-15-mediated control of group 1 ILCs. Nature immunology. 2016;17:1388–1396. doi: 10.1038/ni.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann C, et al. Community-wide response of the gut microbiota to enteropathogenic Citrobacter rodentium infection revealed by deep sequencing. Infection and immunity. 2009;77:4668–4678. doi: 10.1128/IAI.00493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferreira PC, et al. Immunization of mice with Lactobacillus casei expressing a beta-intimin fragment reduces intestinal colonization by Citrobacter rodentium. Clinical and vaccine immunology: CVI. 2011;18:1823–1833. doi: 10.1128/CVI.05262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu, H. Y. et al. Effects of Lactobacillus johnsonii and Lactobacillus reuteri on gut barrier function and heat shock proteins in intestinal porcine epithelial cells. Physiological reports3, doi:10.14814/phy2.12355 (2015). [DOI] [PMC free article] [PubMed]
- 50.Bergstrom KS, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS pathogens. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morampudi V, et al. The goblet cell-derived mediator RELM-beta drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal immunology. 2016;9:1218–1233. doi: 10.1038/mi.2015.140. [DOI] [PubMed] [Google Scholar]
- 52.Chan JM, et al. CD4+ T cells drive goblet cell depletion during Citrobacter rodentium infection. Infection and immunity. 2013;81:4649–4658. doi: 10.1128/IAI.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflammatory bowel diseases. 2006;12:382–388. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 54.Fletcher T, et al. Associations between PFOA, PFOS and changes in the expression of genes involved in cholesterol metabolism in humans. Environment international. 2013;57-58:2–10. doi: 10.1016/j.envint.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 55.Nelson JW, Hatch EE, Webster TF. Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population. Environmental health perspectives. 2010;118:197–202. doi: 10.1289/ehp.0901165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Artis D, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13596–13600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vonarbourg, C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity33, 736–751, doi:S1074-7613(10)00403-6 [pii]10.1016/j.immuni.2010.10.017 (2010). [DOI] [PMC free article] [PubMed]
- 58.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 59.Eberl G, et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nature immunology. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 60.Burkett PR, Meyer zu Horste G, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. The Journal of clinical investigation. 2015;125:2211–2219. doi: 10.1172/JCI78085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patel DD, Kuchroo VK. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity. 2015;43:1040–1051. doi: 10.1016/j.immuni.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 62.Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17858–17863. doi: 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat + cells. Science. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.